Compatibility of Chitosan in Polymer Blends by Chemical Modification of Bio-based Polyesters



Abstract
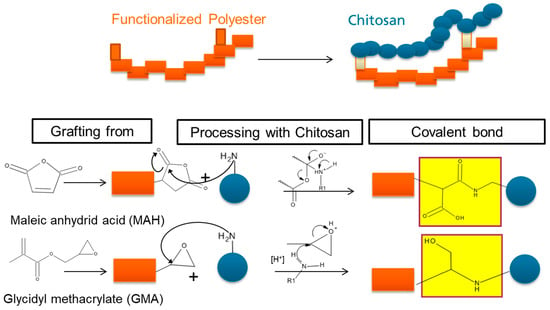
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. PLA and PHBV Grafting in the Melt
2.3. Chitosan Composites Preparation
2.4. Analytical Methods
2.4.1. Determination of the Degree of Grafting of MAH
2.4.2. Determination of the Degree of Grafting of GMA
2.4.3. Determination of Grafting Yield
2.5. Instrumental Methods
2.6. Tensile Testing
3. Results and Discussion
3.1. Grafting
3.1.1. Functionalization in the Internal Mixer
3.1.2. Molecular Weight of Functionalized PLA and PHBV in the Internal Mixer
3.2. Chitosan Composite
3.2.1. Compounding
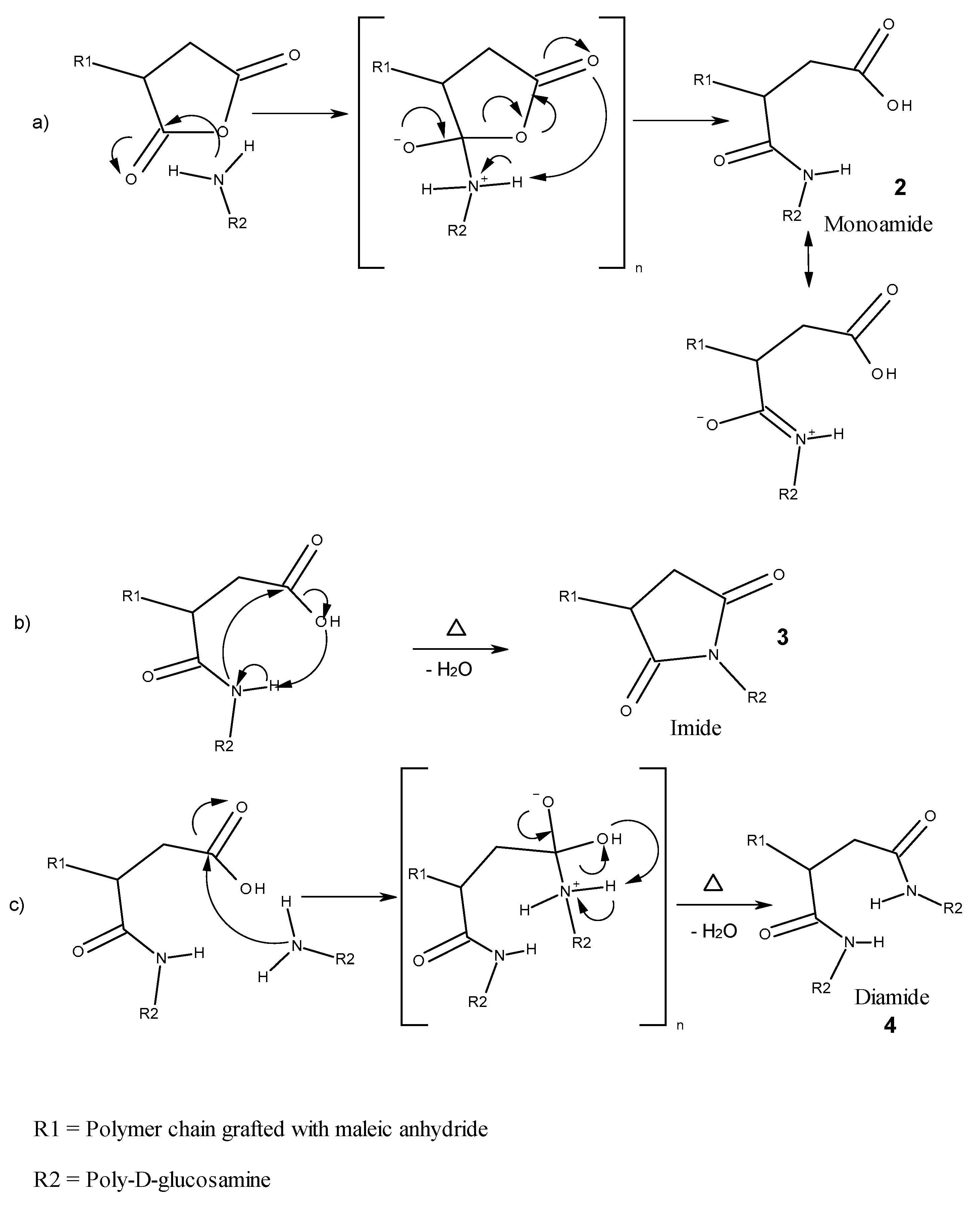
3.2.2. Reaction Mechanism
3.2.3. Reactions after Compounding
3.2.4. Thermal Properties of the Grafted Polymers and Chitosan Blends
3.2.5. FTIR Characterization
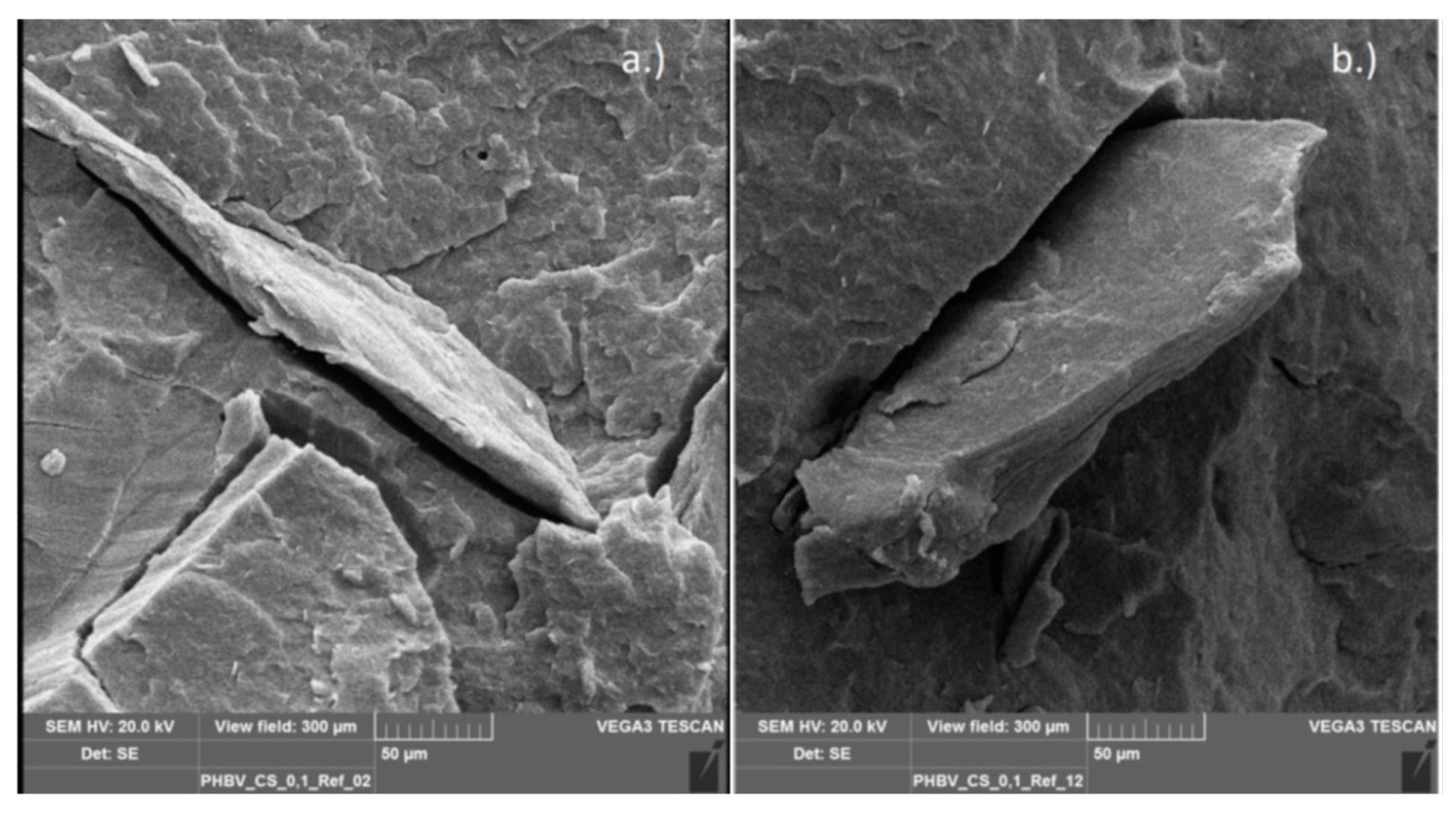
3.2.6. Scanning Electron Microscopy
3.2.7. Mechanical Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

Appendix B



References
- Drumright, R.E.; Gruber, P.R.; Henton, D.E. Polylactic Acid Technology. Adv. Mater. 2000, 12, 1841–1846. [Google Scholar] [CrossRef]
- Andres, Y.; Giraud, L.; Gerente, C.; Le Cloirec, P. Antibacterial effects of chitosan powder: Mechanisms of action. Environ. Technol. 2007, 28, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Goy, R.C.; Morais, S.T.B.; Assis, O.B.G. Evaluation of the antimicrobial activity of chitosan and its quaternized derivative on E. coli and S. aureus growth. Rev. Bras. Farmacogn. 2016, 26, 122–127. [Google Scholar] [CrossRef]
- Wu, C.-S. Modulation, functionality, and cytocompatibility of three-dimensional printing materials made from chitosan-based polysaccharide composites. Mat. Sci. Eng. C 2016, 69, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Sébastien, F.; Stéphane, G.; Copinet, A.; Coma, V. Novel biodegradable films made from chitosan and poly(lactic acid) with antifungal properties against mycotoxinogen strains. Carbohydr. Polym. 2006, 65, 185–193. [Google Scholar] [CrossRef]
- Raquez, J.-M.; Narayan, R.; Dubois, P. Recent Advances in Reactive Extrusion Processing of Biodegradable Polymer-Based Compositions. Macromol. Mater. Eng. 2008, 293, 447–470. [Google Scholar] [CrossRef]
- Gaylord, N.G.; Mehta, M. Role of homopolymerization in the peroxide-catalyzed reaction of maleic anhydride and polyethylene in the absence of solvent. J. Polym. Sci. B Polym. Lett. Ed. 1982, 20, 481–486. [Google Scholar] [CrossRef]
- Mani, R.; Bhattacharya, M.; Tang, J. Functionalization of polyesters with maleic anhydride by reactive extrusion. J. Polym. Sci. A Polym. Chem. 1999, 37, 1693–1702. [Google Scholar] [CrossRef]
- Hwang, S.W.; Lee, S.B.; Lee, C.K.; Lee, J.Y.; Shim, J.K.; Selke, S.E.M.; Soto-Valdez, H.; Matuana, L.; Rubino, M.; Auras, R. Grafting of maleic anhydride on poly(L-lactic acid). Effects on physical and mechanical properties. Polym. Test. 2012, 31, 333–344. [Google Scholar] [CrossRef]
- Detyothin, S.; Selke, S.E.M.; Narayan, R.; Rubino, M.; Auras, R. Reactive functionalization of poly(lactic acid), PLA: Effects of the reactive modifier, initiator and processing conditions on the final grafted maleic anhydride content and molecular weight of PLA. Polym. Degrad. Stab. 2013, 98, 2697–2708. [Google Scholar] [CrossRef]
- Domenichelli, I.; Coiai, S.; Cicogna, F.; Pinzino, C.; Passaglia, E. Towards a better control of the radical functionalization of poly(lactic acid). Polym. Int. 2015, 64, 631–640. [Google Scholar] [CrossRef]
- Avella, M.; Bogoeva-Gaceva, G.; Buzõarovska, A.; Emanuela Errico, M.; Gentile, G.; Grozdanov, A. Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)-based biocomposites reinforced with kenaf fibers. J. Appl. Polym. Sci. 2007, 104, 3192–3200. [Google Scholar] [CrossRef]
- Chen, C.; Peng, S.; Fei, B.; Zhuang, Y.; Dong, L.; Feng, Z.; Chen, S.; Xia, H. Synthesis and characterization of maleated poly(3-hydroxybutyrate). J. Appl. Polym. Sci. 2003, 88, 659–668. [Google Scholar] [CrossRef]
- Avella, A.; Martuscelli, E.; Raimo, M. Review Properties of blends and composites based on poly(3-hydroxy)butyrate (PHB) and poly(3-hydroxybutyrate-hydroxyvalerate) (PHBV) copolymers. J. Mater. Sci. 2000, 35, 523–545. [Google Scholar] [CrossRef]
- Al-Itry, R.; Lamnawar, K.; Maazouz, A. Improvement of thermal stability, rheological and mechanical properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy. Polym. Degrad. Stab. 2012, 97, 1898–1914. [Google Scholar] [CrossRef]
- Schmidt, U.; Zschoche, S.; Werner, C. Modification of poly(octadecene-alt-maleic anhydride) films by reaction with functional amines. J. Appl. Polym. Sci. 2003, 87, 1255–1266. [Google Scholar] [CrossRef]
- Le Suer, W.M.; Norman, G.R. Reaction Product of High Molecular Weight Succinic Acids and Succinic Anhydrides with an Ethylene Poly-Amine. U.S. Patent 3,172,892A, 9 March 1965. [Google Scholar]
- Li, W.; Li, S.; Zhang, Q.; Zhang, S. Synthesis of Bis(amine anhydride)s for Novel High Tgs and Organosoluble Poly(amine imide)s by Palladium-Catalyzed Amination of 4-Chlorophthalide Anhydride. Macromolecules 2007, 40, 8205–8211. [Google Scholar] [CrossRef]
- Marechal, P.; Coppens, G.; Legrad, R.; Dekoninck, J.-M. Amine/anhydride reaction versus amide/anhydride reaction in polyamide/anhydride carriers. J. Polym. Sci. A Polym. Chem. 1995, 33, 757–766. [Google Scholar] [CrossRef]
- Auras, R.; Lim, L.-T.; Selke, S.E.M.; Tsuji, H. (Eds.) Poly (Lactic Acid): Synthesis, Structures, Properties, Processing and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Jost, V.; Kopitzky, R. Blending of Polyhydroxybutyrate-co-valerate with Polylactic Acid for Packaging Applications—Reflections on Miscibility and Effects on Mechanical and Barrier Properties. Chem. Biochem. Eng. Q. 2015, 29, 221–246. [Google Scholar] [CrossRef]
- Nijenhuis, A.J.; Grijpma, D.W.; Pennings, A.J. Crosslinked poly (L-lactide) and poly (ε-caprolactone). Polymer 1996, 37, 2783–2791. [Google Scholar] [CrossRef]
- Dean, K.M.; Petinakis, E.; Meure, S.; Yu, L.; Chryss, A. Melt Strength and Rheological Properties of Biodegradable Poly(Lactic Aacid) Modified via Alkyl Radical-Based Reactive Extrusion Processes. J. Polym. Environ. 2012, 20, 741–747. [Google Scholar] [CrossRef]
- Carlson, D.; Dubois, P.; Nie, L.; Narayan, R. Free radical branching of polylactide by reactive extrusion. Polym. Eng. Sci. 1998, 38, 311–321. [Google Scholar] [CrossRef]
- Kim, C.-H.; Jung, K.-M.; Kim, J.-S.; Park, J.-K. Modification of Aliphatic Polyesters and Their Reactive Blends with Starch. J. Polym. Environ. 2004, 12, 179–187. [Google Scholar] [CrossRef]
- Hale, A.; Macosko, C.W.; Bair, H.E. DSC and 13C-NMR studies of the imidazole-accelerated reaction between epoxides and phenols. J. Appl. Polym. Sci. 1989, 38, 1253–1269. [Google Scholar] [CrossRef]
- Yang, W.; Dominici, F.; Fortunati, E.; Kenny, J.M.; Puglia, D. Melt free radical grafting of glycidyl methacrylate (GMA) onto fully biodegradable poly(lactic) acid films: Effect of cellulose nanocrystals and a masterbatch process. RSC Adv. 2015, 5, 32350–32357. [Google Scholar] [CrossRef]
- Kangwanwatthanasiri, P.; Suppakarn, N.; Ruksakulpiwat, C.; Ruksakulpiwat, Y. Glycidyl Methacrylate Grafted Polylactic Acid: Morphological Properties and Crystallization Behavior. Macromol. Symp. 2015, 354, 237–243. [Google Scholar] [CrossRef]
- Râpă, M.; Miteluţ, A.C.; Tănase, E.E.; Grosu, E.; Popescu, P.; Popa, M.E.; Rosnes, J.T.; Sivertsvik, M.; Darie-Niţă, R.N.; Vasile, C. Influence of chitosan on mechanical, thermal, barrier and antimicrobial properties of PLA-biocomposites for food packaging. Compos. Part B Eng. 2016, 102, 112–121. [Google Scholar] [CrossRef]
- Correlo, V.M.; Boesel, L.F.; Bhattacharya, M.; Mano, J.F.; Neves, N.M.; Reis, R.L. Properties of melt processed chitosan and aliphatic polyester blends. Mat. Sci. Eng. A 2005, 403, 57–68. [Google Scholar] [CrossRef]
- Gupta, A.; Pal, A.K.; Woo, E.M.; Katiyar, V. Effects of Amphiphilic Chitosan on Stereocomplexation and Properties of Poly(lactic acid) Nano-biocomposite. Sci. Rep. 2018, 8, 4351. [Google Scholar] [CrossRef]
- Fortunati, E.; Puglia, D.; Kenny, J.M.; Minhaz-Ul Haque, M.; Pracella, M. Effect of ethylene-co-vinyl acetate-glycidylmethacrylate and cellulose microfibers on the thermal, rheological and biodegradation properties of poly(lactic acid) based systems. Polym. Degrad. Stab. 2013, 98, 2742–2751. [Google Scholar] [CrossRef]
- Li, J.; Sun, C.R.; Zhang, X.Q. Preparation, thermal properties, and morphology of graft copolymers in reactive blends of PHBV and PPC. Polym. Compos. 2012, 33, 1737–1749. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, H.; Zhang, J. Compatibilizing effects of maleated poly(lactic acid)(PLA) on properties of PLA/Soy protein composites. Ind. Eng. Chem. Res. 2012, 51, 7786–7792. [Google Scholar] [CrossRef]
- Pracella, M.; Haque, M.M.-U.; Puglia, D. Morphology and properties tuning of PLA/cellulose nanocrystals bio-nanocomposites by means of reactive functionalization and blending with PVAc. Polymer 2014, 55, 3720–3728. [Google Scholar] [CrossRef]
- Günzler, H.; Gremlich, H.U. IR-Spektroskopie: Eine Einführung; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; ISBN 9783527662876. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Polymer | Grafting Agent | wt.% of Grafting Agent | wt.% of Peroxide |
|---|---|---|---|---|
| PLA_Ref. | PLA | None | 0% | 0% |
| PLA Ref. PHX | PLA | None | 0% | 0.30% |
| PLA3M1P | PLA | MAH | 3% | 0.30% |
| PLA10M1P | PLA | MAH | 10% | 0.30% |
| PLA3M2P | PLA | MAH | 3% | 0.60% |
| PLA10M2P | PLA | MAH | 10% | 0.60% |
| PLA3G1P | PLA | GMA | 3% | 0.30% |
| PLA10G1P | PLA | GMA | 10% | 0.30% |
| PLA3G2P | PLA | GMA | 3% | 0.60% |
| PLA10G2P | PLA | GMA | 10% | 0.60% |
| PHBV_Ref. | PHBV | None | 0% | 0% |
| PHBV_Ref. PHX | PHBV | None | 0% | 0.30% |
| PHBV_3M1P | PHBV | MAH | 3% | 0.30% |
| PHBV_10M1P | PHBV | MAH | 10% | 0.30% |
| PHBV_3M2P | PHBV | MAH | 3% | 0.60% |
| PHBV_10M2P | PHBV | MAH | 10% | 0.60% |
| PHBV_3G1P | PHBV | GMA | 3% | 0.30% |
| PHBV_10G1P | PHBV | GMA | 10% | 0.30% |
| PHBV_3G2P | PHBV | GMA | 3% | 0.60% |
| PHBV_10G2P | PHBV | GMA | 10% | 0.60% |
| Sample Name | % Peroxide | % Chitosan | nNH2:nMAH or nNH2:nGMA |
|---|---|---|---|
| PLA-g-MAH_CS 1:10 | 0.6 | 12.4% | 1:10 |
| PLA-g-MAH/Chitosan 1:1 | 0.6 | 1.2% | 1:1 |
| PHBV-g-MAH/Chitosan 1:10 | 0.3 | 20.1% | 1:10 |
| PHBV-g-MAH/Chitosan 1:1 | 0.3 | 2.0% | 1:1 |
| PLA-g-GMA/Chitosan 1:10 | 0.6 | 21.7% | 1:10 |
| PLA-g-GMA/Chitosan 1:1 | 0.6 | 2.2% | 1:1 |
| PHBV-g-GMA/Chitosan 1:10 | 0.3 | 44.8% | 1:10 |
| PHBV-g-GMA/Chitosan 1:1 | 0.3 | 4.5% | 1:1 |
| Sample Name | MAH in Composite | MAH without Chitosan | Reacted MAH |
|---|---|---|---|
| PLA-g-MAH/Chitosan 1:10 | 0.51% | 5.70% | 91% |
| PLA-g-MAH/Chitosan 1:1 | 0.47% | 6.50% | 93% |
| PHBV-g-MAH/Chitosan 1:10 | 0.56% | 6.30% | 91% |
| PHBV-g-MAH/Chitosan 1:1 | 0.56% | 6.50% | 91% |
| Probename | Tg * [°C] | Tm [°C] | Xc |
|---|---|---|---|
| PLA_Ref. | 62.1 | 151.1 | 0.4% |
| PLA_PHX_Ref. | 63.3 | 150.6 | 28.6% |
| PLA_3M1P | 56.7 | 152.9 | −0.4% |
| PLA_10M1P | 57.5 | 148.3 | 0.5% |
| PLA_3M2P | 59.5 | 152.8 | 1.8% |
| PLA_10M2P | 46.3 | 149.4 | 2.3% |
| PLA_3G1P | 55.5 | 147.4 | 24.0% |
| PLA_10G1P | 61 | 148.9 | −0.4% |
| PLA_3G2P | 62.5 | 148.9 | 10.1% |
| PLA_10G2P | 60.5 | 148.6 | −0.4% |
| PHBV_Ref. | 6.2 | 173.6 | 66.5% |
| PHBV_PHX_Ref. | 8.2 | 168 | 61.0% |
| PHBV_3M1P | 8.9 | 166.5 | 60.5% |
| PHBV_10M1P | 7.5 | 168.5 | 60.7% |
| PHBV_3M2P | 7.4 | 165.8 | 63.9% |
| PHBV_10M2P | 9.1 | 162.8 | 57.0% |
| PHBV_3G1P | 4.6 | 171.5 | 55.4% |
| PHBV_10G1P | 3.7 | 171.4 | 57.9% |
| PHBV_3G2P | 4.6 | 170.1 | 61.4% |
| PHBV_10G2P | 4.3 | 168.1 | 68.8% |
| PLA/Chitosan (Ref.) 1:10 | 58.8 | 151.7 | 4% |
| PLA-g-MAH/Chitosan 1:10 | 59.5 | 149.6 | 0% |
| PLA-g-MAH/Chitosan 1:1 | 59.7 | 149.6 | 0% |
| PLA-g-GMA/Chitosan 1:10 | 60.1 | 148.6 | 0% |
| PLA-g-GMA/Chitosan 1:1 | 57.8 | 148.5 | 0% |
| PHBV/Chitosan (Ref.) 1:10 | - | 175.6 | 65% |
| PHBV-g-MAH/Chitosan 1:10 | 4.5 | 164.4 | 65% |
| PHBV-g-MAH/Chitosan 1:1 | 8.1 | 166.2 | 60% |
| PHBV-g-GMA/Chitosan 1:10 | 6.8 | 168.5 | 83% |
| PHBV-g-GMA/Chitosan 1:1 | 5 | 170.1 | 61% |
| Sample | Wavenumber Range [cm−1] | Vibration | Functional Group |
|---|---|---|---|
| PLA-g-MAH/CS | 3500–3100 | ν(N–H) | Secondary amide |
| 1640 | ν(C=O) | Secondary or tertiary amide | |
| 1550 | ν(C–N) + δ(N–H) | Secondary amide | |
| PHBV-g-MAH/CS | 3600–3200 | ν(N–H) | Secondary amide |
| 1600 | ν(C=O) | Secondary or tertiary amide | |
| PLA-g-GMA/CS | 3600–2500 | ν(O–H) | Hydroxide stretching from carboxilic acid |
| 1550 796 | ν(C–N) + δ(N–H) wag (N–H) | Secondary amine Primary and secondary amine | |
| PHBV-g-GMA/CS | 3600–3100 | ν(O–H) | Hydroxide |
| 1640 | ν(C=O) | Secondary amide |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vernaez, O.; Neubert, K.J.; Kopitzky, R.; Kabasci, S. Compatibility of Chitosan in Polymer Blends by Chemical Modification of Bio-based Polyesters. Polymers 2019, 11, 1939. https://doi.org/10.3390/polym11121939
Vernaez O, Neubert KJ, Kopitzky R, Kabasci S. Compatibility of Chitosan in Polymer Blends by Chemical Modification of Bio-based Polyesters. Polymers. 2019; 11(12):1939. https://doi.org/10.3390/polym11121939
Chicago/Turabian StyleVernaez, Oscar, Katharina Julia Neubert, Rodion Kopitzky, and Stephan Kabasci. 2019. "Compatibility of Chitosan in Polymer Blends by Chemical Modification of Bio-based Polyesters" Polymers 11, no. 12: 1939. https://doi.org/10.3390/polym11121939
APA StyleVernaez, O., Neubert, K. J., Kopitzky, R., & Kabasci, S. (2019). Compatibility of Chitosan in Polymer Blends by Chemical Modification of Bio-based Polyesters. Polymers, 11(12), 1939. https://doi.org/10.3390/polym11121939