1. Introduction
The first hybrid cyclolinear (CL) ladder polyphenylsilsesquioxanes (LPPSSO) with unique physical and chemical characteristics were synthesized more than 50 years ago [
1,
2,
3,
4]. In one of the articles, the authors assumed that
cis-
anti-
cis-tetraphenylcyclolsiloxane formed CL LPPSSO with the
cis-syndiotactic “double-chain” structure, while the
cis-
syn-
cis isomer under equilibrium polymerization gave cage-like structures consisting of 8–12 PhSiO
1.5 groups [
5]. This indicates that steric parameters of a monomer can substantially affect the results of polymerization or polycondensation processes of organocyclosiloxanes. Due to the hydrodynamic and electro-optical characteristics of LPPSSO solutions, the differences in the α-values in the Mark–Kuhn–Houwink equation and the Kuhn segment were revealed depending on the ladder polymer structure, production method, organic substituents at the silicon atoms, and the structure of the prepolymer [
6,
7,
8,
9]. Despite the fact that 50 years have passed, interest in the steric characteristics of the polymers, these hybrid CL LPPSSO and other CL ladder polyorganosilsesquioxanes (LPOSSO), persists. This is because LPOSSO are known to be highly thermostable, to possess good mechanical parameters, and to be capable of forming thin films [
1,
7,
10]. These properties apparently depend on the structure and the steric characteristics of the polymers.
Stereoregular LPOSSO were synthesized using organotrifunctional silanes (organotrichloro- or organotrialkoxysilanes) [
10,
11,
12].
Cis-isotactic CL LPPSO were prepared by polycondensation of 1,1,3,3-dihydroxy-1,3-diphenyl-1,3-disiloxane either on a glass surface or in acetonitrile solution [
13]. The authors also obtained
cis-isotactic LPOSSO via polycondensation of 1,1,3,3-tetrahydroxy-1,3-diorgano-1,3-disiloxane in the presence of Me
4NOH as a catalyst, a π-stacking and H-bonding superstructure of the monomer promoting the reaction [
14]. Polycondensation of
cis-
trans-
cis-tetrahydroxytetramethylcyclosiloxane in the presence of K
2CO
3 furnished
cis-syndiotactic polymethylsesquioxanes with the MeSIOH groups located in the chain between the polymethylsilsesquioxane blocks [
15].
The processes of polycondensation of
cis-
anti-
cis-(tetrahydroxy)(tetramethyl)cyclotetrasiloxane and all-
cis-tetrahydroxy)(tetravinyl)cyclotetrasiloxane were also investigated [
15,
16]. Attention was paid to the study of the influence of the conditions of polycondensation and stepwise coupling polymerization of these organocyclotetrasiloxanes upon the composition and structure of the CL polymethylsilsesquioxanes and ladder polyvinylsilsesquioxanes (LPViSSO) formed [
17,
18,
19]. The reactions of stepwise condensation of 2,4,6,8-(tetrahydroxy)(tetra-
i-propyl)cyclotetrasiloxanes and 1,3-dichloro-1,3-di-
i-propyl-1,3-diphenyldisiloxane were employed to obtain stereoregular penta-, hepta-, and nona-cycle ladder siloxanes [
20,
21,
22].
We recently reported polycondensation of all four isomers: all-
cis,
cis-
trans-
cis-,
cis-
cis-
trans-, and all-
trans-(tetrahydroxy)(tetraphenyl)cyclotetrasiloxanes [PhSi(OH)O]
4 by another method, namely, in the presence of layered-architecture compounds. The formation of LPPSSO with different unit structures and column-type cyclolinear polyphenylsilsesquioxanes was observed [
23,
24]. For example, by means of copolycondensation of the all-
cis- and
cis-
trans-
cis-stereoisomers of [PhSi(OH)O]
4 in solution in the presence of montmorillonite (MMT) in various ratios to the isomers, LPPSSO copolymers were obtained [
25] Only two isomers of the four possible isomers, all-
cis- and
cis-
trans-
cis-[PhSi(OH)O]
4, out formed the
cis-isotactic and
cis-syndiotactic CL PPSSO structures, respectively. However, in the case of the all-
cis isomer when the reactions were conducted both with and without MMT cage-like oligomers also formed. With that, the
cis-
trans-
cis isomer formed CL LPPSO with a higher M
w than the all-
cis one. The all-trans isomer furnished column-type CL PSSO (
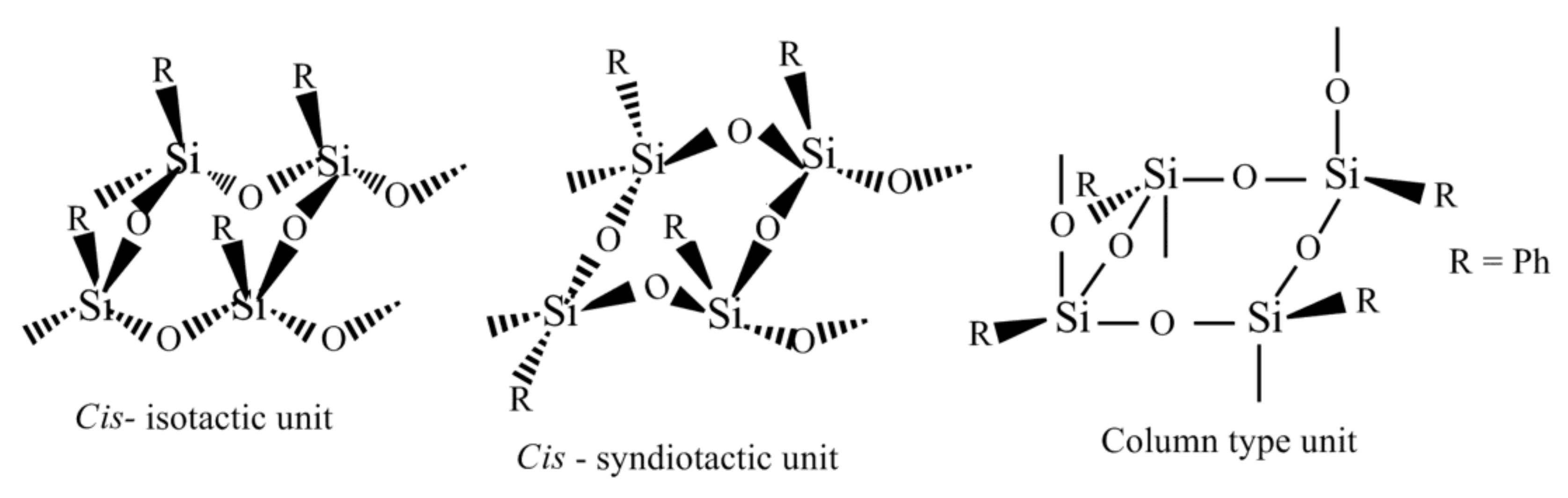
Figure 1).
Thus, the configuration of the starting monomer affected the structure of the units of the polymer formed. The introduction of a substituent into the phenyl ring can impact on the configuration in which the starting (tetrahydroxy)(tetraaryl)cyclotetrasiloxane is formed when synthesized and thus can influence the structures of oligomers and polymers obtained via the polycondensation reaction. It is especially so when the reaction implies intercalation of the monomer into a layered-architecture compound like MMT.
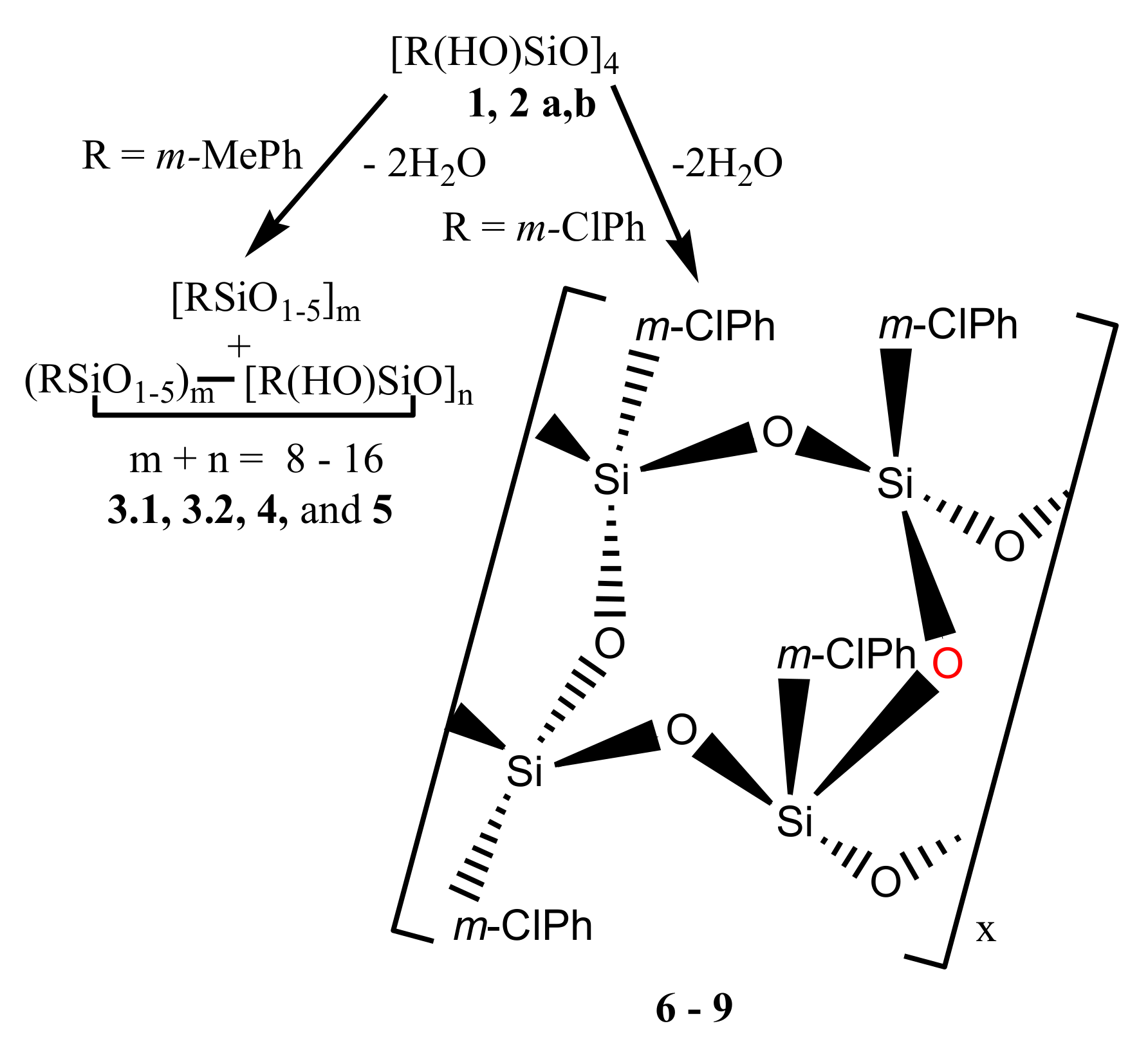
With this in mind, we have synthesized two compounds, stereoisomers of 2,4,6,8-tetrahydroxy-2,4,6,8-tetra-m-tolyl- and 2,4,6,8-tetrahydroxy-2,4,6,8-tetra-m-chlorophenyl-cyclotetrasiloxanes (1 and 2a, respectively) containing substituents at phenyls of different types, donating (Me) and mixed (Cl), and report here on their polycondensation reaction performed under different conditions and in the presence and absence of MMT. A mixture of two isomers of the latter compound (2b) was also prepared and polycondensation of it was investigated.
3. Results and Discussion
Polycondensations of compounds
1,
2a, and
2b were carried out in equilibrium (under atmospheric pressure) and non-equilibrium (in vacuum) conditions in toluene, anisole or ditolylmethane in quartz or molybdenum glass flasks in the absence and in the presence of MMT (see
Note 1 in the SM).
The reactions were monitored by GPC, PI APCI MS, IR, and 1H NMR spectroscopy methods.
Products
3.1,
3.2, and
4 obtained after the same time period of 2 h from polycondensation of
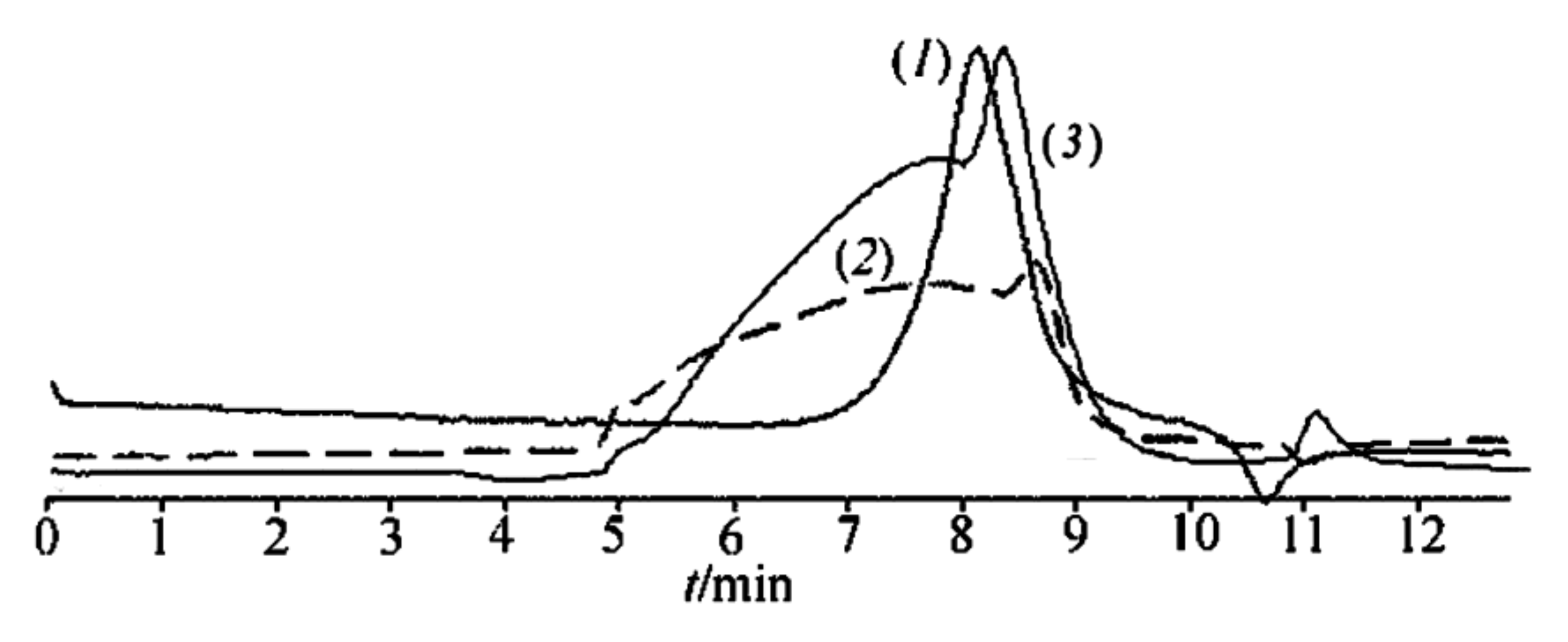
1 in toluene solution at atmospheric pressure or in vacuum in ditolylmethane, respectively, had similar values of molecular weight
Mw = 1900–2800 and
Mw/
Mn = 1.2–1.9 according to GPC (
Figure 2). For product
5 obtained in a molybdenum glass flask at atmospheric pressure in the presence of MMT, the same above values were observed (see
Figure 2, curve (
1) for the experimental GPC track). These data show that the reactions of polycondensation of
1 in equilibrium and non-equilibrium conditions afforded oligomeric products.
These results are in good agreement with those previously reported for polycondensation of the all-
cis-isomer of [PhSi(OH)O]
4 under analogous conditions in the absence of MMT [
23].
Moreover, curve (
2) in
Figure 2 totally coincided in shape and molecular weight interval with that for CL LPPSSO obtained from polycondensation of
cis-
trans-
cis-[Ph(OH)SiO]
4 in anisole in the presence of MMT [
23]. This provided an additional argument in favor of the aforesaid suggestion that
2a was the
cis-
trans-
cis-isomer.
Polycondensation of compound 2a in the absence of MMT under both equilibrium and non-equilibrium conditions led to polymers 6 and 7, respectively, with Mw = 5200; Mw/Mn = 1.7.
In contrast to these products, polymer
8 obtained by polycondensation of compound
2a in anisole in the presence of 29% MMT had
Mw in the range 2000–75,000 after the same time period of 2 h (
Figure 2, curve (
2)). Polycondensation of compounds
2b under identical conditions over the same time period led to the formation of polymer
9 (see
Figure 2, curve (
3) for the experimental GPC track). A comparison of curves (
2) and (
3) in
Figure 2 showed that the use of the
2b mixture for thermal polycondensation led to an increase in the weight fraction of the oligomeric product and to a slight decrease in the molecular weight of the polymer. Moreover, the presence in
2b of another isomer with
Rf = 0.05 besides that with
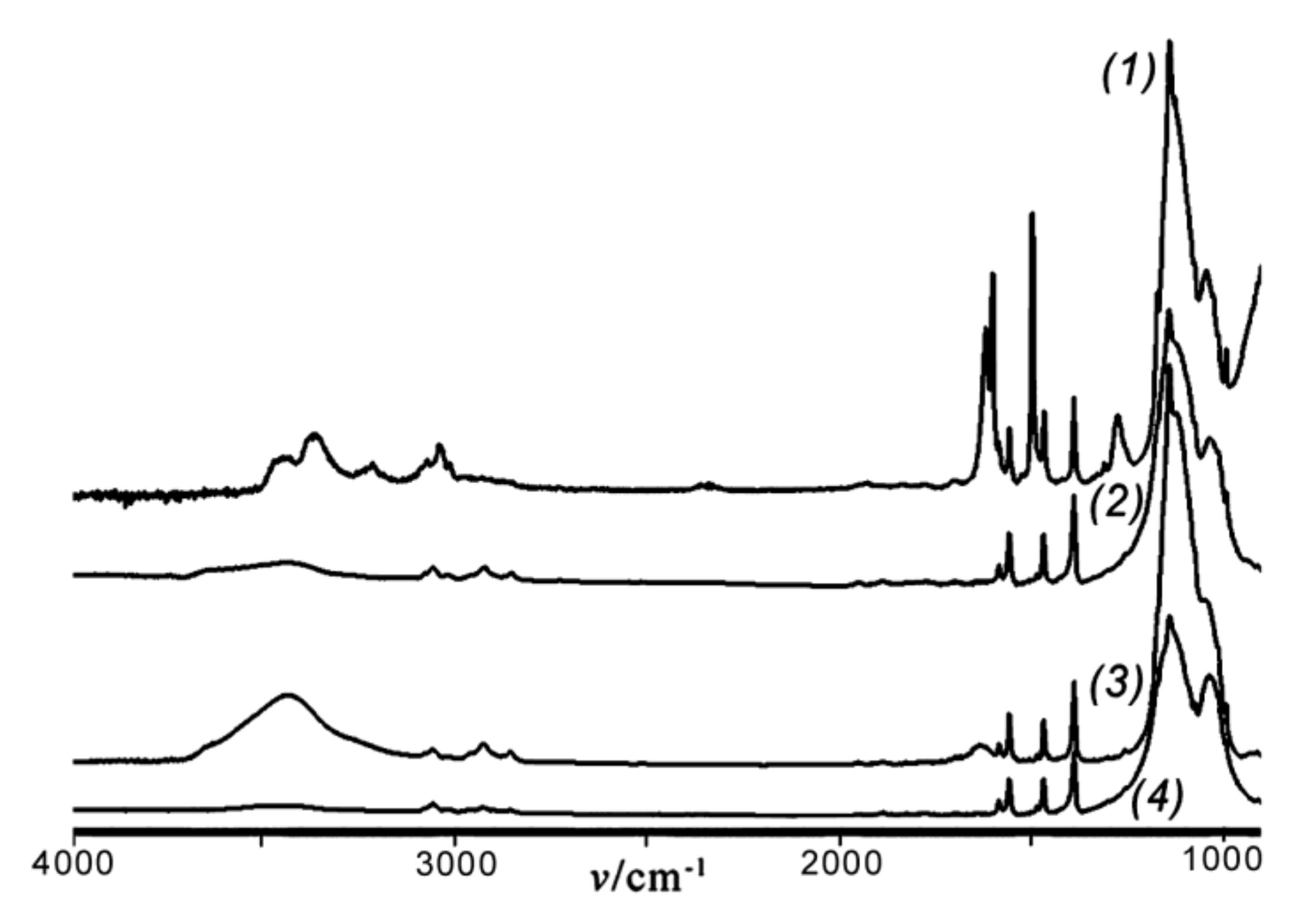
Rf = 0.50 (see the Experimental section) gave rise to the formation of a cyclolinear ladder polymer with different configuration of the units. This is seen from the IR spectra of polymers
8 and
9 ((
2) and (
3) in
Figure 3, respectively), the different configuration of the units in
9 as compared to that of
8 being indicated by different forms of (
3) and (
2) in the 1070–1000 cm
−1 region. As for polymer
8, the IR spectrum of polymer
7 (curve (
1) in
Figure 3) obtained by polycondensation of
2a in vacuum and in the absence of MMT displays two bands at 1050~1055 and 1150~1155 cm
−1 which indicate the formation of CL PClPSSO also in this case.
No reaction was observed, when 2a was heated with MMT at 110 °C without solvent. Low-molecular-weight polymers were obtained from the reaction in toluene at 110 °C in the presence of 5–20% w/w % of MMT in respect to 2a. Only when the addition of MMT was increased to 29%, the polycondensation (in anisole at 110 °C) afforded polymer 8 with Mw up to 75,000 (see above). Thus, the reaction in the presence of these amounts of MMT allowed obtaining a polymer the molecular weight of which was higher than the molecular weight of polymer 6 obtained in the absence of MMT.
The role of MMT lay probably in the fact that intercalation of hydroxycyclotetrasiloxane 2a from the solution into galleries between MMT layers resulted in the disruption of intermolecular hydrogen bonds in 2a. This led to self-organization of the 2a molecules on the MMT surface that facilitated polycondensation, thus providing the increased length of the polymer chain in 8 as compared with that in 6.
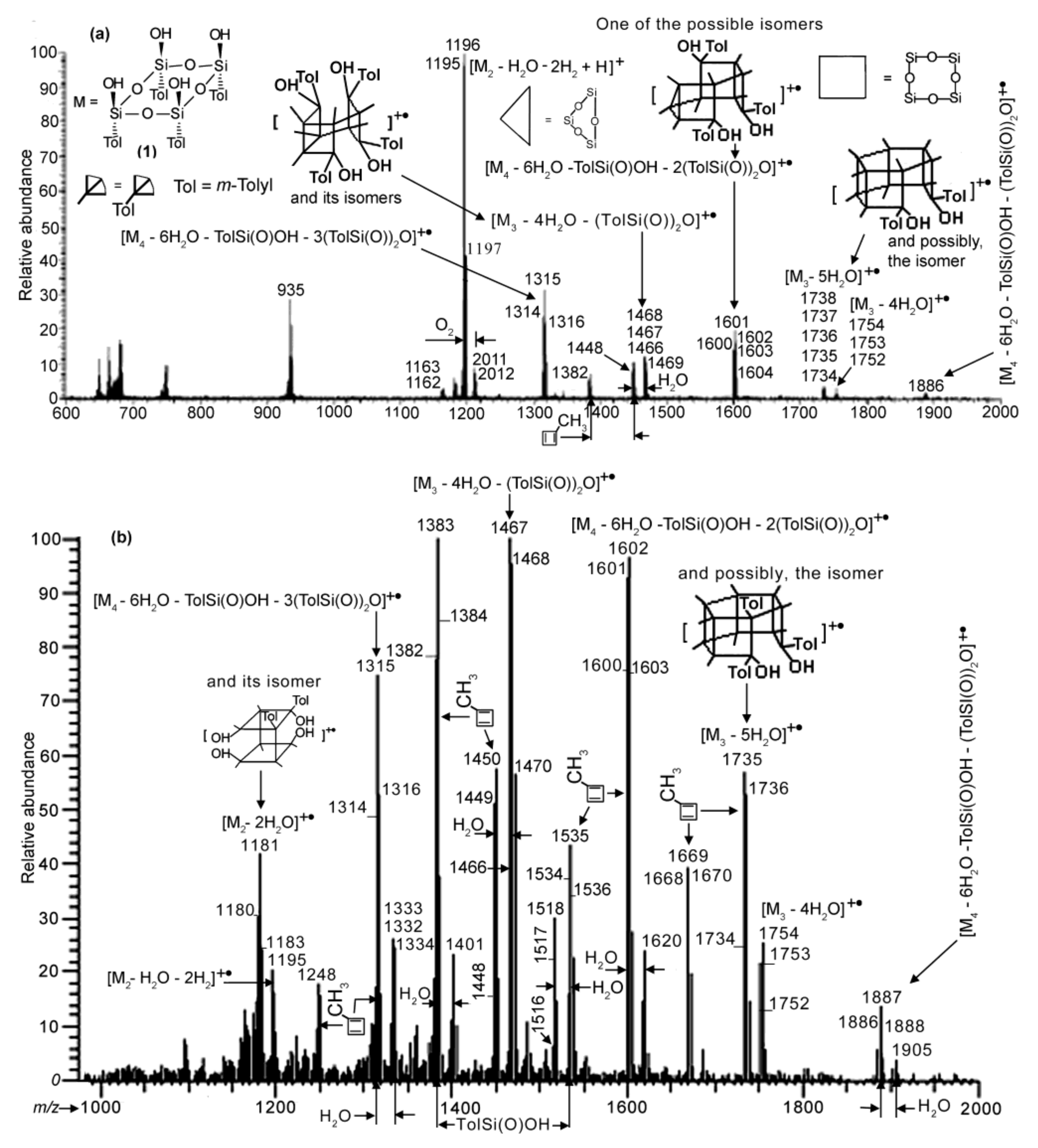
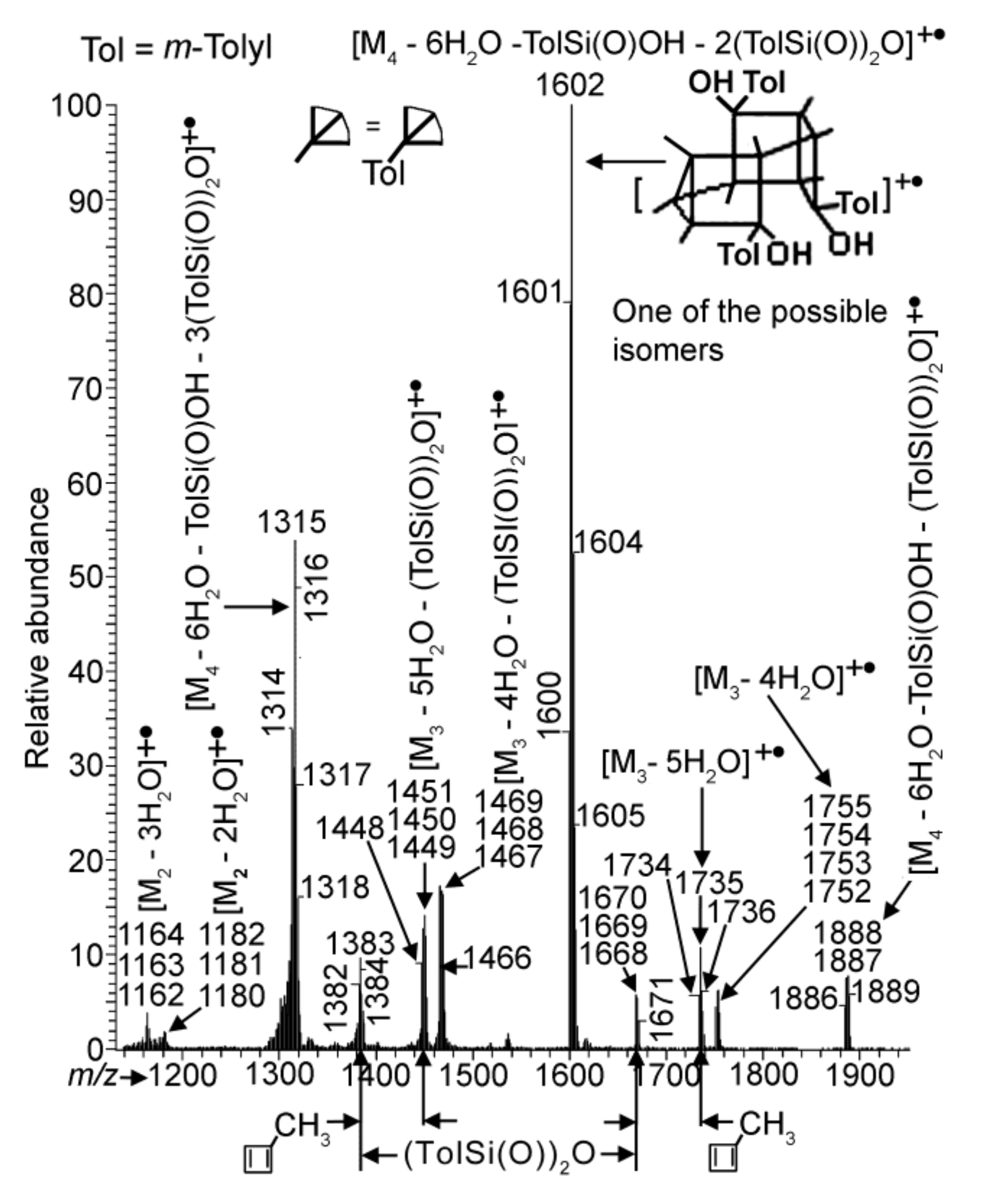
As mentioned above, polycondensation of compound 1 under different conditions (at atmospheric pressure or in vacuum with or without MMT) was monitored by APCI MS.
Figure 4 and
Figure 5 demonstrate the APCI mass spectra of products
3.2,
3.1 and
5 obtained after heating of compound
1 in quartz and molybdenum glass flasks, and in a molybdenum glass flask in the presence of MMT, respectively, all at atmospheric pressure and 110 °C for comparable periods of time. All three spectra show species formed via condensation between two and four molecules of compound
1. With that, the spectra demonstrate the loss of (TolSi(O))
2O (Tol =
m-tolyl) and sometimes of TolSi(O)OH, the former being preliminarily reported for the phenyl analogue [
17,
24]. This loss most likely occurred in the course of the reactions rather than in the mass spectrometer as fragmentations. Ion peaks corresponding to compounds with the nominal masses 1180 Da for
3.1 and 1162 and 1180 Da for
5 appeared owing to the condensation of two molecules of
1 (single condensation) could also be considered as belonging to the species formed via the decomposition of double condensation products (products of condensation of three molecules of
1) according the schemes (M
3 − 5H
2O) − 2(TolSi(O))
2O and (M
3 − 4H
2O) − 2(TolSi(O))
2O. However, the relative abundances of the peak group with the
m/
z 1180 ion decreased strongly in time (
cf. those in
Figure S2a,b in the SM, and
Figure 4b). This suggests that the compounds discussed were formed via the condensation of two molecules of
1.
Nevertheless, spectra (a) and (b) of
Figure 4 and that of
Figure 5 differ from each other. In the first spectrum, the most abundant group of peaks is at
m/
z 1195–1197. It belongs to a species formed due to the single condensation process, while ion peaks of the species obtained via double condensation and condensation of four molecules of
1 (triple condensation) are significantly less intensive. Moreover,
spectra S1a–c in the SM and spectrum 4a give the formation dynamics of product
3.2. In the spectrum recorded after heating of
1 for 30 min, the most abundant peaks turned out to be those of species formed owing to the triple condensation process (at
m/
z 1315 and 1601). When
1 was heated for 60 min, a peak at
m/
z 1196 of a single condensation product became maximal and among the less-abundant peaks of the double and triple condensation species the peak of the former species at
m/
z 1383 was the greatest in intensity. This pattern was retained for the 90 min spectrum, while at last after 130 min, as mentioned above, the single condensation species became strongly dominant (see
Figure 4a). A reasonable explanation for such a behavior is that, initially, the reaction was kinetically controlled. Then, after water accumulated in sufficient amounts in the reaction mixture due to the removal from the reacting species, the thermodynamic control came into play with the single condensation species as the major product.
Another situation was realized when the reaction was carried out in a molybdenum glass flask. It was previously shown that polymerization (and apparently, condensation) could be catalyzed by ions from glass [
30,
31]. Probably, the equilibrium was achieved here at higher rates of the direct reactions and as a result, double and triple condensation products proved to prevail (see
Figure 4b and
Figure S2a,b in the SM).
For the reaction in the molybdenum glass flask in the presence of MMT, the following should be highlighted. The process gave species of triple condensation as the main components of the overall product
5 (
Figure 5). Contrary to spectrum (b) of
Figure 4 (for the reaction without MMT), spectrum 5 exhibits peaks of only two relatively abundant ionic groups, one with the nominal mass ion 1314 Da and another with 1600 Da, peaks of all other ionic groups being significantly less intensive. The most abundant group in the spectrum is of 1600 Da ion, and a possible structure of the corresponding compound is furnished in
Figure 5. It is worth noting that no essential changes were found when the reaction in the presence of MMT was conducted in anisole at 150 °C for 120 min (see
Figure S3a in the SM).
An interesting point of the spectra is worth discussion here. Monomer
1 was registered in the PI APCI spectrum as (M + H
2O)
+• rather than M
+• (see the Experimental section), i.e., water was added immediately in the mass spectrometer. Whether or not this is valid for other compounds registered remains questionable. To throw some light on this, product
5 was treated with an excess of Me
3SiCl and a mass spectrum recorded (
Figure S3b in the SM). A shift by 72 Da to the higher masses was observed for the ionic groups with the nominal mass ions of 1314 and 1600 Da to give those of 1386 and 1672 Da. This indicated that the corresponding compounds contained a free OH group. At the same time, the groups with the nominal mass ions 1448 and 1734 proved to be unshifted. This speaks in favor of these compounds being closed-cage species bearing no OH groups, and water was added during the mass spectral analysis. The corresponding ions could have either hydrate structures: [((M
3 − 6H
2O) − (TolSi(O))
2O) + H
2O]
+• and [(M
3 − 6H
2O) + H
2O]
+•, respectively, or hydrolysis of a Si–O bond occurred to furnish [M
3 − 5H
2O − (TolSi(O))
2O)]
+• and [M
3 − 5H
2O]
+• with two adjacent OH groups. The same is probably valid for many other ions. For simplicity, the second variant is mostly employed in the labels in the mass spectra and the structures depicted therein.
As could be anticipated, the reaction conducted in a vacuum (under non-equilibrium conditions) afforded a mixture of compounds formed due to single, double, and triple condensations of
1, the two last-mentioned groups prevailing with many their components being of relatively high abundance in the APCI mass spectra (see
Figure S4 in the SM). Another interesting point is that an ionic group containing the nominal mass ion of 1734 Da is of a pronounced abundance in all vacuum reaction spectra. This belongs to a species (M
3 − 5H
2O). In all other APCI mass spectra its peaks turned out to be of relatively less abundance. This is consistent with the fact that the decomposition of the compound occurred with the participation of water, whereas most of the evolving water was removed from the reaction in the vacuum process.
Peaks of the compound with the nominal mass of 1448 Da are also of interest. In
Figure 4 and
Figure 5 they are not among the most abundant. However, when the reaction was performed in a vacuum, this compound turned out to be dominant (see
Figure S4 in the SM). As was revealed before, it could most probably have a closed-cage structure with a water molecule added in the course of the analysis. Moreover, the corresponding radical cation could again be either a closed-cage hydrate, or an open-cage species with two OH groups formed due to hydrolysis. Both variants are depicted in
Figure S4c,d in the SM (also, see
Note 3 in the SM).
Unfortunately, our attempts to collect the APCI mass spectra for the products of the polycondensation of compound
2 failed (see
Note 4 in the SM).
The IR spectra of products
3.1,
4, and
5 (
Figure 6) exhibited an absorption band at 1123 cm
−1 with a width ∆
ν1/2 of 200 cm
−1 in the region of asymmetrical stretching vibrations
νas of the Si–O–Si bond. The IR spectra of products
3.1 and
4 also exhibited noticeable OH bands at 3400–3600 cm
−1, while that of
5 was less pronounced. This supported the above MS results of the presence in these products of open-cage compounds with OH groups. Moreover, this is in good agreement with the findings that cage-like organosilsesquioxanes were characterized by an absorption band at 1122–1128 cm
−1 [
5].
In contrast to the IR spectra of products
3.1,
4 and
5, those of polymers
7 and
8 obtained via polycondensation of compound
2a exhibited two absorption bands
νas of the Si–O–Si bonds at 1037–1040 cm
−1 and 1140–1143 cm
−1 with ∆
ν1/2 of ~200 cm
−1 (
Figure 3). The spectra of
6,
7, and
9 showed OH absorption bands at 3300–3600 cm
−1, whereas that of polymer
8 did not.
The
29Si NMR spectra of products
3.2,
4, and
5, and polymers
8 and
9 were characterized by signals in two regions at δ −67.0~−71.0 (excluding
5) and at δ −76.0~−82.0 ppm with different intensities and half-widths (
Figure 7). The spectra of polymers
6 and
7 were similar to that of
8. For products
3.2 and
4, multiplets at δ −67.0~−71.0 ppm were weak in intensity, whereas those at δ −76.0~−82.0 ppm were more intensive and very broad. The
29Si NMR spectrum of product
5 totally coincided with the
29Si NMR spectrum of the products obtained by polycondensation of all-
cis-2,4,6,8-tetrahydroxy-2,4,6,8-tetraphenylcyclotetrasiloxane [
25]. Note, the above fact supported the assumption that compound
1 was all-
cis-[(Tol(HO)SiO]
4.
The
29Si NMR spectra of polymers
6–
8 in the δ region of −76.0~−82.0 ppm were characterized by a single signal at δ −79.9 ppm with ∆δ
1/2 = 3.0 ppm. The signals at δ −67.0~−71.0 ppm specific for ClC
6H
4Si(OH)O units were present but were very small in the spectra of polymers
8 and
9 (
Figure 7b).
Based on the data of
29Si NMR, IR, and mass spectra, polycondensation of compounds
1 and
2a,
b took place according to
Scheme 1 with the formation of products
3.1,
3.2,
4, and
5 and polymers
6–
9.
A comparison was made of the polymer
8 unit configuration order with that of CL ladder poly-
m-chlorophenylsilsesquioxanes (LPClPSSO) (
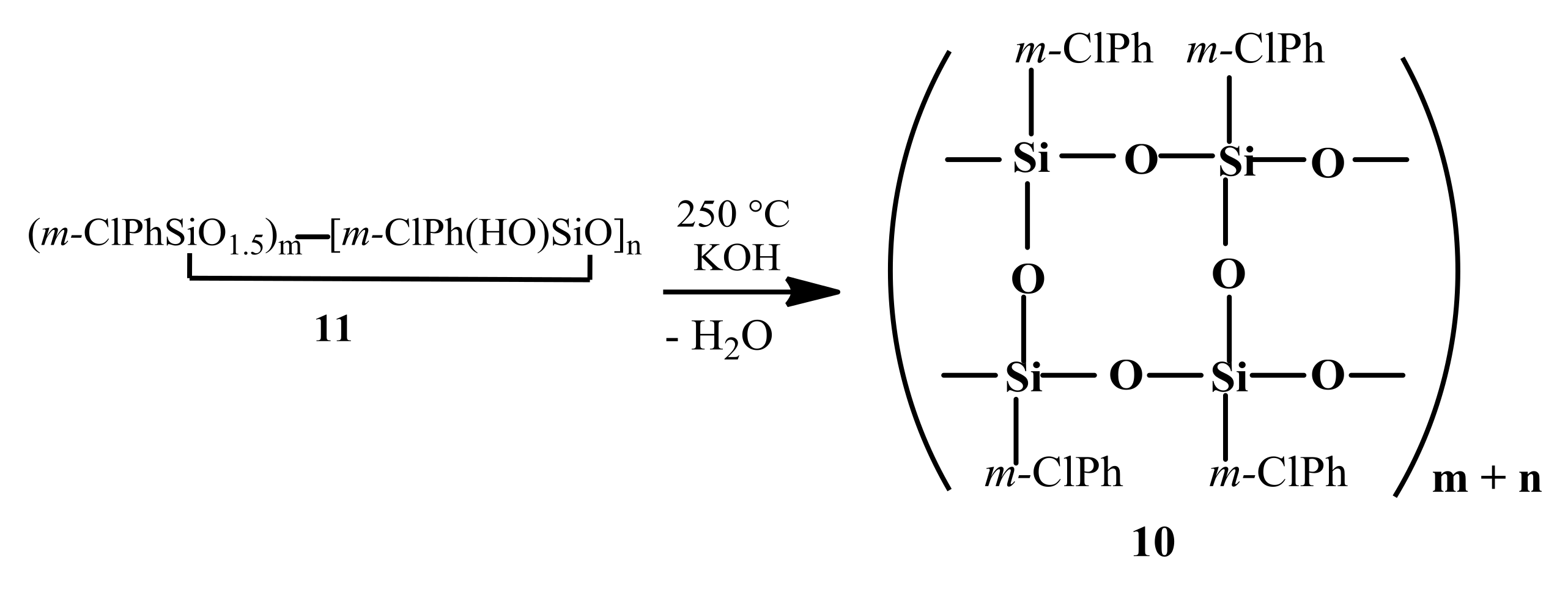
10). This polymer was synthesized by anionic polymerization in the presence of KOH at 250 °C of a prepolymer (
11) obtained by hydrolysis of
m-chlorophenyltrichlorosilane (
Scheme 2) [
1].
After reprecipitation, polymer
10 had
Mw = 5.66 × 10
5,
Mn = 1.15 × 10
5, and
Mw/
Mn = 4.90; [η] = 1.40 dL·g
−1 (20 °C, benzene). The IR spectrum of polymer
10 was characterized by two bands at 1050~1055 and 1140~1145 cm
−1 with comparable intensities (
Figure 3, curve (
4)). This confirmed a CL structure of the main siloxane chain in
10. It is noteworthy that no OH bands were present in the spectrum. As mentioned above, polymer
8 showed a broadened singlet at δ –79.9 ppm in its
29Si NMR spectrum. At the same time, a multiplet with the main maxima at δ −79.3 and −81.5~−82.0 ppm was observed for polymer
10 (
Figure 7b). With that, the ∆δ
1/2 values for both polymers were the same. This splitting in the case of
10 was apparently due to different unit configurations in the polymer chain. These
29Si NMR data are in good accordance with those previously published for LPPSSO copolymers obtained by copolycondensation of all-
cis- and
cis-
trans-
cis-(tetrahydroxy)(tetraphenyl)cyclotetrasiloxanes in different proportions. Their
29Si NMR spectra showed two signals with δ −78.24 and −79.06 ppm [
25].
Thus, sequences of units having different configurations were present in the structures of LPClPSSO synthesized by anionic polymerization, while the configuration of 2a was retained throughout product 8 since only one singlet was detected in the 29Si NMR spectrum of this polymer.
Powder X-ray diffraction was used to determine the inter-chain distance (d1) in polymer 8. It gave d1 = 14.40 Å. This value agrees with the data for low and high molecular-mass LPClPSSO obtained by anionic polymerization. However, this method is inappropriate for detecting configurational differences between chain units of CL LPClPSSO 8 and 10.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








