1. Introduction
Polymer composite materials, or fiber-reinforced plastics, are rapidly being introduced into many areas of our lives and dramatically changing all constructional material industries [
1]. The demands placed on filler materials are more-or-less understandable (they should be lightweight and strong, such as carbon fiber). At the same time, the properties of polymer matrices are much more sophisticated, and their influence is often underestimated. Nevertheless, matrix mechanical properties and their coupling with filler stiffness (which is usually hard to change) primarily determine the overall composite properties for the consumer, including static and dynamic moduli, elastic and brittle behavior, etc. Moreover, on the practical side, it is much easier to tune matrices than it is the filler, because the filler is mostly “as is” material produced by very few manufacturers on the basis of closed technologies [
2]. The other two important contributors to composite material properties are the filler arrangement inside the matrix and filler–matrix interface properties, which are also very sophisticated and widely studied nowadays, but these are out of the scope of our research.
The polymer matrix itself is a percolated three-dimensional network of monomer units connected by covalent bonds, as shown in
Figure 1. These bonds are formed during a chemical polymerization reaction, usually called the curing process. In addition to monomer units, some other species can be added, like hardeners and plasticizers, to control the reaction speed and crosslink density. Depending on the monomer’s chemical structure, the nature of the polymerization reaction can be either a step-growth process (as in the crosslinking of phenolic or epoxy resins) or radical polymerization (as in the crosslinking of polyester resin or acrylic glass). The latter case requires an initiator to start the reaction and usually takes more time. In addition, radical polymerization leads to a more complex topology of the network and contains defects, like dangling ends. In this study, we consider only the case of radical polymerization, although our results are probably also applicable to step-growth polymerization, reduced by the effects of initiator concentration.
In general, there is a vast number of different chemical components which can be polymerized or copolymerized to form a resin. While the chemical nature of the monomers is very important for the details of the curing process and also controls the glass transition temperature and heat resistance, the mechanical properties depend strongly on the topology of the final polymer network. The network topology, in turn, depends on many factors (such as species composition, reaction pathways, and conversion degree) and, in most cases, is not known both a priori and a posteriori.
The influence of the polymer network topology is a well-known problem for polymer elastomers, including rubbers and especially gels [
3,
4]. Usually, the real network topology in elastomers is unknown, except for in cases in which the “network disassembly spectrometry” technique can be applied [
5]. In these cases, a straightforward comparison between the chemical topology and the theoretical model can be done [
6,
7]. However, this approach does not work for resins; thus, only a few attempts to study resin network topology are found in the recent literature [
8,
9]. The variation in polymer network crosslinker functionality and its influence on topology and mechanical properties was studied for the two-dimensional case in [
10]. The study of gel point formation for a universal polymer network model was presented recently [
11]. This model is able to account for loop formation, which is most important for curing in a swollen gel state. The closest to our work is found in [
12], wherein the authors used simulations to s study the radical polymerization of hexanediol diacrylate with varied initiator content. Here, we studied the network topology at a more detailed level and combined both the effects of low-molecular-weight plasticizer and initiator concentration.
A common and very approximate viewpoint is that, on average, a shorter distance between crosslinks (usually called “mesh size”) yields a stiffer and brittler matrix, while larger distances result in softer and more elastic materials. In this sense, it correlates with the classical polymer rubber properties, and the correspondence between soft resin (in the region above the glass transition temperature) and highly crosslinked rubber is obvious.
The phthalonitrile resin is one recent example of various modern resins [
13,
14], and it is thermally much more stable than the classic epoxy resin. It polymerizes via a complex radical process and has a complex network topology. These resins are more expensive compared with the epoxy ones but can be used to create composites with a high temperature load for such uses as turbine blades or hyper-velocity aerodynamic shrouds. With the radical polymerization process, the initiator species should be introduced to start the curing, and the initiator concentration is varied to control the curing time and to increase the final conversion. At the same time, a plasticizer can be used as an inert additive to affect the elastic modulus and fragility.
In parallel to the development of novel chemical species, computer simulation in silico experiments are attracting attention for studying and predicting various material properties, both at the atomic level and the coarse-grained level [
15]. In our recent works, we developed a multiscale simulation methodology that makes it possible to predict various physical properties of highly crosslinked polymer materials [
16,
17,
18], but there is still a lack of understanding such a network topology and the possible connections of the network topology with macroscopic properties.
Taking in mind the polymer matrix properties, let us emphasize once more that the polymer network topology is formed during the curing process, which is usually fully controlled by the final product makers. Thus, the main purpose of our research is to build a computer model and describe the influence of the curing process input parameters on the final polymer matrix topology and mechanical properties. As the input parameters, we used the initiator and plasticizer concentration, which determine the matrix network topology, followed by the corresponding mechanical properties. Because of the substantial polymer resin diversity, we considered the specific example of the phthalonitrile resin, as it is one of the most interesting and promising cases. However, the same conclusions are most likely valid for any thermoset resin.
Please note also that, in our model, there are no real defects, such as dangling ends or large dangling loops. This is because of the monomer and initiator units’ special design (see
Figure 2b), which prohibits the formation of such defects. This means that we study defect-free networks and can measure the influence of network topology solely, disregarding any dangling or other elastically inactive elements.
3. Results and Discussions
3.1. Curing Speed and Gelation Point
First, let us consider the positive effect of the initiator additive to the overall processing time. Indeed, the processing time itself is a very important feature in any industry-related system, because the time needed to leave a fabric unit in a mold form is critical to the entire manufacturing workflow.
Figure 3 gives an overall view of the influence of the initiator concentration on the curing time. We also present the simulation time to reach a 95% conversion degree versus the initiator amount in the inset.
Let us discuss, in more detail, the process of gelation and which properties of the network form near the sol–gel transition (i.e., percolation threshold).
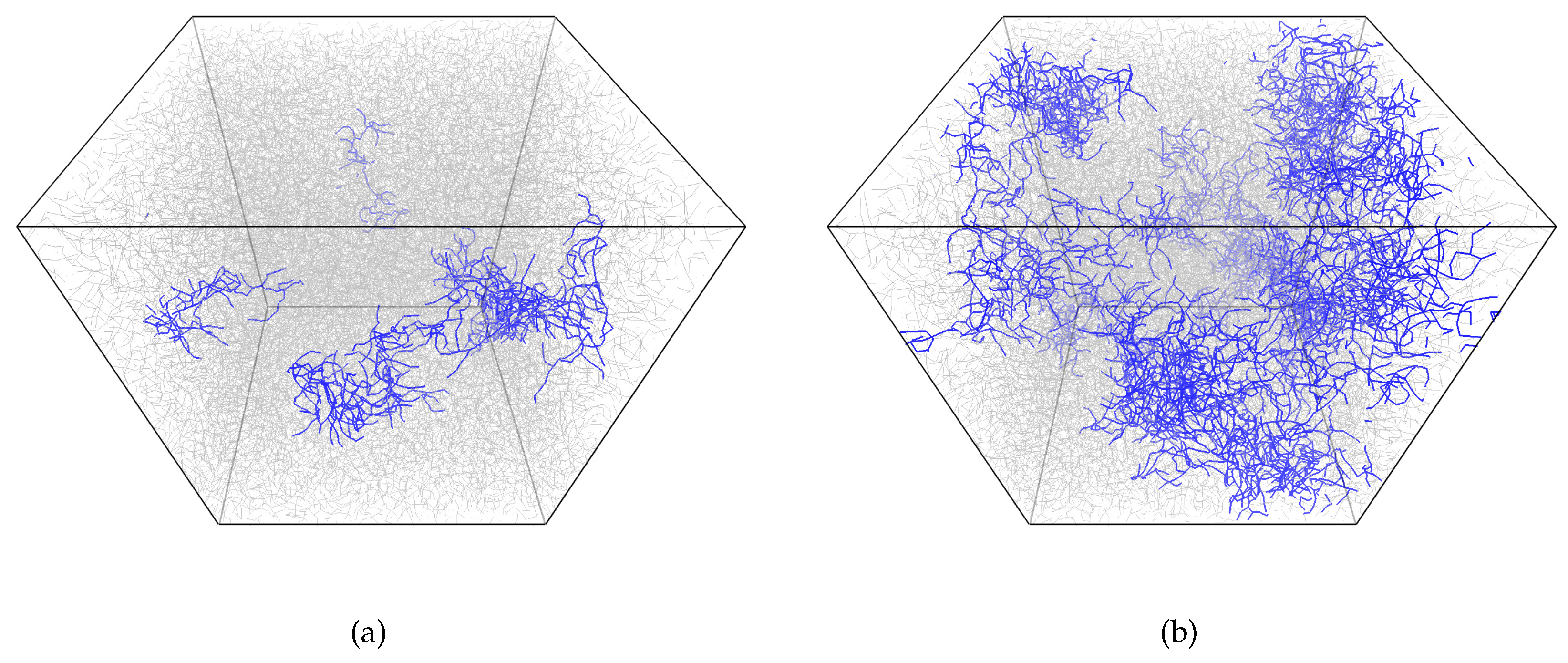
Figure 4 gives snapshots of the network structure below and above the gelation point, while
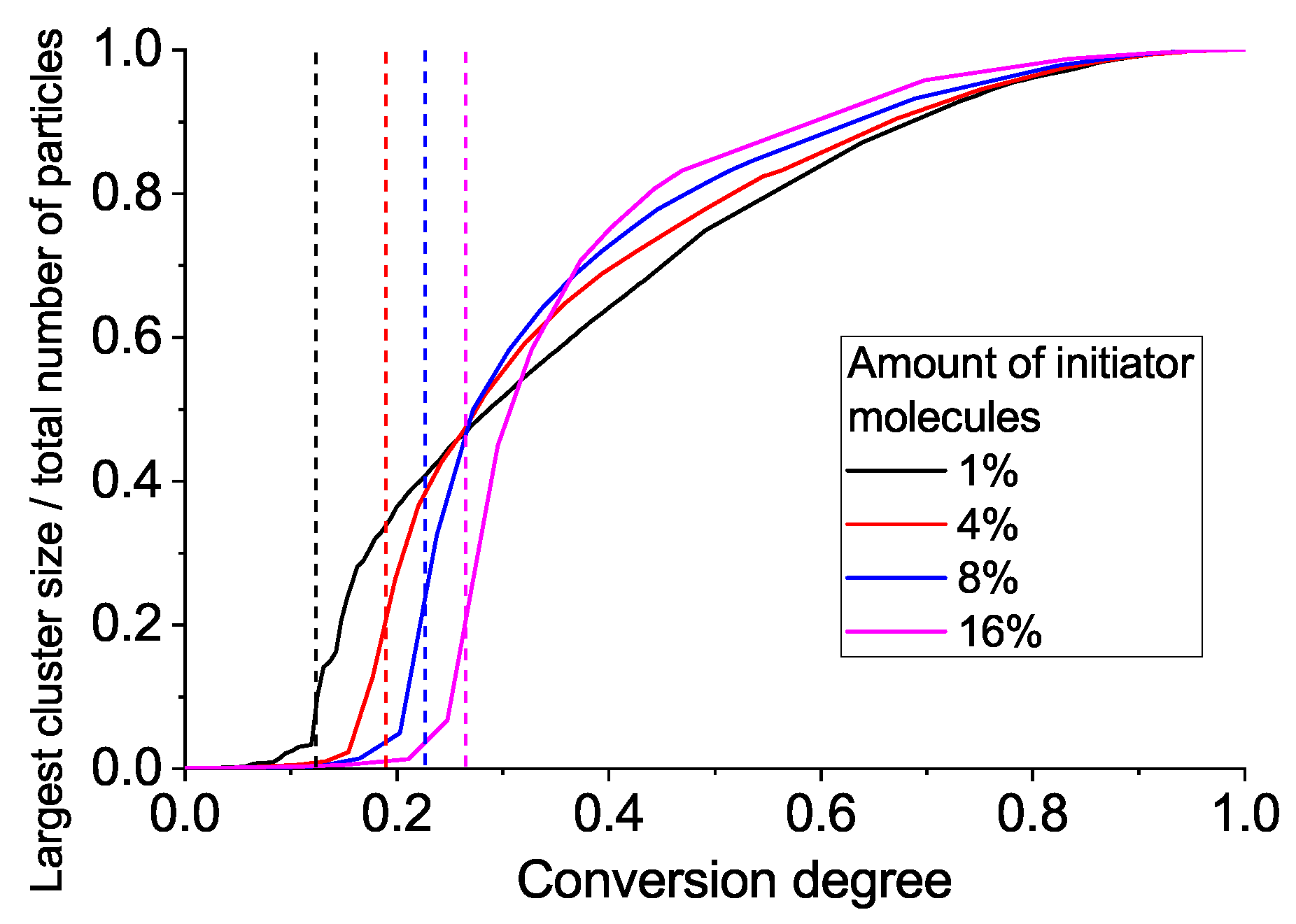
Figure 5 gives the largest cluster size versus conversion plot. We observe a very heterogeneous network with several growing clumps crosslinked with each other upon gelation. Such heterogeneity is, of course, the consequence of each monomer unit’s valence being equal to four, which leads to the formation of a very dense crosslinking network. An interesting conclusion can be made from
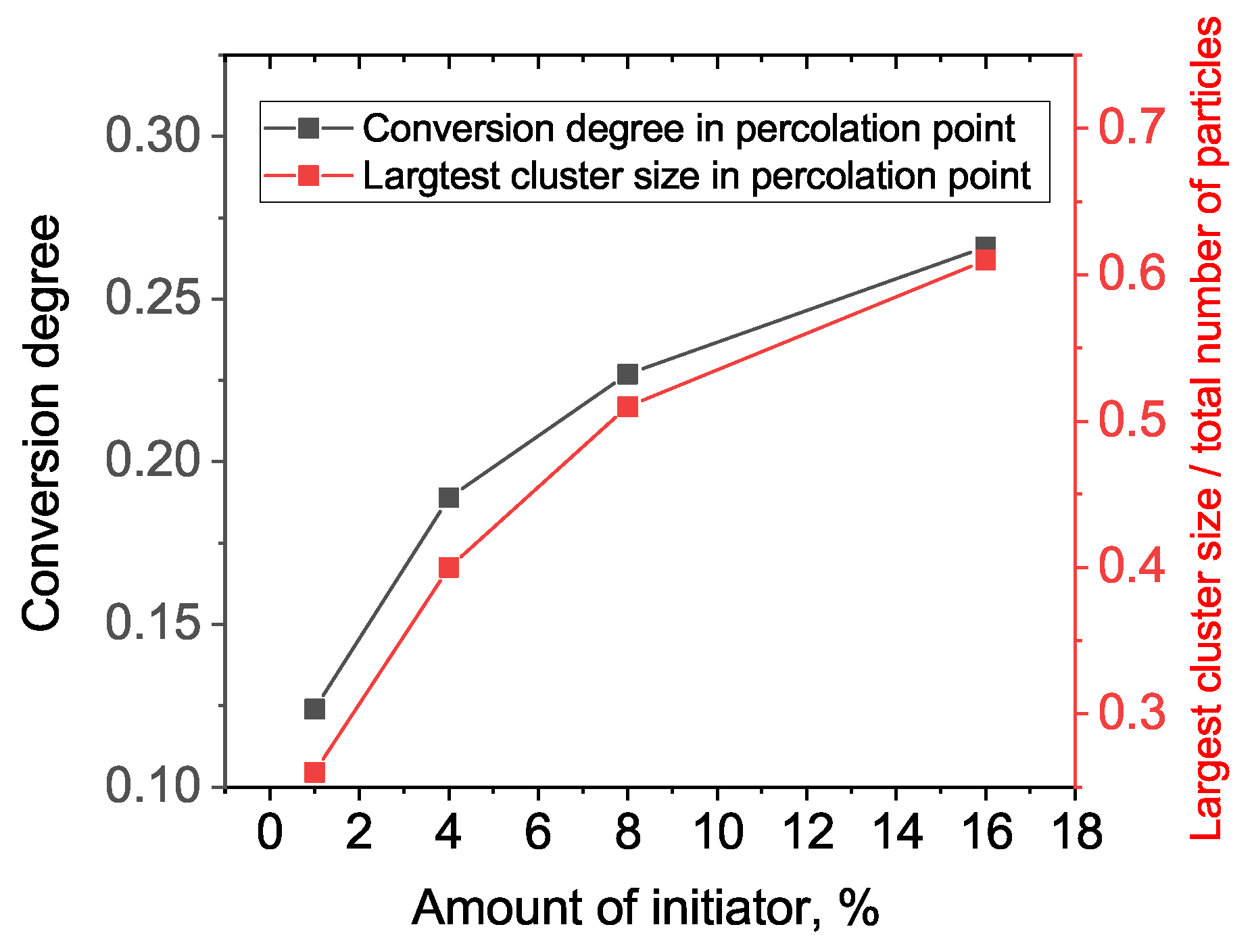
Figure 5: the system with a higher amount of initiator has a more delayed and sharp gelation transition (note: here, we talk in terms of conversion along the abscissa axis, not real time). This results in an increased conversion degree and the largest cluster size at gel point, as can be seen from
Figure 6.
Thus, we have one more important effect of the initiator, in addition to the enhancement of overall reaction speed: the more initiator we have, the greater the fraction of monomer units included in the gel fraction just after the gel point. That is, at 1% of initiator, there is only 25% of all units included in the first percolation cluster, but if we use 16% of initiator, the percolation cluster contains more than 60% of all monomers. So, we propose that a sample with an increased percolation cluster size can be removed from the molding just after the gelation point, while a sample with a smaller percolation cluster should be exposed to additional post-gelation curing to avoid deformation and leakage after molded form removal.
We also tested the percolation cluster properties in the systems with a varied amount of plasticizer, but there was no significant influence of the plasticizer on the percolation threshold or maximum cluster size, so we do not present these results here.
3.2. Mechanical Properties
We performed network uniaxial constant volume deformations to study the system’s mechanical properties, as shown in
Figure 7 (here,
i and
p refer to the amount of initiator and plasticizer, correspondingly). In the upper part of
Figure 7, the 2D slice snapshots are presented in equilibrium and deformed state (
), while, below, the stress–strain curves are shown. One can see slight alignment along the elongation axis in the deformed state. In addition, there is sufficiently great monomer stretching at
(see averaged monomer end-to-end distance in insets in
Figure 7). This is likely responsible for the rapid nonlinear increase in stress during high elongation, which is not predicted by the Flory model. We also note that the non-uniform direction of these fully stretched segments, especially for the system with the plasticizer, shows that the deformation of the internal network structure is non-affine.
We are well aware of the entropic nature of the elastic deformations of amorphous (non-glassy) networks, while most thermoset systems in the working temperature interval are in the glassy state. Nevertheless, such analysis could be useful to compare with standard polymer elastomer models and to give some estimate of general network properties. The comparison with the polymer elasticity model was made via the simpler Flory affine network model [
27] for a region of small deformations
(i) and later with the more sophisticated Three Chain Model over all
values and taking into account the crosslink volume density (
) from the Flory model. The detailed fitting formulas are given in the Methodology section.
To summarize, the standard Flory model works nicely for small deformations, and the value gives the exact measure of the elastic modulus (in units). For the greater deformations, the TCM fit overestimates the nonlinear increase in stress caused by the subchains’ final expansibility, but the fitting parameter n, which is proportional to the average mesh size, is always close to the same value, which is around 0.56. This overestimation most likely comes from the irregularity of our modeled network: there are no equally distributed cubic meshes inside the network topology, which is assumed for the TCM.
3.3. Topology of the Cured Networks
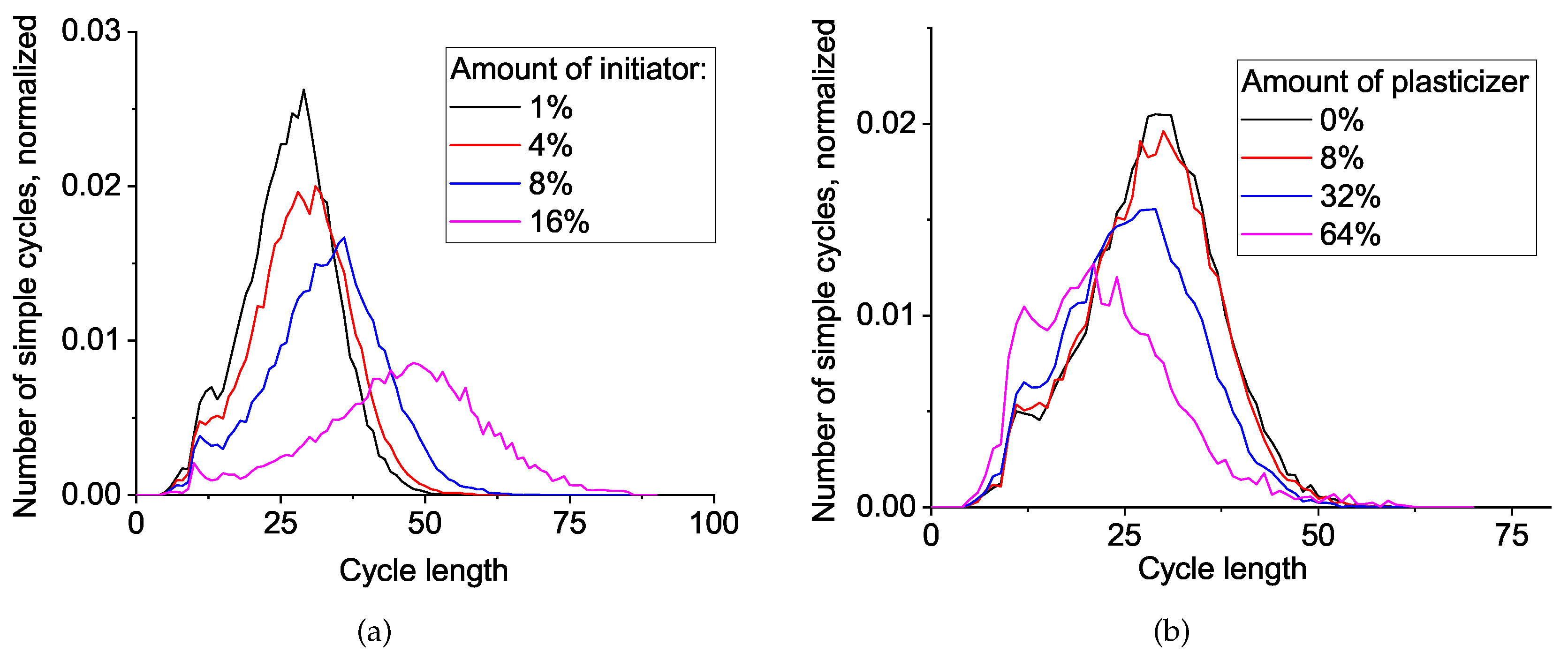
To understand the differences in network topology for systems with various initiator and plasticizer concentrations and to correlate these differences with mechanical properties we studied simple cycles distribution (see
Figure 8). In all cases, the cycles are distributed around a Gaussian bell shape, so we can address the average cycle length value and its distribution width (i.e., dispersity). We can clearly observe an increase in mean cycle length and dispersity upon the increase of initiator concentration. This trend is more or less obvious and can be explained by the inclusion of the valuable fraction of a two-valence initiator into the melt of four-valence monomers.
The more intriguing result comes from the plasticizer influence: there is a noticeable decrease in the mean cycle length upon increasing the plasticizer amount. To some extent, this is caused by the redistribution between the standard network mesh size and self-loops from two neighboring monomer units because of the plasticizer-driven dilution effect of active ends, similar to that observed in [
11]. However, the other contribution is not obvious and should be discussed in more accurate terms.
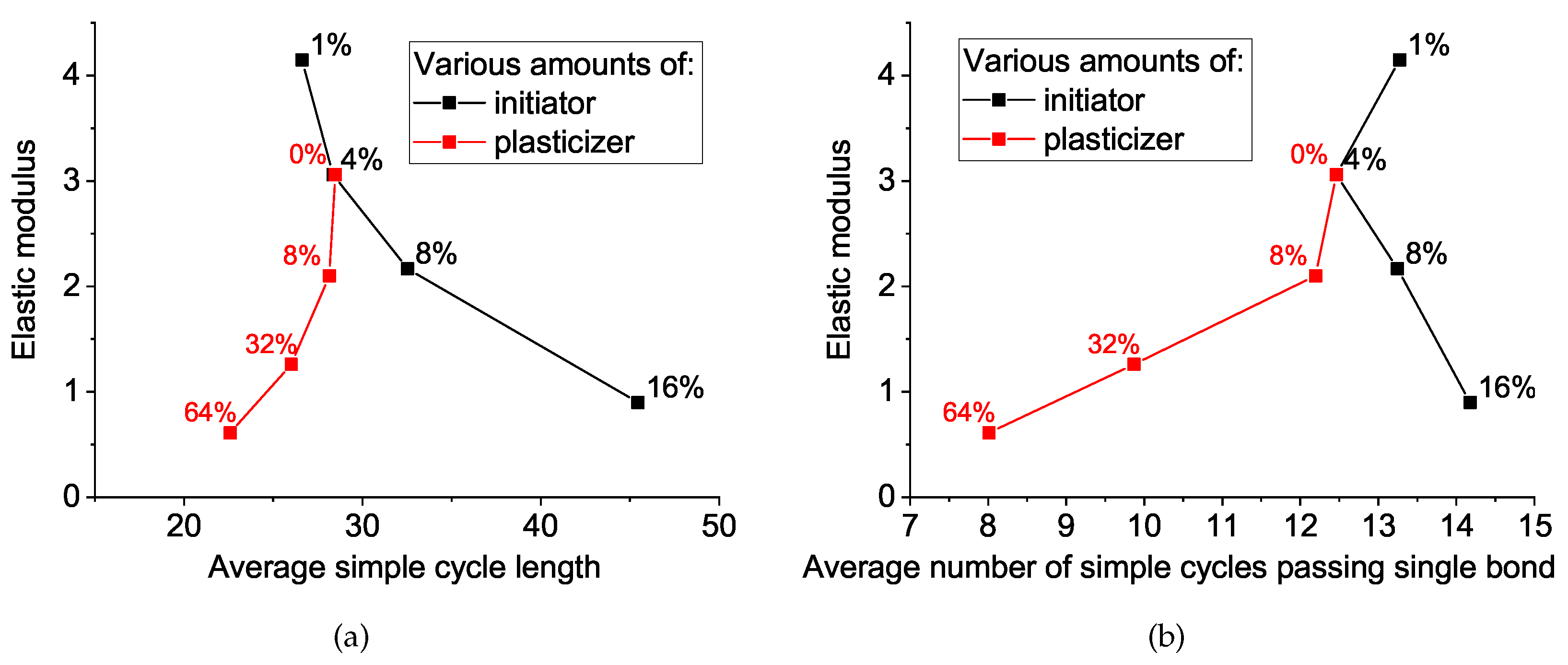
To do this, we calculated one more network property: the average number of cycles passing through the bond, which we correlated with the elastic modulus, together with the average cycle length, as seen in
Figure 9. The elastic modulus decreases during the increase in initiator concentration (black curve) in straightforward accordance with the simple cycle behavior. However, the situation with the plasticizer concentration is the opposite and, at first glance, counterintuitive: the decrease in cycle length yields a decrease in the elastic modulus. The preliminary conclusion here is the following: both additives (initiator and plasticizer) affect the network properties and considerably decrease the elastic modulus, but the mechanisms are transversal. In the next session, we try to rationalize the aforementioned difference in the mechanism of network softening by the initiator and plasticizer.
Additionally, we calculated the conformational properties of the cycles, namely, spatial distance versus distance along the chain , for any pair i and j inside a simple cycle. However, these plots were almost the same for all cases, indicating Gaussian conformation with ; we do not present the corresponding plots here.
4. Conclusions and Outlook
In this paper, we study the thermoset polymer matrix and the influence of plasticizer and initiator on the gelation process and final matrix properties. The gelation process depends primarily on the initiator amount, and gelation time is inversely proportional to the initiator concentration. In addition, we found that the average gel fraction at the gel point is much higher for the case of a higher initiator amount, which could be of practical interest for potential industry workflows. However, the flip side of the coin is that the initiator increases the mesh size and network softness. The plasticizer does not speed up the curing time, but it also increases network softness.
In addition, in this paper, we propose to calculate one new network topology characteristic, namely, the number of simple cycles passing through a single bond. To the best of our knowledge, this the first time this characteristic of network topology is considered in addition to cycle distribution, which is often studied in state-of-the-art papers on polymer networks [
9,
12].
We correlated mechanical and topological network properties and propose a valuable way to characterize network softness based on two topological properties: average simple cycle length and average number of simple cycles passing through a single covalent bond. These values can be treated in the more familiar (although inaccurate) terms of network “mesh size” and “sponginess”, correspondingly. We found that, in the studied case of phthalonitrile resin, the initiator mainly changes the “mesh size”, while the plasticizer only affects “sponginess”, and they do so orthogonally to each other, as we present in
Figure 10. The combination of these two topological parameters allows one to characterize any given network in an implicit but precise way and predict the resulting network properties, including the mechanical modulus. We believe that the same approach could also be useful for other polymer networks, including rubbers and gels.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









