Thermoresponsive Poly(ß-hydroxyl amine)s: Synthesis of a New Stimuli Responsive Amphiphilic Homopolymer Family through Amine-Epoxy ‘Click’ Polymerization



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Linear Polymer Synthesis
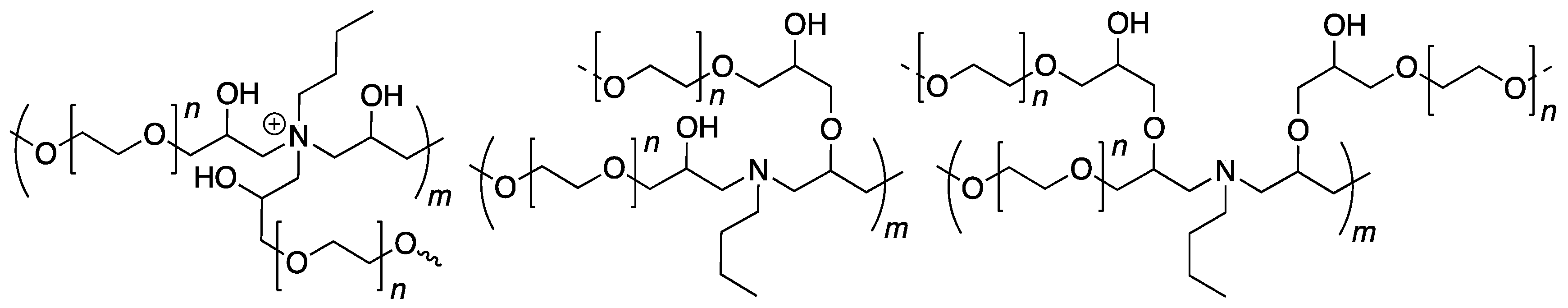
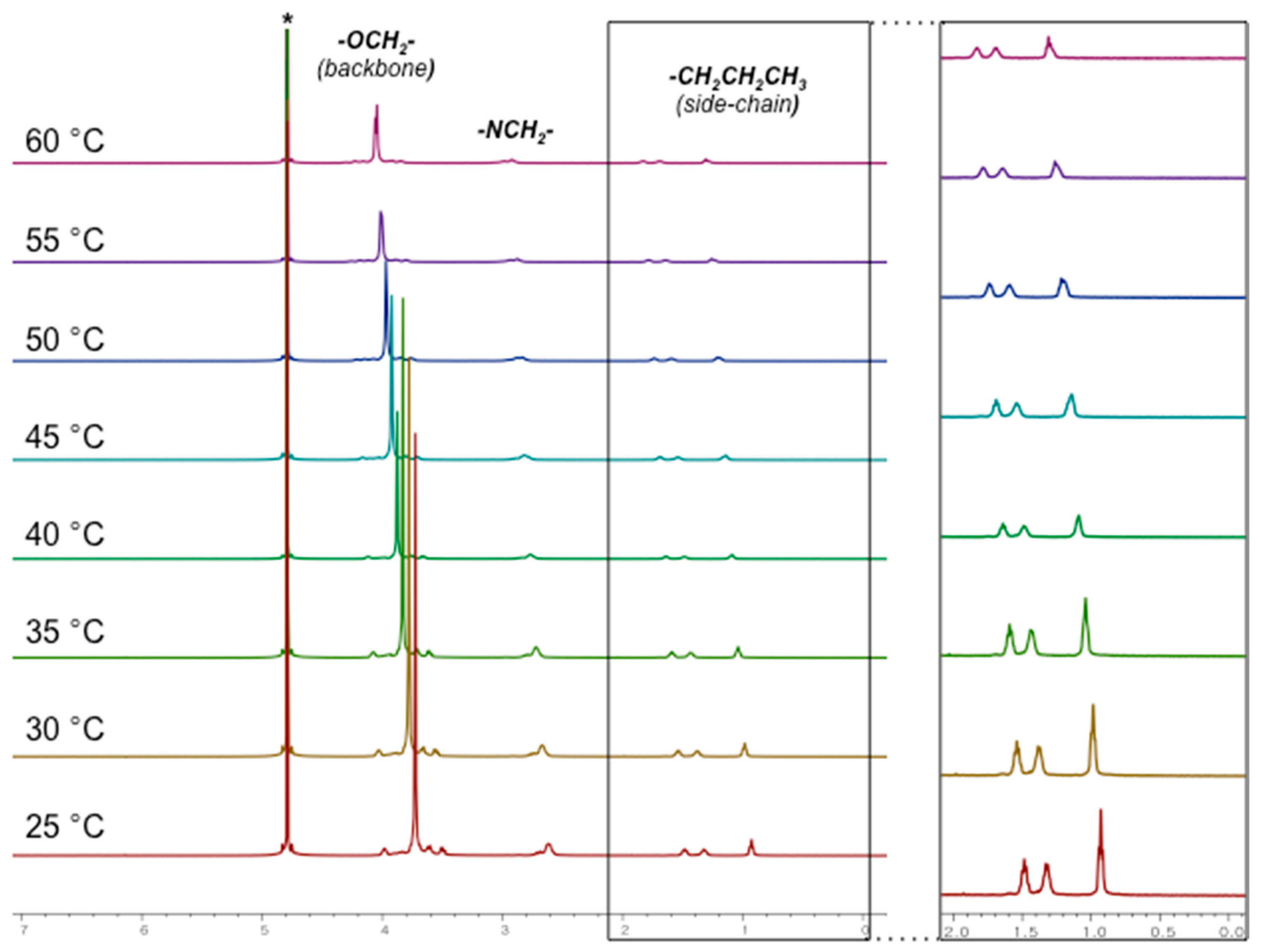
2.2. Absence of Over-Alkylation and Branching in the System
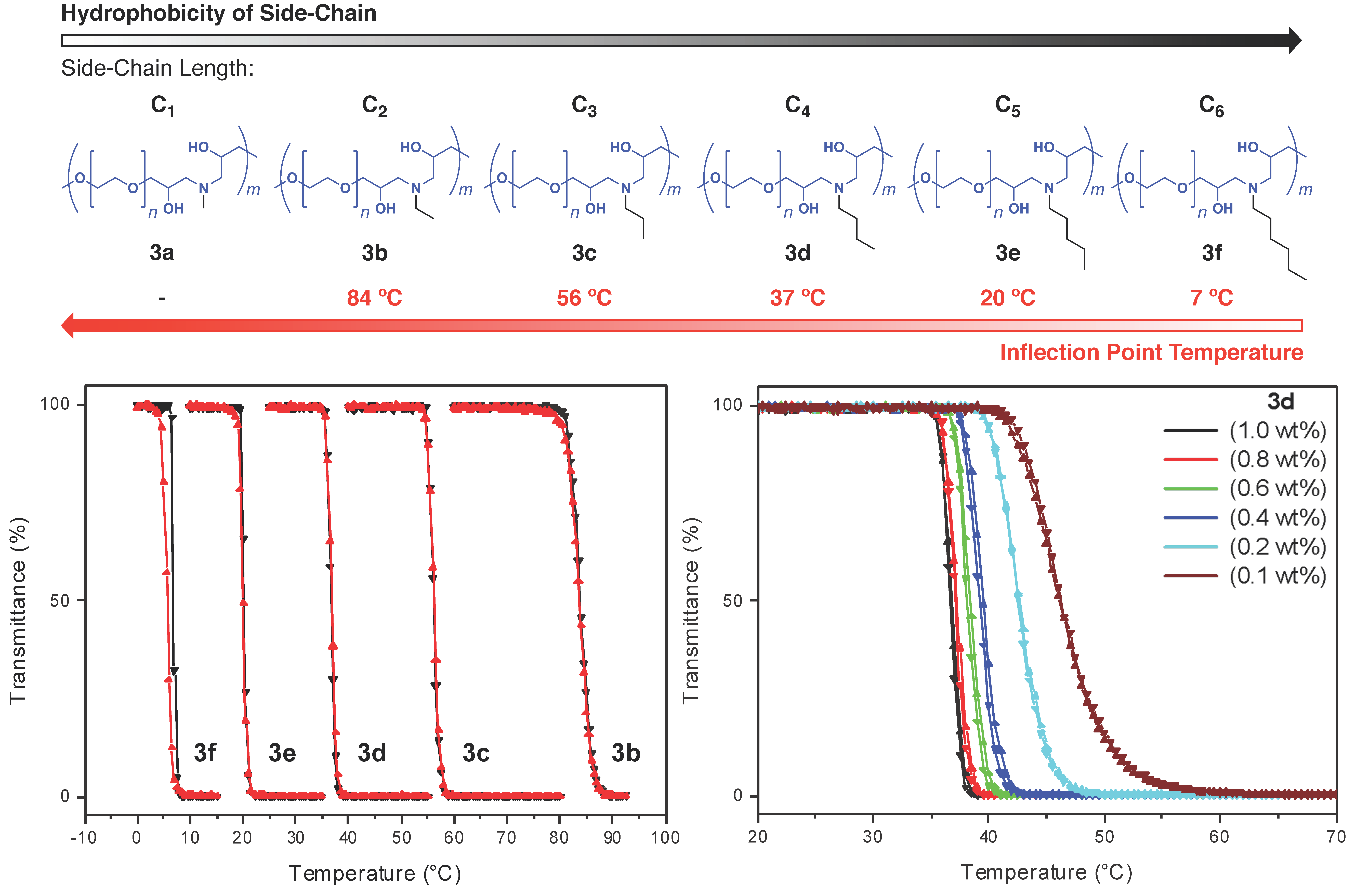
2.3. Thermoresponsive Behavior
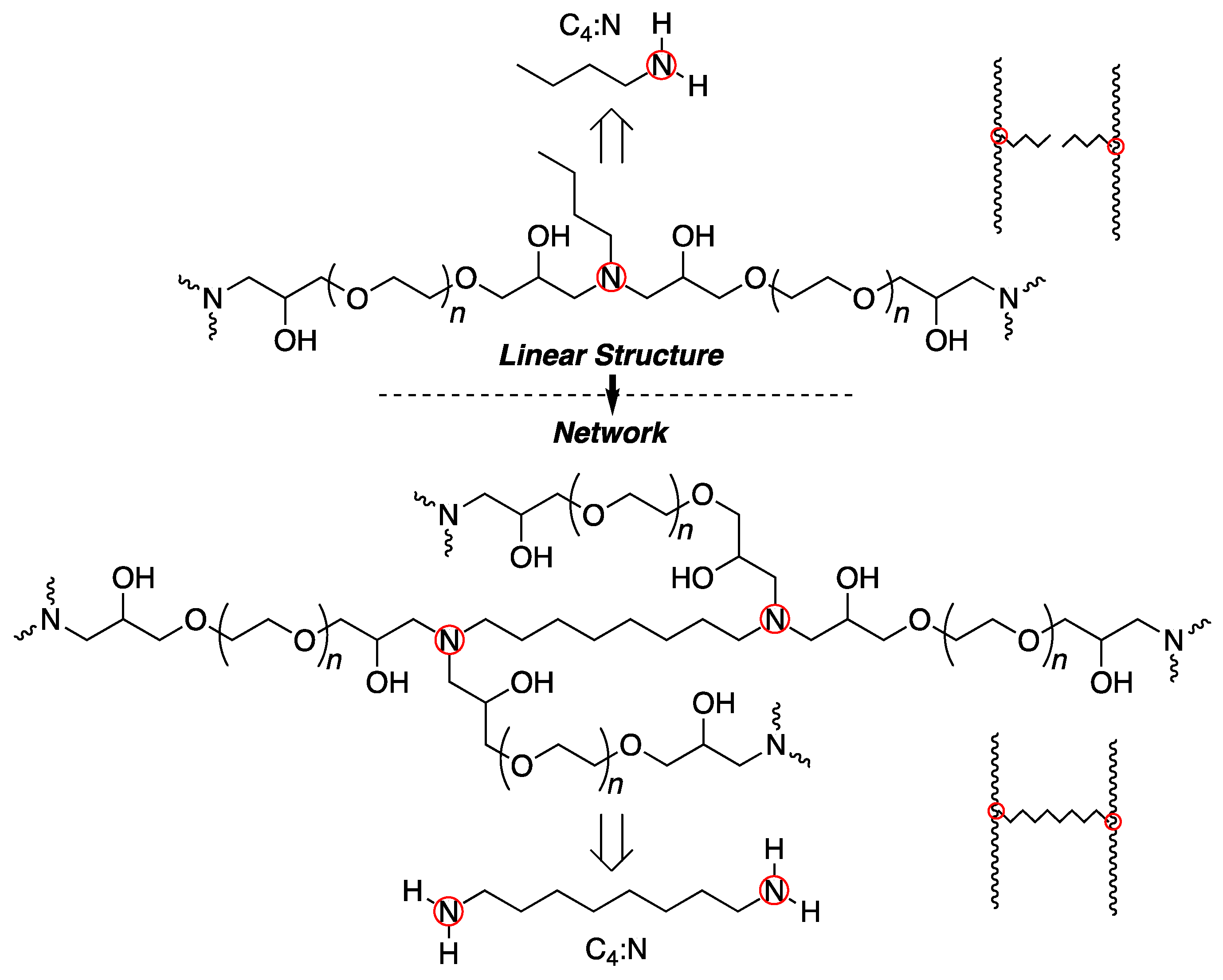
2.4. Hydrogel Synthesis
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Hawker, C.J.; Wooley, K.L.J.S. The convergence of synthetic organic and polymer chemistries. Science 2005, 309, 1200–1205. [Google Scholar] [PubMed]
- Kolb, H.C.; Finn, M.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Hawker, C.J.; Fokin, V.V.; Finn, M.; Sharpless, K.B. Bringing efficiency to materials synthesis: The philosophy of click chemistry. Aust. J. Chem. 2007, 60, 381–383. [Google Scholar] [CrossRef]
- Qin, A.; Lam, J.W.; Tang, B.Z. Click polymerization: Progresses, challenges, and opportunities. Macromolecules 2010, 43, 8693–8702. [Google Scholar] [CrossRef]
- Shi, Y.; Sun, J.Z.; Qin, A. Click polymerization: The aurora of polymer synthetic methodology. J. Polym. Sci. A 2017, 55, 616–621. [Google Scholar] [CrossRef]
- Huang, D.; Liu, Y.; Qin, A.; Tang, B.Z. Recent advances in alkyne-based click polymerizations. Polym. Chem. 2018, 9, 2853–2867. [Google Scholar] [CrossRef]
- Li, B.; Huang, D.; Qin, A.; Tang, B.Z. Progress on Catalytic Systems Used in Click Polymerization. Macromol. Rapid Commun. 2018, 39, 1800098. [Google Scholar] [CrossRef]
- Lutz, J.-F.; Börner, H.G.; Weichenhan, K. Combining ATRP and “click” chemistry: A promising platform toward functional biocompatible polymers and polymer bioconjugates. Macromolecules 2006, 39, 6376–6383. [Google Scholar] [CrossRef]
- Espeel, P.; Du Prez, F.E. “Click”-inspired chemistry in macromolecular science: Matching recent progress and user expectations. Macromolecules 2014, 48, 2–14. [Google Scholar] [CrossRef]
- Sumerlin, B.S.; Vogt, A.P. Macromolecular Engineering through Click Chemistry and Other Efficient Transformations. Macromolecules 2010, 43, 1–13. [Google Scholar] [CrossRef]
- Sanyal, A. Physics, Diels–Alder Cycloaddition-Cycloreversion: A Powerful Combo in Materials Design. Macromol. Chem. Phys. 2010, 211, 1417–1425. [Google Scholar] [CrossRef]
- Members of public are everyday users of the amine-epoxy chemistry commonly referred to as ‘two-part epoxy’ in the market place. For instance, a construction worker uses it to form a protective layer on wooden floors or to cover cracks or to give a glossy finish to a tabletop in the kitchen. Children use its thermosetting nature to fabricate pendants and other ornamental items.
- Jin, F.-L.; Li, X.; Park, S.-J. Synthesis and application of epoxy resins: A review. J. Ind. Eng. Chem. 2015, 29, 1–11. [Google Scholar] [CrossRef]
- Van der Ende, A.E.; Kravitz, E.J.; Harth, E. Approach to formation of multifunctional polyester particles in controlled nanoscopic dimensions. J. Am. Chem. Soc. 2008, 130, 8706–8713. [Google Scholar] [CrossRef] [PubMed]
- Van der Ende, A.; Croce, T.; Hamilton, S.; Sathiyakumar, V.; Harth, E. Tailored polyester nanoparticles: Post-modification with dendritic transporter and targeting units via reductive amination and thiol-ene chemistry. Soft Matter 2009, 5, 1417–1425. [Google Scholar] [CrossRef]
- Kang, T.; Amir, R.J.; Khan, A.; Ohshimizu, K.; Hunt, J.N.; Sivanandan, K.; Montanez, M.I.; Malkoch, M.; Ueda, M.; Hawker, C.J. Facile access to internally functionalized dendrimers through efficient and orthogonal click reactions. Chem. Commun. 2010, 46, 1556–1558. [Google Scholar] [CrossRef]
- Amir, R.J.; Albertazzi, L.; Willis, J.; Khan, A.; Kang, T.; Hawker, C.J. Multifunctional trackable dendritic scaffolds and delivery agents. Angew. Chem. Int. Ed. 2011, 50, 3425–3429. [Google Scholar] [CrossRef]
- Damiron, D.; Mazzolini, J.; Cousin, F.; Boisson, C.; D’Agosto, F.; Drockenmuller, E. Poly (ethylene) brushes grafted to silicon substrates. Polym. Chem. 2012, 3, 1838–1845. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Khan, A.; Stuparu, M.C. Post-polymerization modification reactions of poly (glycidyl methacrylate) s. RSC Adv. 2017, 7, 55874–55884. [Google Scholar] [CrossRef]
- Ren, Y.; Jiang, X.; Yin, J. Poly (ether tert-amine): A novel family of multiresponsive polymer. J. Polym. Sci. A 2009, 47, 1292–1297. [Google Scholar] [CrossRef]
- Saha, A.; De, S.; Stuparu, M.C.; Khan, A. Facile and general preparation of multifunctional main-chain cationic polymers through application of robust, efficient, and orthogonal click chemistries. J. Am. Chem. Soc. 2012, 134, 17291–17297. [Google Scholar] [CrossRef]
- Si, G.; Elzes, M.; Engbersen, J.; Paulusse, J. Modular Synthesis of Bioreducible Gene Vectors through Polyaddition of N, N′-Dimethylcystamine and Diglycidyl Ethers. Polymers 2018, 10, 687. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Bu, Y.; Zhang, L.; Wang, X.; Yang, Y.; Zhuang, Y.; Yang, F.; Shen, H.; Wu, D. Facile construction of pH-and redox-responsive micelles from a biodegradable poly (β-hydroxyl amine) for drug delivery. Biomacromolecules 2015, 17, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Liu, J.; Wang, X.; Li, M.; Cui, X.; Zhang, L.; Wu, D.; Tang, P. Fabrication of dual-sensitive poly(β-hydroxyl amine) micelles for controlled drug delivery. Eur. Polym. J. 2019, 114, 338–345. [Google Scholar]
- Su, Z.; Jiang, X. Multi-stimuli responsive amine-containing polyethers: Novel building blocks for smart assemblies. Polymer 2016, 93, 221–239. [Google Scholar] [CrossRef]
- Liu, S.; Oderinde, O.; Hussain, I.; Yao, F.; Fu, G. Dual ionic cross-linked double network hydrogel with self-healing, conductive, and force sensitive properties. Polymer 2018, 144, 111–120. [Google Scholar] [CrossRef]
- Yu, J.; Su, Z.; Xu, H.; Ma, X.; Yin, J.; Jiang, X. One-pot approach to synthesize hyperbranched poly(thiol–ether amine) (hPtEA) through sequential “thiol–ene” and “epoxy–amine” click reactions. Polym. Chem. 2015, 6, 6946–6954. [Google Scholar] [CrossRef]
- Xu, L.Q.; Pranantyo, D.; Neoh, K.-G.; Kang, E.-T.; Teo, S.L.-M.; Fu, G.D. Synthesis of catechol and zwitterion-bifunctionalized poly (ethylene glycol) for the construction of antifouling surfaces. Polym. Chem. 2016, 7, 493–501. [Google Scholar]
- Hamid, Z.A.; Blencowe, A.; Ozcelik, B.; Palmer, J.A.; Stevens, G.W.; Abberton, K.M.; Morrison, W.A.; Penington, A.J.; Qiao, G.G. Epoxy-amine synthesised hydrogel scaffolds for soft-tissue engineering. Biomaterials 2010, 31, 6454–6467. [Google Scholar]
- Ozcelik, B.; Brown, K.D.; Blencowe, A.; Daniell, M.; Stevens, G.W.; Qiao, G.G. Ultrathin chitosan–poly (ethylene glycol) hydrogel films for corneal tissue engineering. Acta Biomater. 2013, 9, 6594–6605. [Google Scholar]
- Stevens, L.; Calvert, P.; Wallace, G.G.; Panhuis, M.I.H. Ionic-covalent entanglement hydrogels from gellan gum, carrageenan and an epoxy-amine. Soft Matter 2013, 9, 3009–3012. [Google Scholar] [CrossRef]
- Krüger, A.J.; Köhler, J.; Cichosz, S.; Rose, J.C.; Gehlen, D.B.; Haraszti, T.; Möller, M.; De Laporte, L. A catalyst-free, temperature controlled gelation system for in-mold fabrication of microgels. Chem. Commun. 2018, 54, 6943–6946. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Truong, V.X.; Qu, Y.; Lithgow, T.; Fu, G.; Forsythe, J.S. Antibacterial poly (ethylene glycol) hydrogels from combined epoxy-amine and thiol-ene click reaction. J. Polym. Sci. A 2016, 54, 656–667. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, C.; Xu, H.; Ma, X.; Shi, Z.; Yin, J. A Facile Method Synthesizing Hydrogel Using Hybranched Polyether Amine (hPEA) as Coinitiator and Crosslinker. Macromol. Chem. Phys. 2017, 218, 1700251. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Z.; Zhang, X.; Shi, Z.; Xu, H.; Ma, X.; Yin, J.; Tian, M. Polyetheramine (PEA): A versatile platform to tailor the properties of hydrogels via H-bonding interactions. Polym. Chem. 2017, 8, 5367–5373. [Google Scholar] [CrossRef]
- Bakarich, S.E.; Balding, P.; Gorkin, R., III; Spinks, G.M. Printed ionic-covalent entanglement hydrogels from carrageenan and an epoxy amine. RSC Adv. 2014, 4, 38088–38092. [Google Scholar] [CrossRef]
- Ding, J.; Zhou, C.; Li, K.; Zhang, A.; Yao, F.; Xu, L.; Fu, G. Preparation of well-defined fibrous hydrogels via electrospinning and in situ “click chemistry”. RSC Adv. 2016, 6, 27871–27878. [Google Scholar] [CrossRef]
- Qian, S.; Zhou, C.; Xu, L.; Yao, F.; Cen, L.; Fu, G. High strength biocompatible PEG single-network hydrogels. RSC Adv. 2014, 4, 25241–25250. [Google Scholar] [CrossRef]
- Oh, J.; Jung, K.I.; Jung, H.W.; Khan, A. A Modular and Practical Synthesis of Zwitterionic Hydrogels through Sequential Amine-Epoxy “Click” Chemistry and N-Alkylation Reaction. Polymers 2019, 11, 1491. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive Polymers for Biomedical Applications. Polymers 2011, 3, 1215–1242. [Google Scholar] [CrossRef]
- Aseyev, V.; Tenhu, H.; Winnik, F. Self organized nanostructures of amphiphilic block copolymers II. Adv. Polym. Sci. 2011, 242, 29–89. [Google Scholar]
- Gibson, M.I.; O’Reilly, R.K. To aggregate, or not to aggregate? considerations in the design and application of polymeric thermally-responsive nanoparticles. Chem. Soc. Rev. 2013, 42, 7204–7213. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Brooks, W.L.; Sumerlin, B.S. New directions in thermoresponsive polymers. Chem. Soc. Rev. 2013, 42, 7214–7243. [Google Scholar] [CrossRef] [PubMed]
- Jochum, F.D.; Theato, P. Temperature-and light-responsive smart polymer materials. Chem. Soc. Rev. 2013, 42, 7468–7483. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Weber, C.; Schubert, U.S.; Hoogenboom, R. Thermoresponsive polymers with lower critical solution temperature: From fundamental aspects and measuring techniques to recommended turbidimetry conditions. Mater. Horiz. 2017, 4, 109–116. [Google Scholar] [CrossRef]
- Atanase, L.I.; Desbrieres, J.; Riess, G. Micellization of synthetic and polysaccharides-based graft copolymers in aqueous media. Prog. Polym. Sci. 2017, 73, 32–60. [Google Scholar] [CrossRef]
- Atanase, L.I.; Riess, G. Self-Assembly of Block and Graft Copolymers in Organic Solvents: An Overview of Recent Advances. Polymers 2018, 10, 62–88. [Google Scholar] [CrossRef]
- Lutz, J.-F.; Hoth, A. Preparation of ideal PEG analogues with a tunable thermosensitivity by controlled radical copolymerization of 2-(2-methoxyethoxy) ethyl methacrylate and oligo (ethylene glycol) methacrylate. Macromolecules 2006, 39, 893–896. [Google Scholar] [CrossRef]
- Lutz, J.-F.; Akdemir, Ö.; Hoth, A. Point by point comparison of two thermosensitive polymers exhibiting a similar LCST: Is the age of poly (NIPAM) over? J. Am. Chem. Soc. 2006, 128, 13046–13047. [Google Scholar] [CrossRef]
- Lutz, J.-F.; Weichenhan, K.; Akdemir, Ö.; Hoth, A. About the phase transitions in aqueous solutions of thermoresponsive copolymers and hydrogels based on 2-(2-methoxyethoxy) ethyl methacrylate and oligo (ethylene glycol) methacrylate. Macromolecules 2007, 40, 2503–2508. [Google Scholar] [CrossRef]
- Skrabania, K.; Kristen, J.; Laschewsky, A.; Akdemir, Ö.; Hoth, A.; Lutz, J.-F. Design, synthesis, and aqueous aggregation behavior of nonionic single and multiple thermoresponsive polymers. Langmuir 2007, 23, 84–93. [Google Scholar] [CrossRef]
- De, P.; Gondi, S.R.; Sumerlin, B.S. Folate-conjugated thermoresponsive block copolymers: Highly efficient conjugation and solution self-assembly. Biomacromolecules 2008, 9, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.P.; Sumerlin, B.S. Tuning the temperature response of branched poly (N-isopropylacrylamide) prepared by RAFT polymerization. Macromolecules 2008, 41, 7368–7373. [Google Scholar] [CrossRef]
- Li, H.; Bapat, A.P.; Li, M.; Sumerlin, B.S. Protein conjugation of thermoresponsive amine-reactive polymers prepared by RAFT. Polym. Chem. 2011, 2, 323–327. [Google Scholar] [CrossRef]
- De, P.; Sumerlin, B.S. Precision control of temperature response by copolymerization of di (ethylene glycol) acrylate and an acrylamide comonomer. Macromol. Chem. Phys. 2013, 214, 272–279. [Google Scholar] [CrossRef]
- Li, M.; Li, H.; De, P.; Sumerlin, B.S. Thermoresponsive Block Copolymer–Protein Conjugates Prepared by Grafting-from via RAFT Polymerization. Macromol. Rapid Commun. 2011, 32, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Cambre, J.N.; Sumerlin, B.S. Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci. 2010, 35, 278–301. [Google Scholar] [CrossRef]
- Schattling, P.; Jochum, F.D.; Theato, P. Multi-stimuli responsive polymers–the all-in-one talents. Polym. Chem. 2014, 5, 25–36. [Google Scholar] [CrossRef]
- Wei, M.; Gao, Y.; Li, X.; Serpe, M.J. Stimuli-responsive polymers and their applications. Polym. Chem. 2017, 8, 127–143. [Google Scholar] [CrossRef]
- Kubo, T.; Easterling, C.P.; Olson, R.A.; Sumerlin, B.S. Synthesis of multifunctional homopolymers via sequential post-polymerization reactions. Polym. Chem. 2018, 9, 4605–4610. [Google Scholar] [CrossRef]
- Kale, T.S.; Klaikherd, A.; Popere, B.; Thayumanavan, S. Supramolecular assemblies of amphiphilic homopolymers. Langmuir 2009, 25, 9660–9670. [Google Scholar] [CrossRef]
- Vasilevskaya, V.V.; Govorun, E.N. Hollow and Vesicle Particles from Macromolecules with Amphiphilic Monomer Units. Polym. Rev. 2019, 59, 625–650. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, K.; Müllen, K.; Yin, M. Self-assemblies of amphiphilic homopolymers: Synthesis, morphology studies and biomedical applications. Chem. Commun. 2015, 51, 11541–11555. [Google Scholar] [CrossRef] [PubMed]
- Carothers, W.H. Polymerization. Chem. Rev. 1931, 8, 353–426. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymer Chemistry; Wiley: New York, NY, USA, 1991. [Google Scholar]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, J.; Khan, A. Thermoresponsive Poly(ß-hydroxyl amine)s: Synthesis of a New Stimuli Responsive Amphiphilic Homopolymer Family through Amine-Epoxy ‘Click’ Polymerization. Polymers 2019, 11, 1941. https://doi.org/10.3390/polym11121941
Hong J, Khan A. Thermoresponsive Poly(ß-hydroxyl amine)s: Synthesis of a New Stimuli Responsive Amphiphilic Homopolymer Family through Amine-Epoxy ‘Click’ Polymerization. Polymers. 2019; 11(12):1941. https://doi.org/10.3390/polym11121941
Chicago/Turabian StyleHong, Jeonghui, and Anzar Khan. 2019. "Thermoresponsive Poly(ß-hydroxyl amine)s: Synthesis of a New Stimuli Responsive Amphiphilic Homopolymer Family through Amine-Epoxy ‘Click’ Polymerization" Polymers 11, no. 12: 1941. https://doi.org/10.3390/polym11121941
APA StyleHong, J., & Khan, A. (2019). Thermoresponsive Poly(ß-hydroxyl amine)s: Synthesis of a New Stimuli Responsive Amphiphilic Homopolymer Family through Amine-Epoxy ‘Click’ Polymerization. Polymers, 11(12), 1941. https://doi.org/10.3390/polym11121941