Nanoparticle-Hydrogel Composites: From Molecular Interactions to Macroscopic Behavior



Abstract
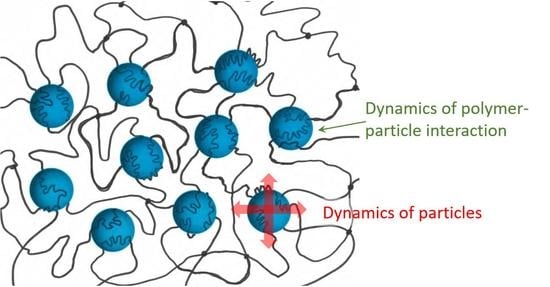
:
1. Introduction
2. Design of Nanoparticle–Hydrogel Composites
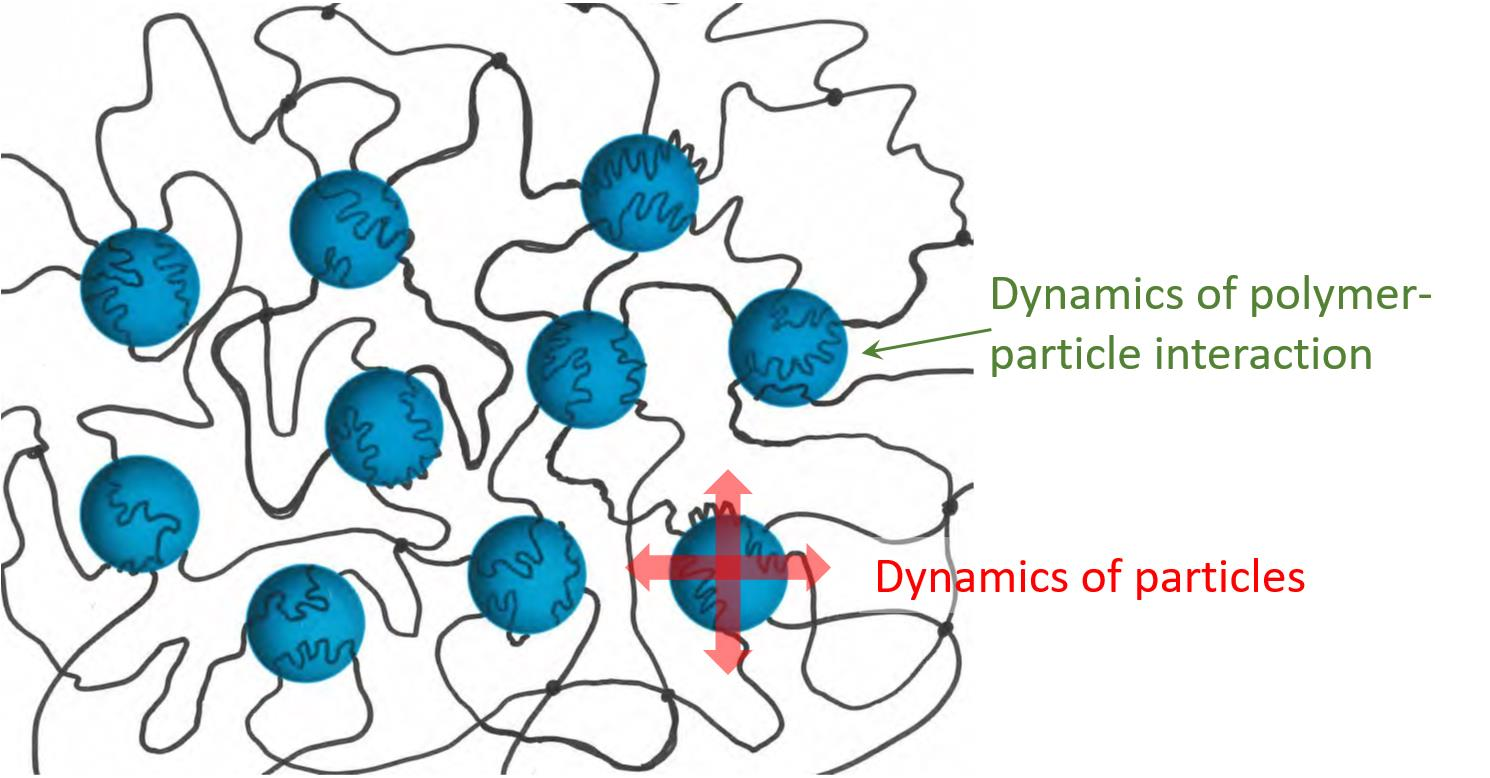
2.1. Nano- and Micro-Gel Composites
2.2. Macroscopic Hydrogel Composites
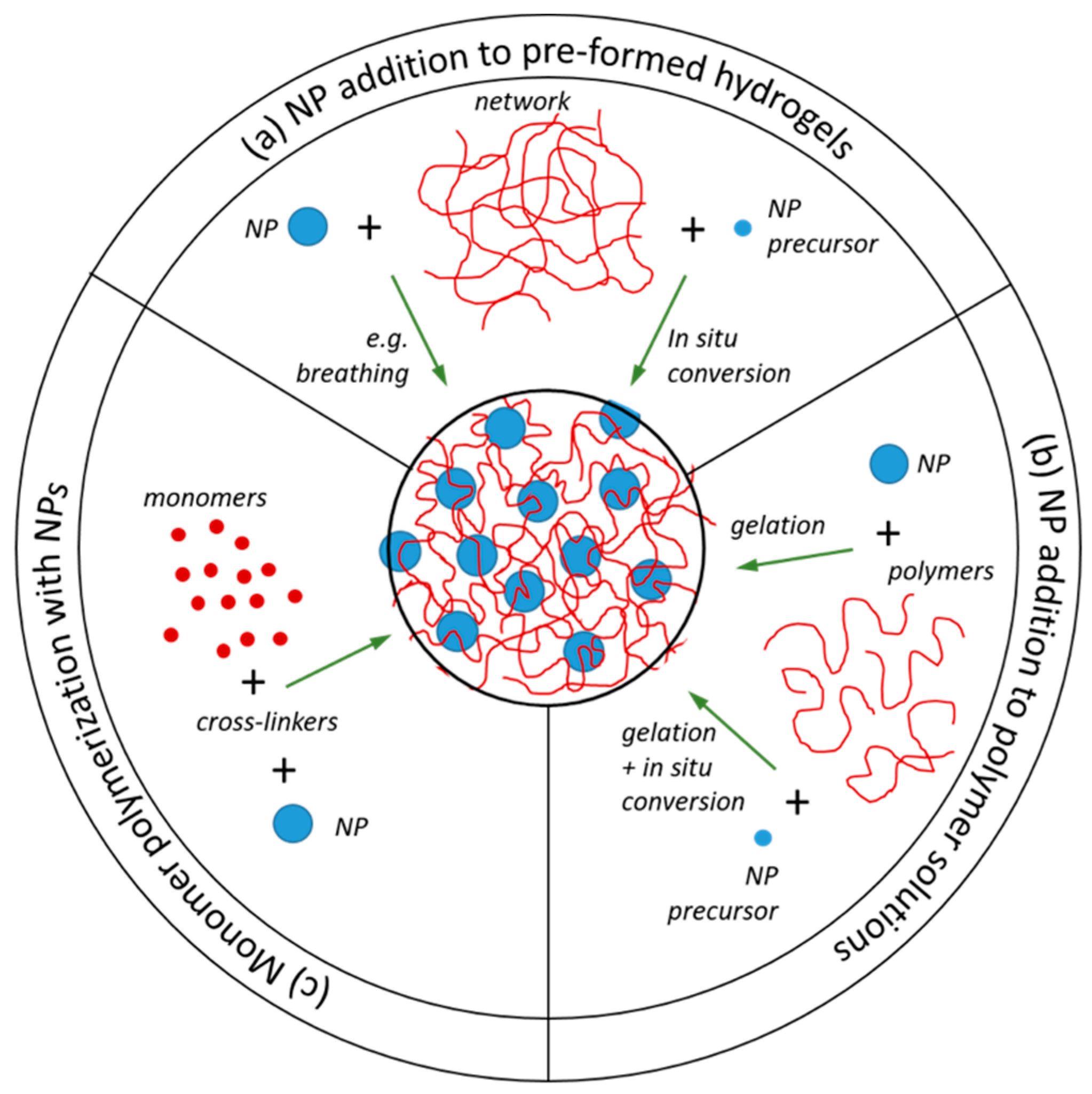
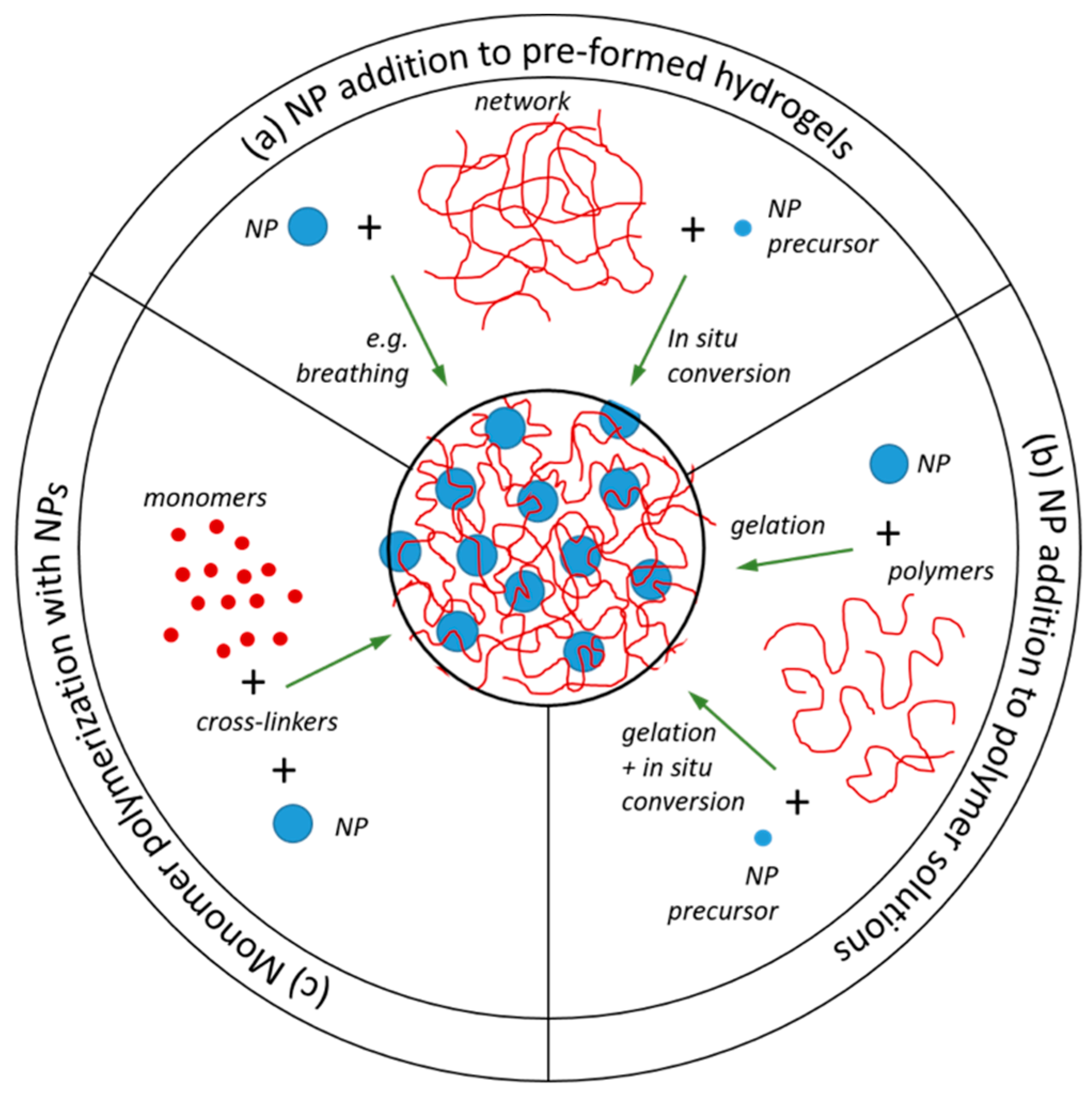
2.3. Nanoparticle Addition to Pre-Formed Hydrogels
2.4. NP Addition to Polymer Solutions Prior to Network Formation
2.5. Monomer Polymerization in the Presence of Nanoparticles
3. Overall Mechanism and Forces Involved in Hybrid Composite Formation
4. Nanoparticles for Improving Hydrogel Properties
4.1. Optical Properties
4.2. Mechanical Properties
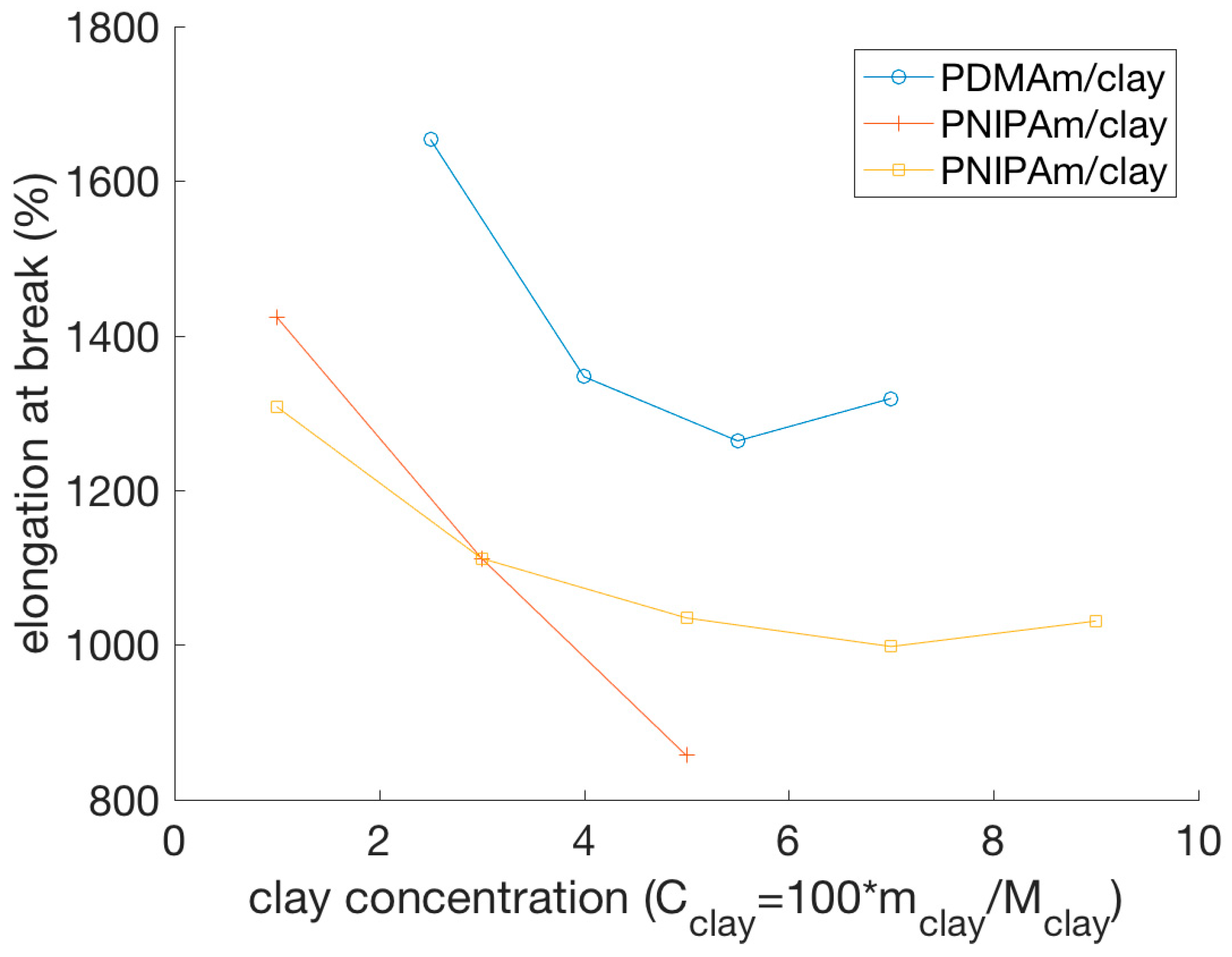
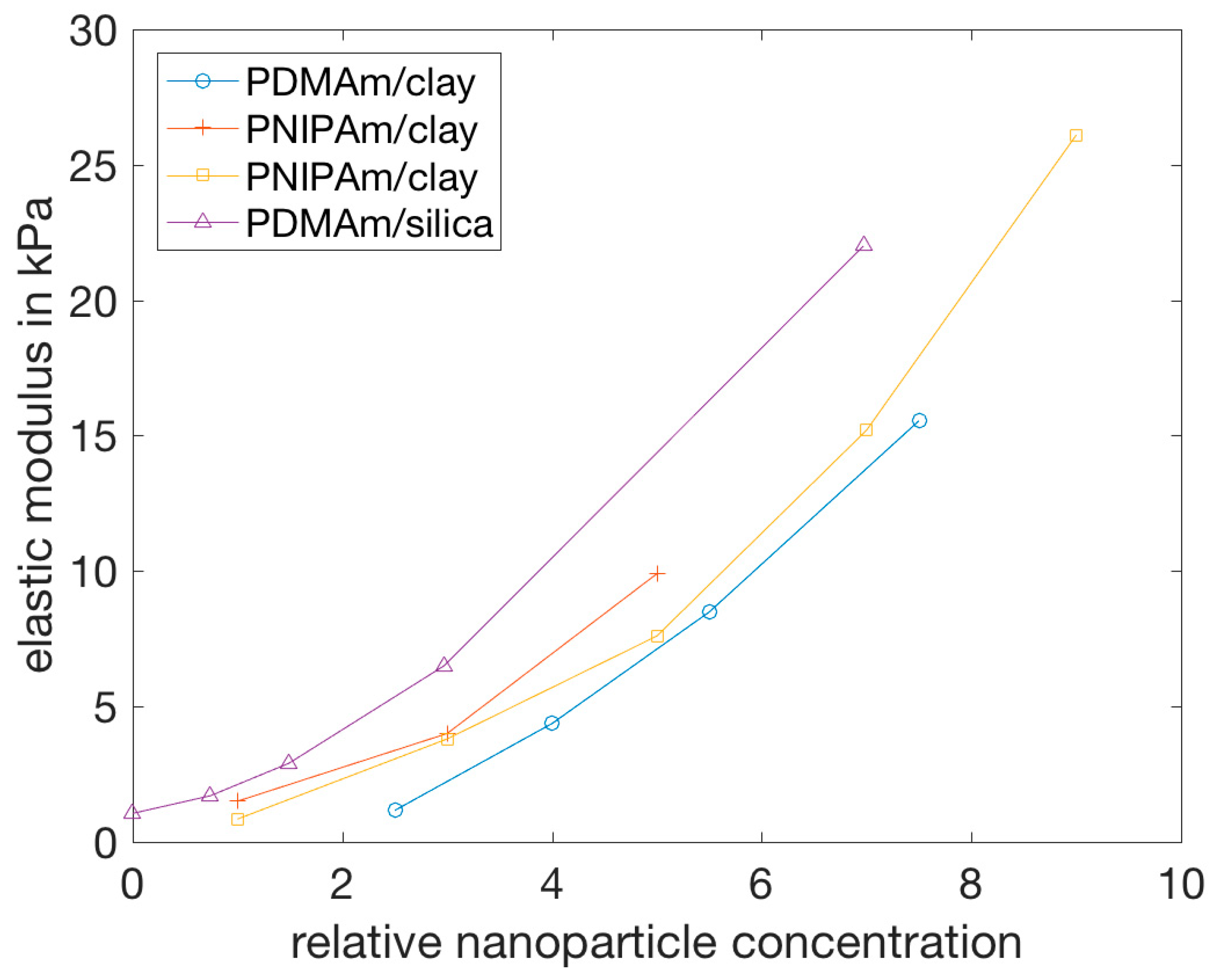
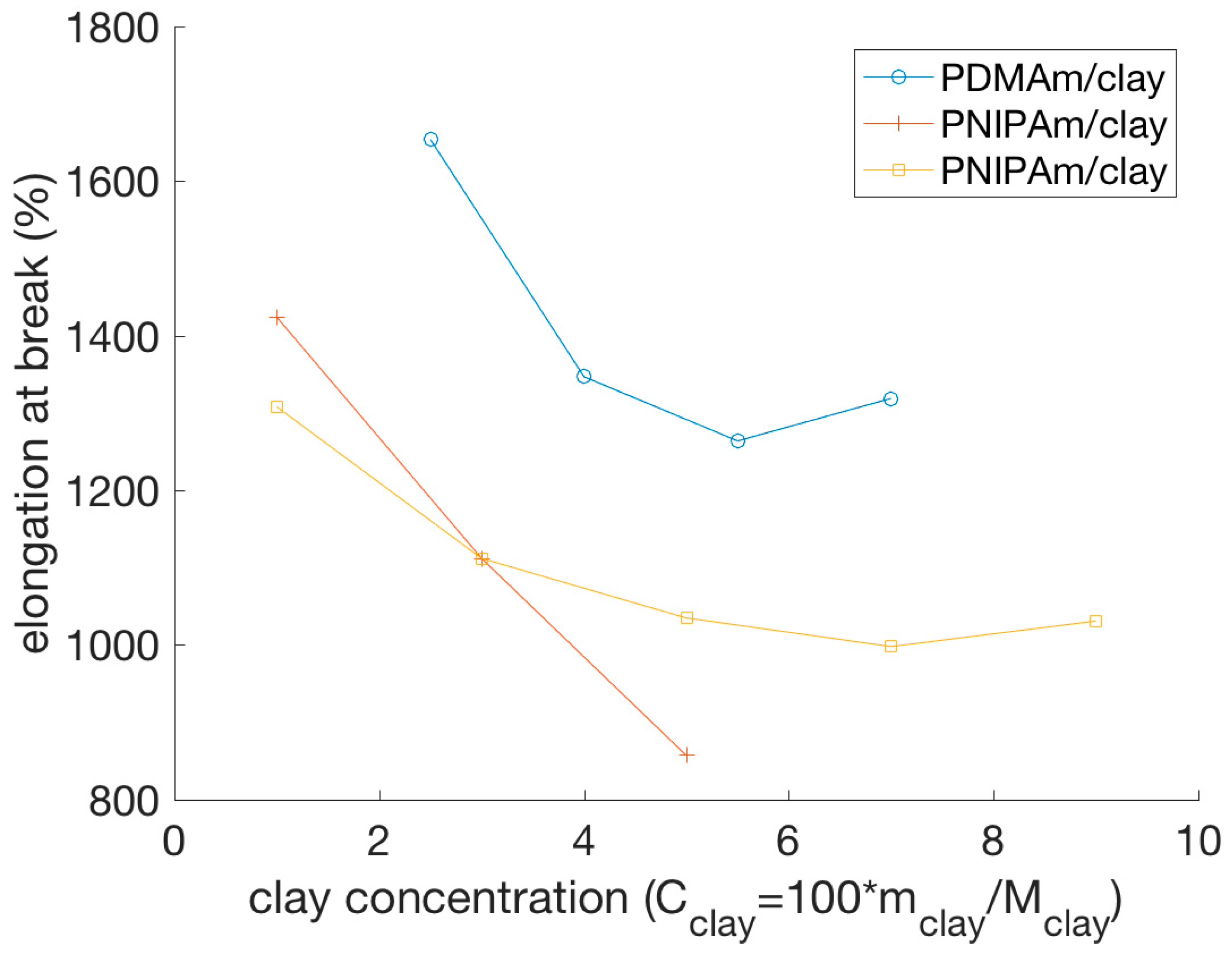
4.2.1. Stress-Strain Behavior
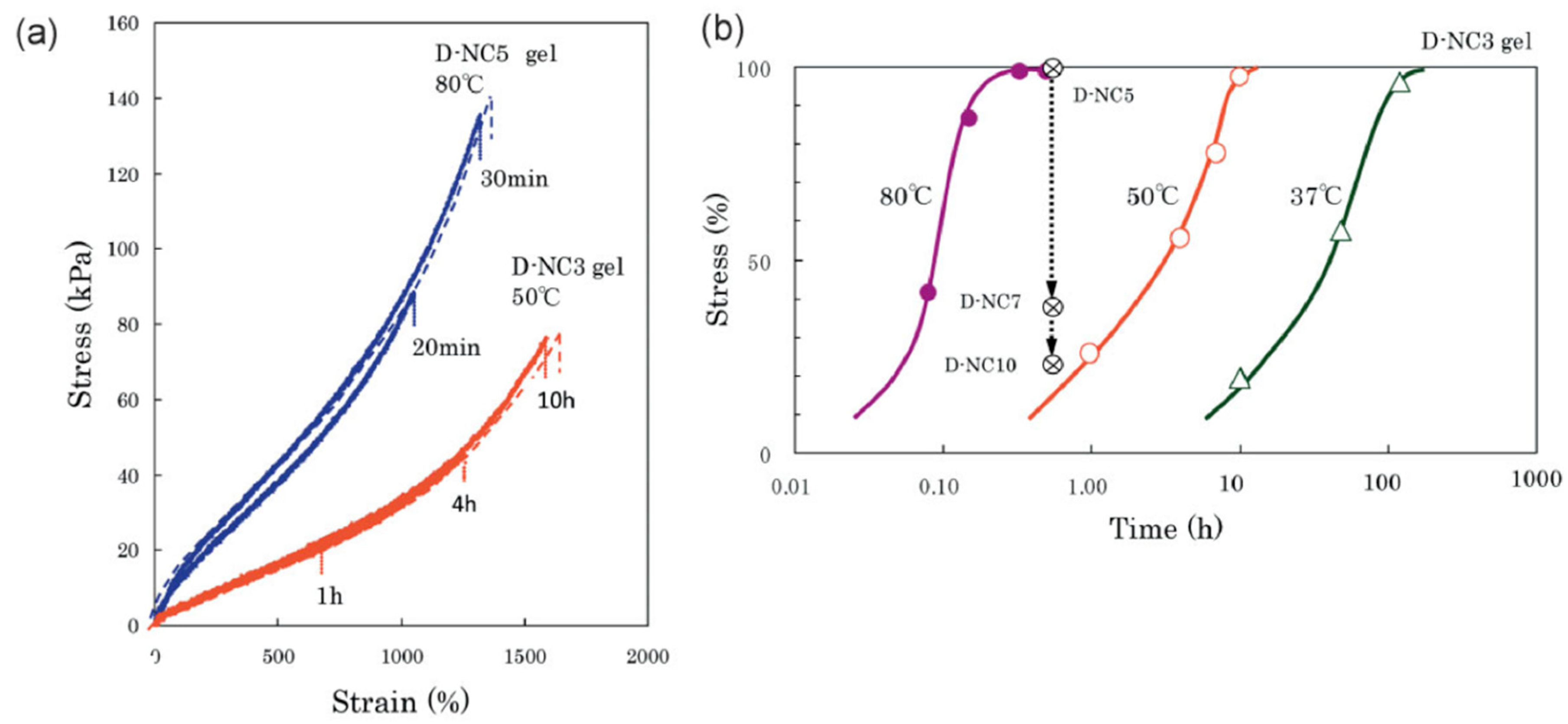
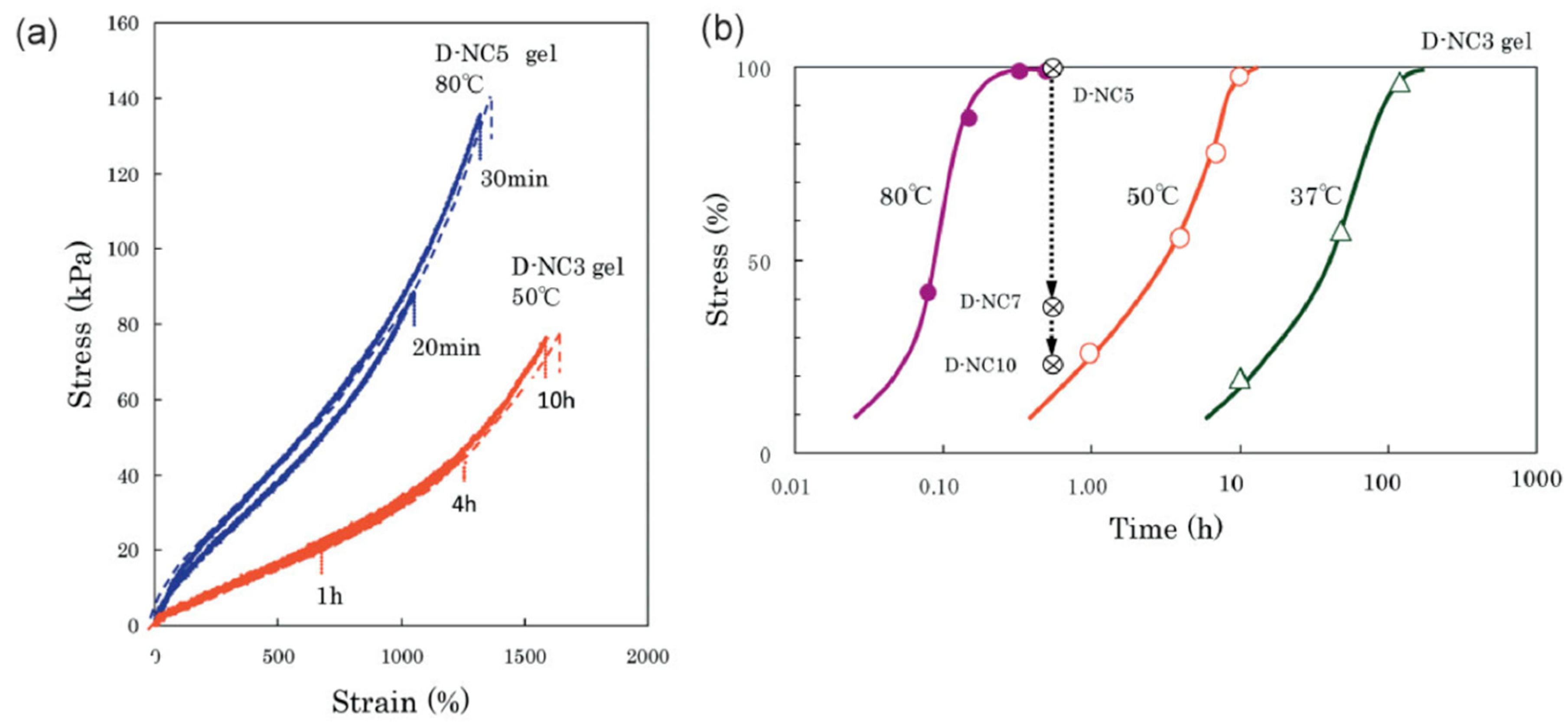
4.2.2. Stress Relaxation
5. Novel Functionalities
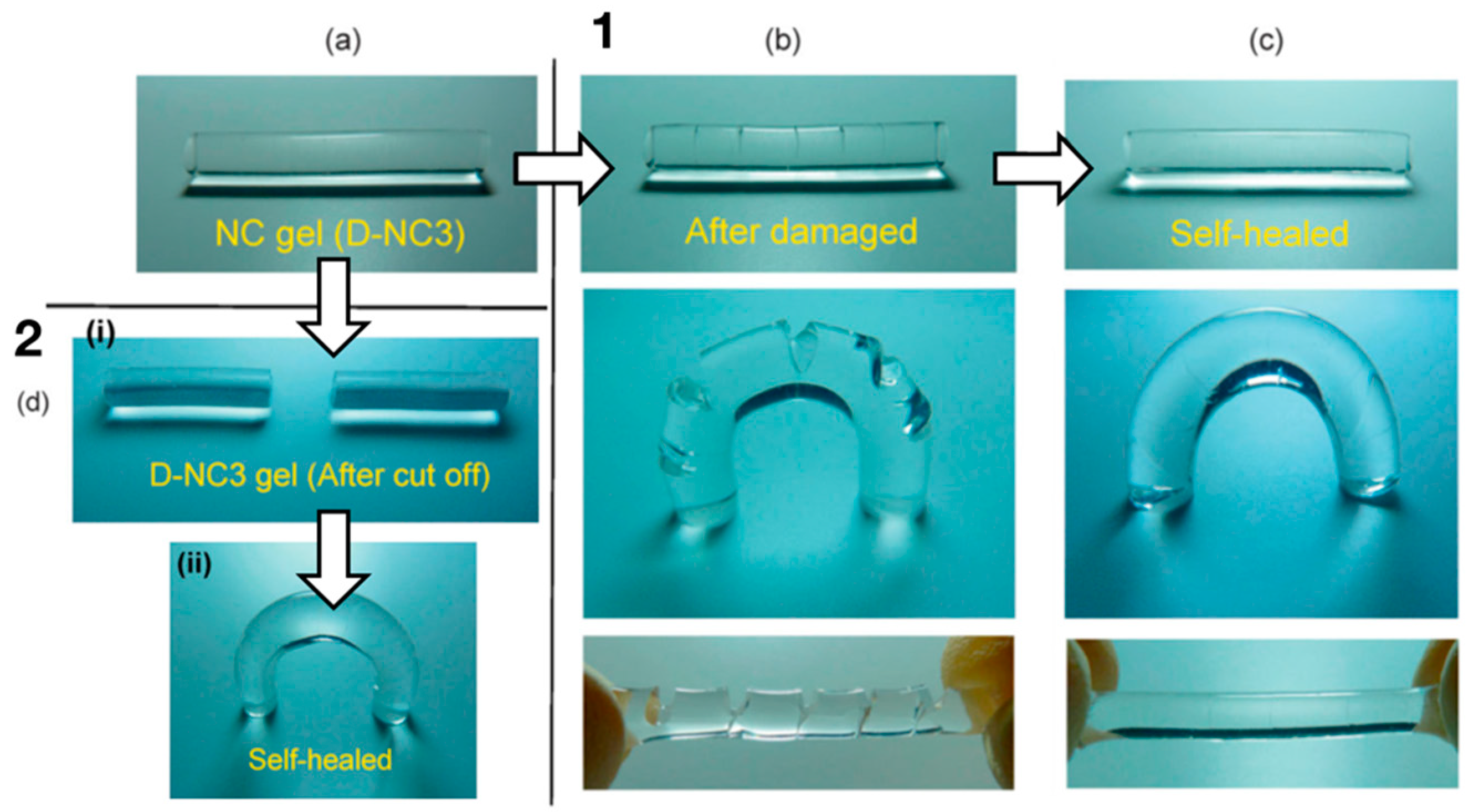
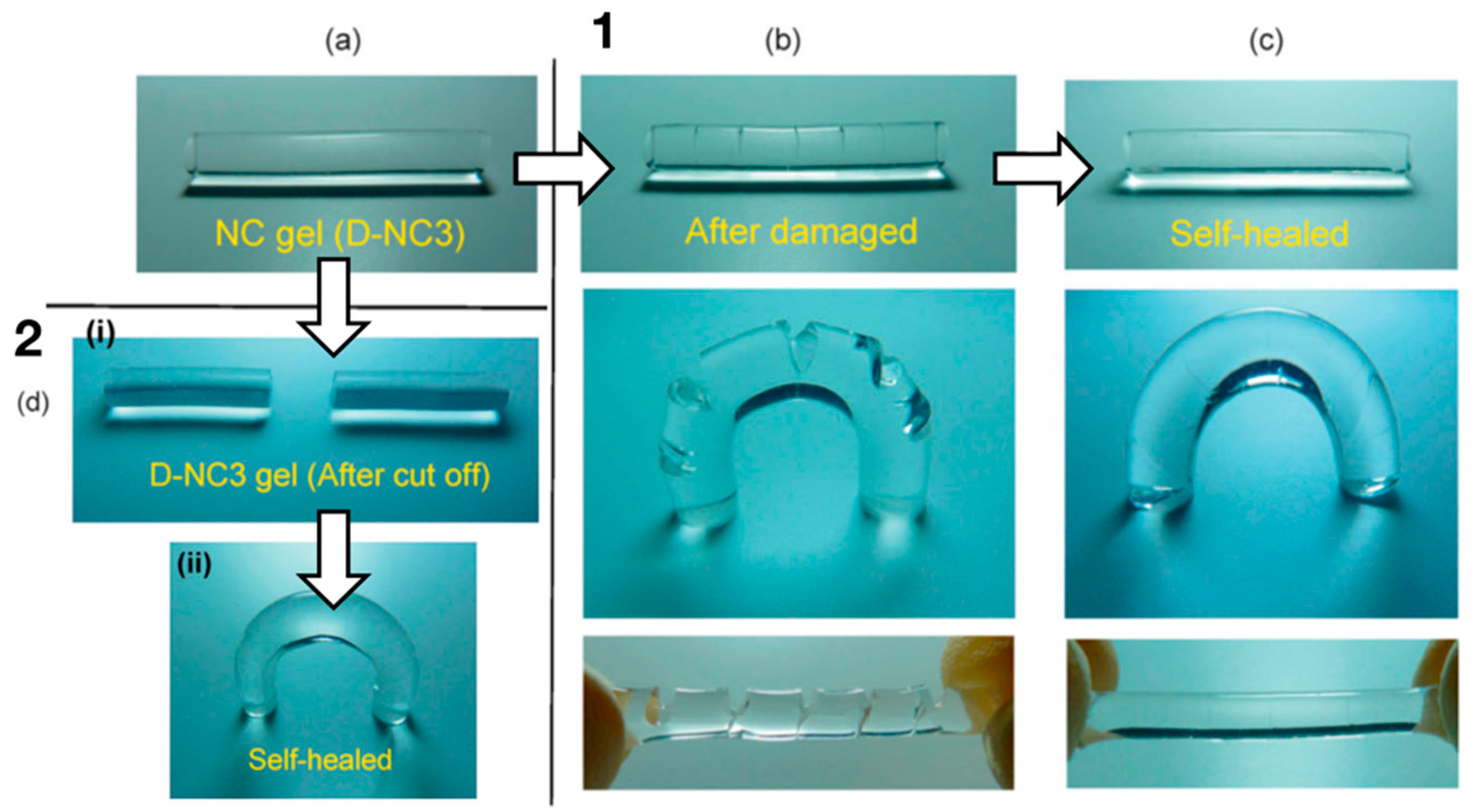
5.1. Self-Healing Hydrogels
5.2. Adhesive Materials
5.3. Nanoparticles for Gluing Hydrogels
5.4. Hydrogels for Improved Nanoparticle Performance
6. Nanoparticles as Tools for Studying and Monitoring Hydrogel Properties
6.1. Crystalline Colloidal Arrays in Hydrogels
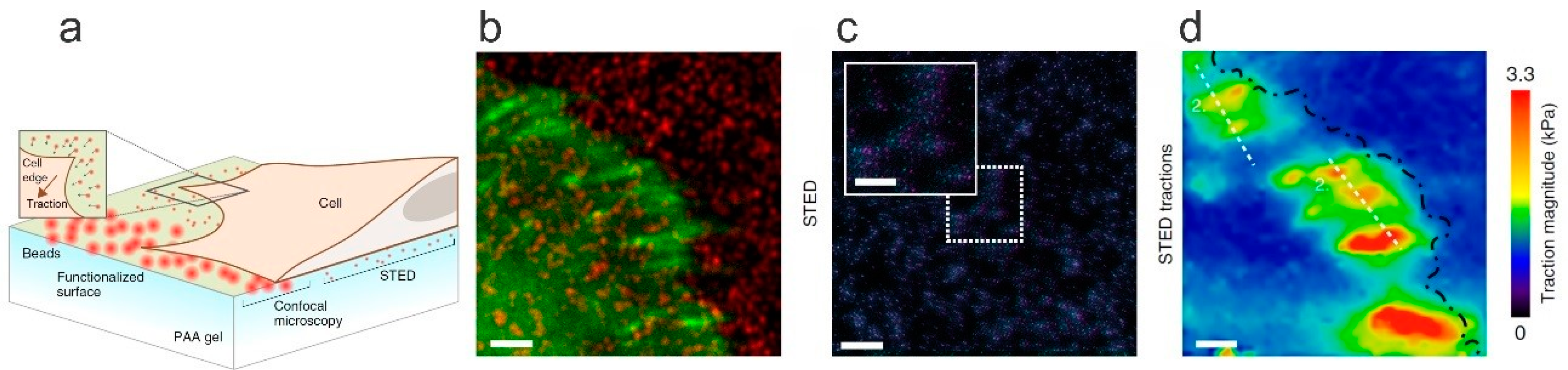
6.2. Traction Force Microscopy
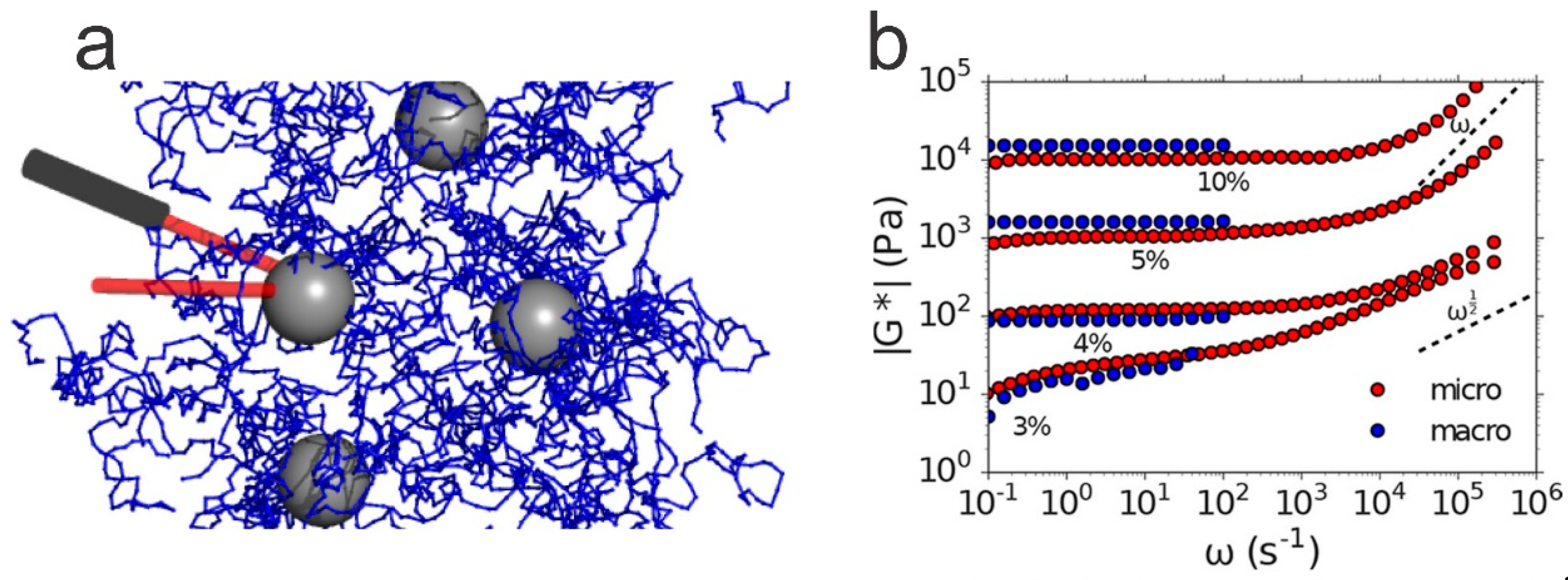
6.3. Micro-Rheology
7. General Comments and Outlook
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barbucci, R. (Ed.) Hydrogels Biological Properties and Applications; Springer: Milan, Italy, 2010. [Google Scholar]
- Bohidar, H.B.; Dubin, P.; Osade, Y. (Eds.) Polymer Gels: Fundamentals and Applications; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Huglin, M.R. Hydrogels in Medicine and Pharmacy; Peppas, N.A., Ed.; CRC Press Inc.: Boca Raton, FL, USA, 1986. [Google Scholar]
- Wichterle, O. Soft Contact Lenses; Ruben, M., Ed.; Wiley: New York, NY, USA, 1978; pp. 3–5. [Google Scholar]
- Haider, M.; Megeed, Z.; Ghandehari, H. Genetically engineered polymers: Status and prospects for controlled release. J. Controlled Release 2004, 95, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.; Gutowska, A. Lessons from nature: Stimuli-responsive polymers and their biomedical applications. Trends Biotech. 2002, 20, 305–311. [Google Scholar] [CrossRef]
- Langer, R.; Tirrell, D.A. Designing materials for biology and medicine. Nature 2004, 428, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Megeed, Z.; Cappello, J.; Ghandehari, H. Genetically engineered silk-elastinlike protein polymers for controlled drug delivery. Adv. Drug Deliv. Rev. 2002, 54, 1075–1091. [Google Scholar] [CrossRef]
- Chacko, R.T.; Ventura, J.; Zhuang, J.M.; Thayumanavan, S. Polymer nanogels: A versatile nanoscopic drug delivery platform. Adv. Drug Deliv. Rev. 2012, 64, 836–851. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Muller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Merino, S.; Martín, C.; Kostarelos, K.; Prato, M.; Vázquez, E. Nanocomposite Hydrogels: 3D Polymer-Nanoparticle Synergies for On-Demand Drug Delivery. ACS Nano 2015, 9, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Okumura, Y.; Ito, K. The polyrotaxane gel: A topological gel by figure-of-eight cross-links. Adv. Mater. 2001, 13, 485–487. [Google Scholar] [CrossRef]
- Karino, T.; Shibayama, M.; Ito, K. Slide-ring gel: Topological gel with freely movable cross-links. Phys. B Condens. Matter 2006, 385, 692–696. [Google Scholar] [CrossRef]
- Ito, K. Novel cross-linking concept of polymer network: Synthesis, structure, and properties of slide-ring gels with freely movable junctions. Polym. J. 2007, 39, 489. [Google Scholar] [CrossRef]
- Bin Imran, A.; Esaki, K.; Gotoh, H.; Seki, T.; Ito, K.; Sakai, Y.; Takeoka, Y. Extremely stretchable thermosensitive hydrogels by introducing slide-ring polyrotaxane cross-linkers and ionic groups into the polymer network. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takehisa, T. Nanocomposite hydrogels: A unique organic-inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties. Adv. Mater. 2002, 14, 1120–1124. [Google Scholar] [CrossRef]
- Creton, C. 50th Anniversary Perspective: Networks and Gels: Soft but Dynamic and Tough. Macromolecules 2017, 50, 8297–8316. [Google Scholar] [CrossRef]
- Haraguchi, K. Nanocomposite hydrogels. Curr. Opin. Solid State Mater. Sci. 2007, 11, 47–54. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Fu, Y.Q.; Jiang, R.; Yao, J.; Xiao, L.; Zeng, G.M. Novel magnetic chitosan/poly(vinyl alcohol) hydrogel beads: Preparation, characterization and application for adsorption of dye from aqueous solution. Bioresour. Technol. 2012, 105, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Gaharwar, A.K.; Peppas, N.A.; Khademhosseini, A. Nanocomposite hydrogels for biomedical applications. Biotechnol. Bioeng. 2014, 111, 441–453. [Google Scholar] [CrossRef]
- Jung, S.; Kaar, J.L.; Stoykovich, M.P. Design and functionalization of responsive hydrogels for photonic crystal biosensors. Mol. Syst. Des. Eng. 2016, 1, 225–241. [Google Scholar] [CrossRef]
- Yetisen, A.K.; Naydenova, I.; Vasconcellos, F.D.; Blyth, J.; Lower, C.R. Holographic Sensors: Three-Dimensional Analyte-Sensitive Nanostructures and Their Applications. Chem. Rev. 2014, 114, 10654–10696. [Google Scholar] [CrossRef]
- Quesada-Perez, M.; Maroto-Centeno, J.; Forcada, J.; Hidalgo-Alvarez, R. Gel swelling theories: The classical formalism and recent approaches. Soft Matter 2011, 7, 10536–10547. [Google Scholar] [CrossRef]
- Schröder, U.P.; Oppermann, W. Properties of polyelectrolyte gels. In Physical Properties of Polymeric Gels; Wiley: Chichester, West Sussex, England, 1996; pp. 19–38. [Google Scholar]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Raemdonck, K.; Demeester, J.; De Smedt, S. Advanced nanogel engineering for drug delivery. Soft Matter 2009, 5, 707–715. [Google Scholar] [CrossRef]
- Lin, C.H.; Sheng, D.K.; Liu, X.D.; Xu, S.B.; Ji, F.; Dong, L.; Zhou, Y.; Yang, Y.M. A self-healable nanocomposite based on dual-crosslinked Graphene Oxide/Polyurethane. Polymer 2017, 127, 241–250. [Google Scholar] [CrossRef]
- Qiao, Z.Y.; Zhang, R.; Du, F.S.; Liang, D.H.; Li, Z.C. Multi-responsive nanogels containing motifs of ortho ester, oligo(ethylene glycol) and disulfide linkage as carriers of hydrophobic anti-cancer drugs. J. Controlled Release 2011, 152, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Kobitski, A.Y.; Nienhaus, G.U.; Delaittre, G. A simple route to highly active single-enzyme nanogels. Chem. Sci. 2018, 9, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, A.V.; Vinogradov, S.V. Nanogels as Pharmaceutical Carriers: Finite Networks of Infinite Capabilities. Angew. Chem. 2009, 48, 5418–5429. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Hayashi, H.; Michihiro, I.D.; Nagasaki, Y. Endosomal release and intracellular delivery of anticancer drugs using pH-sensitive PEGylated nanogels. J. Mater. Chem. 2007, 17, 3720–3725. [Google Scholar] [CrossRef]
- Nazli, C.; Demirer, G.S.; Yar, Y.; Acar, H.Y.; Kizilel, S. Targeted delivery of doxorubicin into tumor cells via MMP-sensitive PEG hydrogel-coated magnetic iron oxide nanoparticles (MIONPs). Colloids Surf. B 2014, 122, 674–683. [Google Scholar] [CrossRef]
- Chen, T.; Hou, K.; Ren, Q.Y.; Chen, G.Y.; Wei, P.L.; Zhu, M.F. Nanoparticle-Polymer Synergies in Nanocomposite Hydrogels: From Design to Application. Macromol. Rapid Commun. 2018, 39. [Google Scholar] [CrossRef]
- Ma, Y.K.; Ge, Y.X.; Li, L.B. Advancement of multifunctional hybrid nanogel systems: Construction and application in drug co-delivery and imaging technique. Mater. Sci. Eng. C 2017, 71, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.F.; Yang, W.L.; Min, K.; Zha, L.S.; Wang, C.C.; Fu, S.K. Thermosensitive poly (N-isopropylacrylamide) nanocapsules with controlled permeability. Polymer 2005, 46, 1087–1093. [Google Scholar] [CrossRef]
- Palioura, D.; Armes, S.P.; Anastasiadis, S.H.; Vamvakaki, M. Metal nanocrystals incorporated within pH-responsive microgel particles. Langmuir 2007, 23, 5761–5768. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kwon, J.E.; Lee, K.; Koh, W.-G. Signal-amplifying nanoparticle/hydrogel hybrid microarray biosensor for metal-enhanced fluorescence detection of organophosphorus compounds. Biofabrication 2018, 10, 035002. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.W.; Rodarte, A.L.; Som, M.; Arya, G.; Tao, A.R. Colloidal Plasmonic Nanocomposites: From Fabrication to Optical Function. Chem. Rev. 2018, 118, 3100–3120. [Google Scholar] [CrossRef] [PubMed]
- Wahid, F.; Zhong, C.; Wang, H.S.; Hu, X.H.; Chu, L.Q. Recent Advances in Antimicrobial Hydrogels Containing Metal Ions and Metals/Metal Oxide Nanoparticles. Polymers 2017, 9, 636. [Google Scholar] [CrossRef]
- Strong, L.E.; West, J.L. Thermally responsive polymer-nanoparticle composites for biomedical applications. Wiley Interdiscip. Rev.-Nanomed. Nanobiotech. 2011, 3, 307–317. [Google Scholar] [CrossRef]
- Hezaveh, H.; Muhamad, II. Effect of MgO nanofillers on burst release reduction from hydrogel nanocomposites. J. Mater. Sci. 2013, 24, 1443–1453. [Google Scholar] [CrossRef]
- Zhang, J.G.; Xu, S.Q.; Kumacheva, E. Polymer microgels: Reactors for semiconductor, metal, and magnetic nanoparticles. J. Am. Chem. Soc. 2004, 126, 7908–7914. [Google Scholar] [CrossRef]
- Wang, C.; Flynn, N.T.; Langer, R. Controlled structure and properties of thermoresponsive nanoparticle-hydrogel composites. Adv. Mater. 2004, 16, 1074–1079. [Google Scholar] [CrossRef]
- Pardo-Yissar, V.; Gabai, R.; Shipway, A.N.; Bourenko, T.; Willner, I. Gold nanoparticle/hydrogel composites with solvent-switchable electronic properties. Adv. Mater. 2001, 13, 1320–1323. [Google Scholar] [CrossRef]
- Jones, C.D.; Lyon, L.A. Photothermal patterning of microgel/gold nanoparticle composite colloidal crystals. J. Am. Chem. Soc. 2003, 125, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Huang, S.J.; Li, X.J.; Zhang, L.H.; Tan, Y.Y.; Wei, C.; Lv, J. Facile fabrication of novel polyhedral oligomeric silsesquioxane/carboxymethyl cellulose hybrid hydrogel based on supermolecular interactions. Mater. Lett. 2013, 90, 142–144. [Google Scholar] [CrossRef]
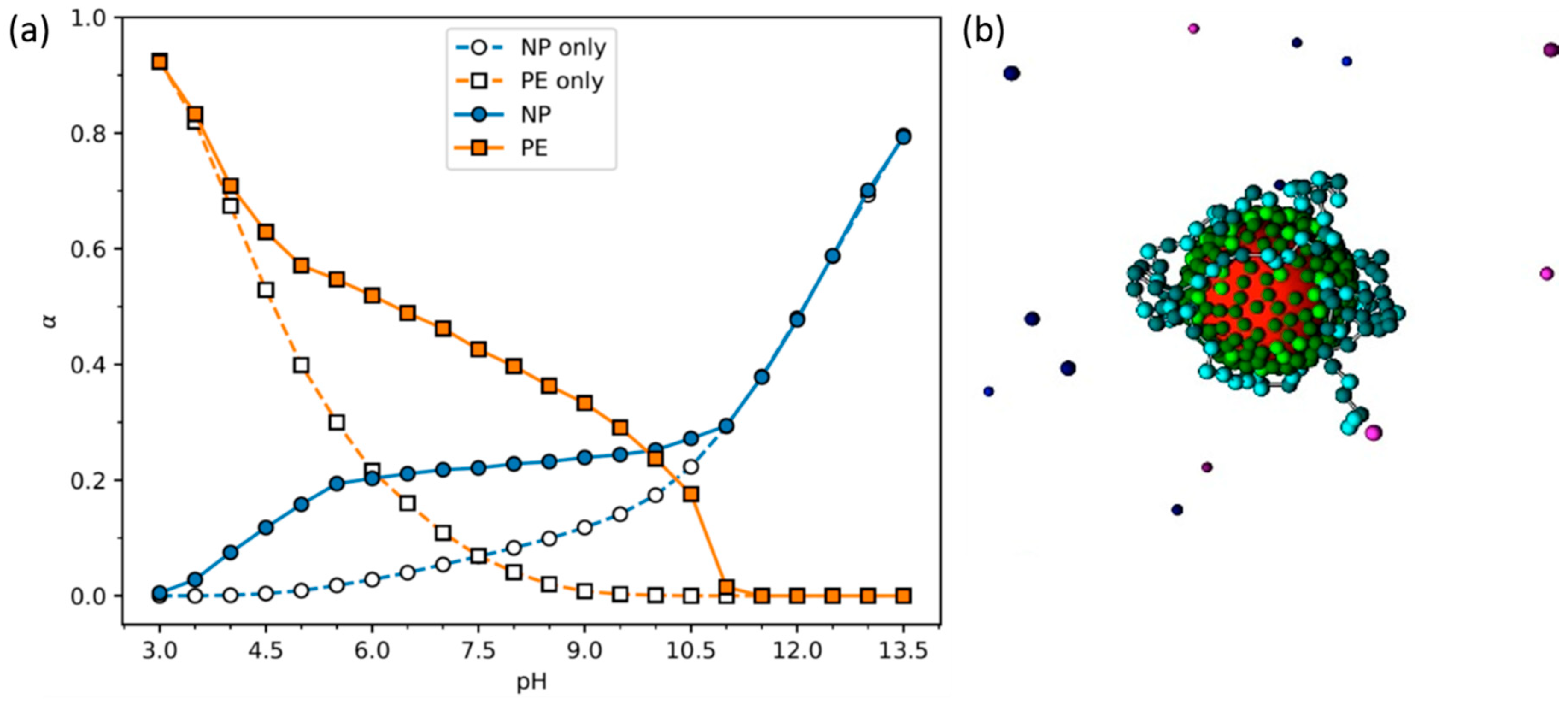
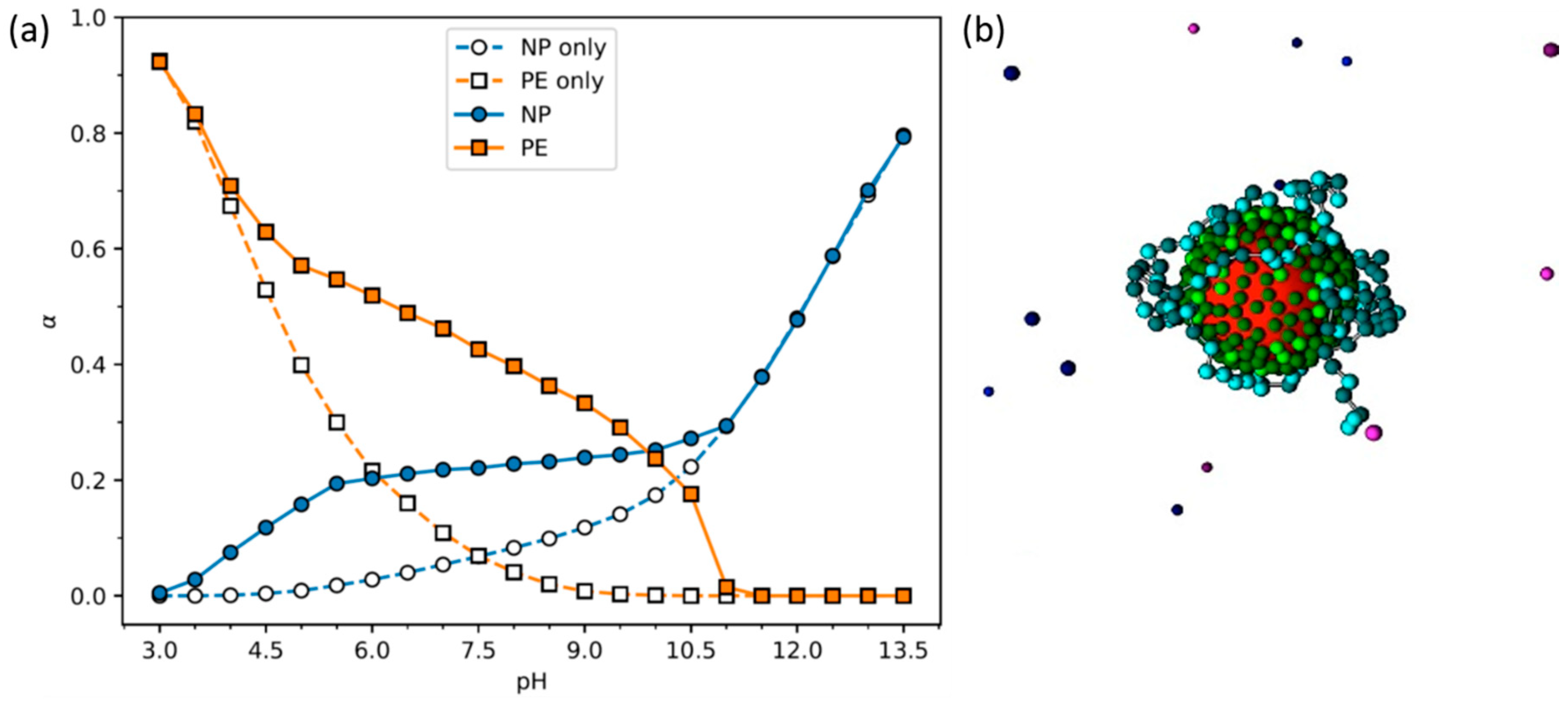
- Stornes, M.; Linse, P.; Dias, R.S. Monte Carlo Simulations of Complexation between Weak Polyelectrolytes and a Charged Nanoparticle. Influence of Polyelectrolyte Chain Length and Concentration. Macromolecules 2017, 50, 5978–5988. [Google Scholar] [CrossRef]
- Stornes, M.; Shrestha, B.; Dias, R.S. pH-Dependent Polyelectrolyte Bridging of Charged Nanoparticles. J. Phys. Chem. B 2018, 122, 10237–10246. [Google Scholar] [CrossRef] [PubMed]
- Portehault, D.; Petit, L.; Pantoustier, N.; Ducouret, G.; Lafuma, F.; Hourdet, D. Hybrid thickeners in aqueous media. Colloids Surf. A 2006, 278, 26–32. [Google Scholar] [CrossRef]
- Carlsson, L.; Rose, S.; Hourdet, D.; Marcellan, A. Nano-hybrid self-crosslinked PDMA/silica hydrogels. Soft Matter 2010, 6, 3619–3631. [Google Scholar] [CrossRef]
- Pasqui, D.; Atrei, A.; Giani, G.; De Cagna, M.; Barbucci, R. Metal oxide nanoparticles as cross-linkers in polymeric hybrid hydrogels. Mater. Lett. 2011, 65, 392–395. [Google Scholar] [CrossRef]
- Garcia-Astrain, C.; Chen, C.; Buron, M.; Palomares, T.; Eceiza, A.; Fruk, L.; Corcuera, M.A.; Gabilondo, N. Biocompatible Hydrogel Nanocomposite with Covalently Embedded Silver Nanoparticles. Biomacromolecules 2015, 16, 1301–1310. [Google Scholar] [CrossRef]
- da Silva, E.P.; Guilherme, M.R.; Garcia, F.P.; Nakamura, C.V.; Cardozo, L.; Alonso, C.G.; Rubira, A.F.; Kunita, M.H. Drug release profile and reduction in the in vitro burst release from pectin/HEMA hydrogel nanocomposites crosslinked with titania. RSC Adv. 2016, 6, 19060–19068. [Google Scholar] [CrossRef]
- Garcia-Astrain, C.; Hernandez, R.; Guaresti, O.; Fruk, L.; Mijangos, C.; Eceiza, A.; Gabilondo, N. Click Crosslinked Chitosan/Gold Nanocomposite Hydrogels. Macromol. Mater. Eng. 2016, 301, 1295–1300. [Google Scholar] [CrossRef]
- Bardajee, G.R.; Hooshyar, Z.; Asli, M.J.; Shahidi, F.E.; Dianatnejad, N. Synthesis of a novel supermagnetic iron oxide nanocomposite hydrogel based on graft copolymerization of poly((2-dimethylamino)ethyl methacrylate) onto salep for controlled release of drug. Mater. Sci. Eng. C 2014, 36, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, B.Z.; Zhang, F. Facile One-Pot Synthesis of Iron Oxide Nanoparticles Cross-linked Magnetic Poly(vinyl alcohol) Gel Beads for Drug Delivery. ACS Appl. Mater. Interfaces 2012, 4, 192–199. [Google Scholar] [CrossRef] [PubMed]
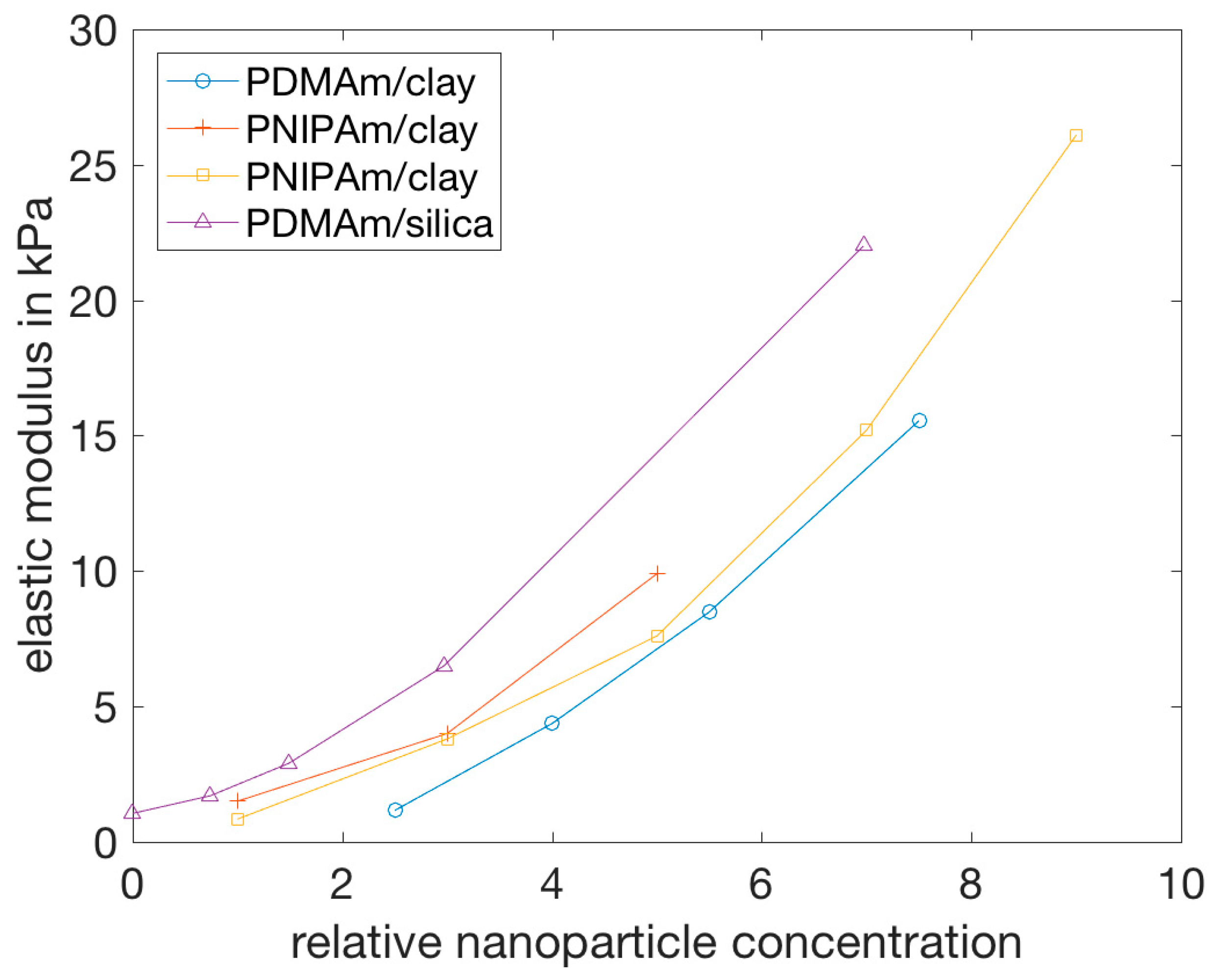
- Haraguchi, K.; Takehisa, T.; Fan, S. Effects of clay content on the properties of nanocomposite hydrogels composed of poly(N-isopropylacrylamide) and clay. Macromolecules 2002, 35, 10162–10171. [Google Scholar] [CrossRef]
- Miyazaki, S.; Karino, T.; Endo, H.; Haraguchi, K.; Shibayama, M. Clay concentration dependence of microstructure in deformed poly(N-isopropylacrylamide)-clay nanocomposite gels. Macromolecules 2006, 39, 8112–8120. [Google Scholar] [CrossRef]
- Tan, C.L.; Cao, X.H.; Wu, X.J.; He, Q.Y.; Yang, J.; Zhang, X.; Chen, J.Z.; Zhao, W.; Han, S.K.; Nam, G.H.; et al. Recent Advances in Ultrathin Two-Dimensional Nanomaterials. Chem. Rev. 2017, 117, 6225–6331. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.J.; Ishida, Y.; Ebina, Y.; Sasaki, T.; Hikima, T.; Takata, M.; Aida, T. An anisotropic hydrogel with electrostatic repulsion between cofacially aligned nanosheets. Nature 2015, 517, 68–72. [Google Scholar] [CrossRef]
- Sun, J.W.; Schmidt, B.; Wang, X.; Shalom, M. Self-Standing Carbon Nitride-Based Hydrogels with High Photocatalytic Activity. ACS Appl. Mater. Interfaces 2017, 9, 2029–2034. [Google Scholar] [CrossRef]
- Hata, Y.; Kojima, T.; Koizumi, T.; Okura, H.; Sakai, T.; Sawada, T.; Serizawa, T. Enzymatic Synthesis of Cellulose Oligomer Hydrogels Composed of Crystalline Nanoribbon Networks under Macromolecular Crowding Conditions. ACS Macro Lett. 2017, 6, 165–170. [Google Scholar] [CrossRef]
- Hata, Y.; Sawada, T.; Serizawa, T. Effect of solution viscosity on the production of nanoribbon network hydrogels composed of enzymatically synthesized cellulose oligomers under macromolecular crowding conditions. Polym. J. 2017, 49, 575–581. [Google Scholar] [CrossRef]
- Ko, J.W.; Choi, W.S.; Kim, J.; Kuk, S.K.; Lee, S.H.; Park, C.B. Self-Assembled Peptide-Carbon Nitride Hydrogel as a Light-Responsive Scaffold Material. Biomacromolecules 2017, 18, 3551–3556. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Prevoteau, A.; Elziere, P.; Hourdet, D.; Marcellan, A.; Leibler, L. Nanoparticle solutions as adhesives for gels and biological tissues. Nature 2014, 505, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mynar, J.L.; Yoshida, M.; Lee, E.; Lee, M.; Okuro, K.; Kinbara, K.; Aida, T. High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 2010, 463, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Tamesue, S.; Ohtani, M.; Yamada, K.; Ishida, Y.; Spruell, J.M.; Lynd, N.A.; Hawker, C.J.; Aida, T. Linear versus Dendritic Molecular Binders for Hydrogel Network Formation with Clay Nanosheets: Studies with ABA Triblock Copolyethers Carrying Guanidinium Ion Pendants. J. Am. Chem. Soc. 2013, 135, 15650–15655. [Google Scholar] [CrossRef] [PubMed]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, H.; Choi, Y.; Lee, D.S.; Kim, J.; Yi, G.R. Colloidal Mesoporous Silica Nanoparticles as Strong Adhesives for Hydrogels and Biological Tissues. ACS Appl. Mater. Interfaces 2017, 9, 31469–31477. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Dobrynin, A. Nanoparticles as Adhesives for Soft Polymeric Materials. Macromolecules 2016, 49, 3586–3592. [Google Scholar] [CrossRef]
- Meddahi-Pelle, A.; Legrand, A.; Marcellan, A.; Louedec, L.; Letourneur, D.; Leibler, L. Organ Repair, Hemostasis, and In Vivo Bonding of Medical Devices by Aqueous Solutions of Nanoparticles. Angew. Chem. 2014, 53, 6369–6373. [Google Scholar] [CrossRef]
- Nejadnik, M.R.; Yang, X.; Bongio, M.; Alghamdi, H.S.; Van den Beucken, J.; Huysmans, M.C.; Jansen, J.A.; Hilborn, J.; Ossipov, D.; Leeuwenburgh, S.C.G. Self-healing hybrid nanocomposites consisting of bisphosphonated hyaluronan and calcium phosphate nanoparticles. Biomaterials 2014, 35, 6918–6929. [Google Scholar] [CrossRef]
- Abe, H.; Hara, Y.; Maeda, S.; Hashimoto, S. Adhesion of Gels by Silica Particle. J. Phys. Chem. B 2014, 118, 2518–2522. [Google Scholar] [CrossRef]
- Napper, D. Polymeric Stabilization of Colloidal Particles; Academic Press: London, UK, 1983. [Google Scholar]
- Ottewill, R.H. Stability and Instability in Disperse Systems. J. Colloid Interface Sci. 1977, 58, 357–373. [Google Scholar] [CrossRef]
- Overbeek, J.T.G. Recent Developments in Understanding of Colloid Stability. J. Colloid Interface Sci. 1977, 58, 408–422. [Google Scholar] [CrossRef]
- Amiel, C.; Sebille, B. FTIR Study of Copolymer Adsorption on Silica. J. Colloid Interface Sci. 1992, 149, 481–492. [Google Scholar] [CrossRef]
- Perrin, E.; Schoen, M.; Coudert, F.; Boutin, A. Structure and Dynamics of Solvated Polymers near a Silica Surface: On the Different Roles Played by Solvent. J. Phys. Chem. B 2018, 122, 4573–4582. [Google Scholar] [CrossRef] [PubMed]
- Greenland, D.J. Interactions between organic polymers and inorganic soil particles. In Proceeding Symposium on Fundamentals of Soil Conditioning; De Boodt, M., Ed.; State University of Ghent: Ghent, Belgium, 1972; pp. 897–914. [Google Scholar]
- Rubio, J.; Kitchener, J.A. Mechanism of Adsorption of Poly(Ethylene Oxide) Flocculant on Silica. J. Colloid Interface Sci. 1976, 57, 132–142. [Google Scholar] [CrossRef]
- Mathur, S.; Moudgil, B.M. Adsorption mechanism(s) of poly (ethylene oxide) on oxide surfaces. J. Colloid Interface Sci. 1997, 196, 92–98. [Google Scholar] [CrossRef]
- Tadros, T.F. The interaction of cetyltrimethylammonium bromide and sodium dodecylbenzene sulfonate with polyvinyl alcohol. adsorption of the polymer—Surfactant complexes on silica. I. Colloid Interface Sci. 1974, 46, 528–540. [Google Scholar] [CrossRef]
- Postmus, B.; Leermakers, F.; Stuart, M. Self-consistent field modeling of poly(ethylene oxide) adsorption onto silica: The multiple roles of electrolytes. Langmuir 2008, 24, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, E.; Biesheuvel, P.; de Vos, W. Adsorption of charged and neutral polymer chains on silica surfaces: The role of electrostatics, volume exclusion, and hydrogen bonding. Phys. Rev. E 2015, 91, 012601. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Aoyama, Y.; Yamanaka, J.; Toyotama, A.; Okuzono, T.J. Particle Adsorption on Hydrogel Surfaces in Aqueous Media due to van der Waals Attraction. Sci. Rep. 2017, 7, 6099. [Google Scholar] [CrossRef] [PubMed]
- He, P.S.; Tang, X.W.; Chen, L.; Xie, P.W.; He, L.; Zhou, H.; Zhang, D.; Fan, T.X. Patterned Carbon Nitride-Based Hybrid Aerogel Membranes via 3D Printing for Broadband Solar Wastewater Remediation. Adv. Funct. Mat. 2018, 28, 1801121. [Google Scholar] [CrossRef]
- Nie, L.; Liu, C.H.; Wang, J.; Shuai, Y.; Cui, X.Y.; Liu, L. Effects of surface functionalized graphene oxide on the behavior of sodium alginate. Carbohydr. Polym. 2015, 117, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Li, C.; Wang, X.L.; Shi, G.Q. A pH-sensitive graphene oxide composite hydrogel. Chem. Commun. 2010, 46, 2376–2378. [Google Scholar] [CrossRef]
- Liu, L.Q.; Barber, A.H.; Nuriel, S.; Wagner, H.D. Mechanical properties of functionalized single-walled carbon-nanotube/poly(vinyl alcohol) nanocomposites. Adv. Funct. Mater. 2005, 15, 975–980. [Google Scholar] [CrossRef]
- Reid, M.; Villalobos, M.; Cranston, E. The role of hydrogen bonding in non-ionic polymer adsorption to cellulose nanocrystals and silica colloids. Curr. Opin. Colloid Interface Sci. 2017, 29, 76–82. [Google Scholar] [CrossRef]
- Lindman, B.; Karlstrom, G.; Stigsson, L. On the mechanism of dissolution of cellulose. J. Mol. Liq. 2010, 156, 76–81. [Google Scholar] [CrossRef]
- Medronho, B.; Romano, A.; Miguel, M.; Stigsson, L.; Lindman, B. Rationalizing cellulose (in)solubility: Reviewing basic physicochemical aspects and role of hydrophobic interactions. Cellulose 2012, 19, 581–587. [Google Scholar] [CrossRef]
- Jiang, W.J.; Luo, W.J.; Zong, R.L.; Yao, W.Q.; Li, Z.P.; Zhu, Y.F. Polyaniline/Carbon Nitride Nanosheets Composite Hydrogel: A Separation-Free and High-Efficient Photocatalyst with 3D Hierarchical Structure. Small 2016, 12, 4370–4378. [Google Scholar] [CrossRef]
- Kumru, B.; Shalom, M.; Antonietti, M.; Schmidt, B. Reinforced Hydrogels via Carbon Nitride Initiated Polymerization. Macromolecules 2017, 50, 1862–1869. [Google Scholar] [CrossRef]
- Liu, H.; Peng, Y.; Yang, C.; Wang, M. Silica Nanoparticles as Adhesives for Biological Tissues? Re-Examining the Effect of Particles Size, Particle Shape, and the Unexpected Role of Base. Part. Part. Syst. Charact. 2017, 34. [Google Scholar] [CrossRef]
- Mallam, S.; Horkay, F.; Hecht, A.M.; Geissler, E. Scattering and Swelling Properties of Inhomogeneous Polyacrylamide Gels. Macromolecules 1989, 22, 3356–3361. [Google Scholar] [CrossRef]
- Okajima, T.; Harada, I.; Nishio, K.; Hirotsu, S. Discontinuous crossover between fast and slow kinetics at the volume phase transition in poly-N-isopropylacrylamide gels. Jpn. J. Appl. Phys. 2 2000, 39, L875–L877. [Google Scholar] [CrossRef]
- Zhao, X.; Huebsch, N.; Mooney, D.; Suo, Z. Stress-relaxation behavior in gels with ionic and covalent crosslinks. J. Appl. Phys. 2010, 107. [Google Scholar] [CrossRef] [PubMed]
- Valles-Lluch, A.; Poveda-Reyes, S.; Amoros, P.; Beltran, D.; Pradas, M.M. Hyaluronic Acid-Silica Nanohybrid Gels. Biomacromolecules 2013, 14, 4217–4225. [Google Scholar] [CrossRef] [PubMed]
- Boonmahitthisud, A.; Nakajima, L.; Nguyen, K.D.; Kobayashi, T. Composite effect of silica nanoparticle on the mechanical properties of cellulose-based hydrogels derived from cottonseed hulls. J. Appl. Polym. Sci. 2017, 134, 44557. [Google Scholar] [CrossRef]
- Du, J.; Wu, R.L.; Liu, H.H.; Nie, X.D.; Li, H.L.; Xu, S.M.; Wang, J.D. Synthesis of Amphoteric Nanocomposite Hydrogels with Ultrahigh Tensibility. Polym. Compos. 2015, 36, 538–544. [Google Scholar] [CrossRef]
- Ben Ahmed, N.; Ronsin, O.; Mouton, L.; Sicard, C.; Yepremian, C.; Baumberger, T.; Brayner, R.; Coradin, T. The physics and chemistry of silica-in-silicates nanocomposite hydrogels and their phycocompatibility. J. Mater. Chem. B 2017, 5, 2931–2940. [Google Scholar] [CrossRef]
- Yang, C.C.; Xue, R.; Zhang, Q.S.; Yang, S.L.; Liu, P.F.; Chen, L.; Wang, K.; Zhang, X.Y.; Wei, Y. Nanoclay cross-linked semi-IPN silk sericin/poly(NIPAm/LMSH) nanocomposite hydrogel: An outstanding antibacterial wound dressing. Mater. Sci. Eng. C 2017, 81, 303–313. [Google Scholar] [CrossRef]
- Wang, T.; Zheng, S.; Sun, W.; Liu, X.; Fu, S.; Tong, Z. Notch insensitive and self-healing PNIPAm-PAM-clay nanocomposite hydrogels. Soft Matter 2014, 10, 3506–3512. [Google Scholar] [CrossRef]
- Shibayama, M.; Karino, T.; Miyazaki, S.; Okabe, S.; Takehisa, T.; Haraguchi, K. Small-angle neutron scattering study on uniaxially stretched poly(N-isopropylacrylamide)-clay nanocomposite gels. Macromolecules 2005, 38, 10772–10781. [Google Scholar] [CrossRef]
- Haraguchi, K.; Farnworth, R.; Ohbayashi, A.; Takehisa, T. Compositional effects on mechanical properties of nanocomposite hydrogels composed of poly(N,N-dimethylacrylamide) and clay. Macromolecules 2003, 36, 5732–5741. [Google Scholar] [CrossRef]
- Haraguchi, K.; Uyama, K.; Tanimoto, H. Self-healing in Nanocomposite Hydrogels. Macromol. Rapid Commun. 2011, 32, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Chang, H.; Wang, M.; Xu, F.; Yang, J. High-Strength, Tough, and Self-Healing Nanocomposite Physical Hydrogels Based on the Synergistic Effects of Dynamic Hydrogen Bond and Dual Coordination Bonds. ACS Appl. Mater. Interfaces 2017, 9, 28305–28318. [Google Scholar] [CrossRef] [PubMed]
- Kostina, N.; Sharifi, S.; Pereira, A.; Michalek, J.; Grijpma, D.; Rodriguez-Emmenegger, C. Novel antifouling self-healing poly(carboxybetaine methacrylamide-co-HEMA) nanocomposite hydrogels with superior mechanical properties. J. Mat. Chem. B 2013, 1, 5644–5650. [Google Scholar] [CrossRef]
- Jeong, S.H.; Koh, Y.H.; Kim, S.W.; Park, J.U.; Kim, H.E.; Song, J. Strong and Biostable Hyaluronic Acid-Calcium Phosphate Nanocomposite Hydrogel via in Situ Precipitation Process. Biomacromolecules 2016, 17, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Su, E.; Okay, O. Hybrid cross-linked poly(2-acrylamido-2-methyl-1-propanesulfonic acid) hydrogels with tunable viscoelastic, mechanical and self-healing properties. React. Funct. Polym. 2018, 123, 70–79. [Google Scholar] [CrossRef]
- Pan, C.; Liu, L.; Chen, Q.; Zhang, Q.; Guo, G. Tough, Stretchable, Compressive Novel Polymer/Graphene Oxide Nanocomposite Hydrogels with Excellent Self-Healing Performance. ACS Appl. Mater. Interfaces 2017, 9, 38052–38061. [Google Scholar] [CrossRef]
- Gao, G.; Du, G.; Sun, Y.; Fu, J. Self-Healable, Tough, and Ultrastretchable Nanocomposite Hydrogels Based on Reversible Polyacrylamide/Montmorillonite Adsorption. ACS Appl. Mater. Interfaces 2015, 7, 5029–5037. [Google Scholar] [CrossRef]
- Kokabi, M.; Sirousazar, M.; Hassan, Z.M. PVA-clay nanocomposite hydrogels for wound dressing. Eur. Polym. J. 2007, 43, 773–781. [Google Scholar] [CrossRef]
- Zhong, M.; Liu, Y.; Xie, X. Self-healable, super tough graphene oxide-poly(acrylic acid) nanocomposite hydrogels facilitated by dual cross-linking effects through dynamic ionic interactions. J. Mater. Chem. B 2015, 3, 4001–4008. [Google Scholar] [CrossRef]
- Gonzalez-Dominguez, J.M.; Martin, C.; Dura, O.J.; Merino, S.; Vazquez, E. Smart Hybrid Graphene Hydrogels: A Study of the Different Responses to Mechanical Stretching Stimulus. ACS Appl. Mater. Interfaces 2018, 10, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Binder, W.H. Self-Healing Polymers: From Principles to Applications; Wiley-VCH: Weinheim, Germany, 2013; p. 446. [Google Scholar]
- Yang, J.; Zhu, L.; Yan, X.; Wei, D.; Qin, G.; Liu, B.; Liu, S.; Chen, Q. Hybrid nanocomposite hydrogels with high strength and excellent self-recovery performance. RSC Adv. 2016, 6, 59131–59140. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, G.; Feng, X.; Liu, H.; Li, F.; Wang, M.; Li, H. Room-temperature self-healing tough nanocomposite hydrogel crosslinked by zirconium hydroxide nanoparticles. Compos. Sci. Tech. 2017, 140, 54–62. [Google Scholar] [CrossRef]
- Shao, C.; Wang, M.; Chang, H.; Xu, F.; Yang, J. A Self-Healing Cellulose Nanocrystal-Poly(ethylene glycol) Nanocomposite Hydrogel via Diels-Alder Click Reaction. ACS Sustain. Chem. Eng. 2017, 5, 6167–6174. [Google Scholar] [CrossRef]
- Wu, D.; Xu, J.; Chen, Y.; Yi, M.; Wang, Q. Gum Arabic: A promising candidate for the construction of physical hydrogels exhibiting highly stretchable, self-healing and tensility reinforcing performances. Carbohydr. Polym. 2018, 181, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gao, Z.; Yu, K. Interfacial self-healing of nanocomposite hydrogels: Theory and experiment. J. Mech. Phys. Solids 2017, 109, 288–306. [Google Scholar] [CrossRef]
- Diba, M.; An, J.; Schmidt, S.; Hembury, M.; Ossipov, D.; Boccaccini, A.; Leeuwenburgh, S. Exploiting Bisphosphonate-Bioactive-Glass Interactions for the Development of Self-Healing and Bioactive Composite Hydrogels. Macromol. Rapid Commun. 2016, 37, 1952–1959. [Google Scholar] [CrossRef]
- Peng, R.; Yu, Y.; Chen, S.; Yang, Y.; Tang, Y. Conductive nanocomposite hydrogels with self-healing property. RSC Adv. 2014, 4, 35149–35155. [Google Scholar] [CrossRef]
- Zhang, E.; Wang, T.; Zhao, L.; Sun, W.; Liu, X.; Tong, Z. Fast Self-Healing of Graphene Oxide-Hectorite Clay-Poly(N,N-dimethylacrylamide) Hybrid Hydrogels Realized by Near-Infrared Irradiation. ACS Appl. Mater. Interfaces 2014, 6, 22855–22861. [Google Scholar] [CrossRef]
- Wang, B.; Jeon, Y.; Park, H.; Kim, J. Self-healable mussel-mimetic nanocomposite hydrogel based on catechol-containing polyaspartamide and graphene oxide. Mater. Sci. Eng. C 2016, 69, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Peak, C.W.; Wilker, J.J.; Schmidt, G. A review on tough and sticky hydrogels. Colloid Polym. Sci. 2013, 291, 2031–2047. [Google Scholar] [CrossRef]
- Wu, C.J.; Wilker, J.J.; Schmidt, G. Robust and Adhesive Hydrogels from Cross-Linked Poly(ethylene glycol) and Silicate for Biomedical Use. Macromol. Biosci. 2013, 13, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Gaharwar, A.K.; Rivera, C.P.; Wu, C.J.; Schmidt, G. Transparent, elastomeric and tough hydrogels from poly(ethylene glycol) and silicate nanoparticles. Acta Biomater. 2011, 7, 4139–4148. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Boyer, C.; Gaborit, V.; Rouillon, T.; Guicheux, J.; Tassin, J.F.; Geoffroy, V.; Rethore, G.; Weiss, P. A Cellulose/Laponite Interpenetrated Polymer Network (IPN) Hydrogel: Controllable Double-Network Structure with High Modulus. Polymers 2018, 10, 634. [Google Scholar] [CrossRef]
- Geim, A.K.; Grigorieva, I.V. Van der Waals heterostructures. Nature 2013, 499, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Frisenda, R.; Navarro-Moratalla, E.; Gant, P.; De Lara, D.; Jarillo-Herrero, P.; Gorbachev, R.; Castellanos-Gomez, A. Recent progress in the assembly of nanodevices and van der Waals heterostructures by deterministic placement of 2D materials. Chem. Soc. Rev. 2018, 47, 53–68. [Google Scholar] [CrossRef]
- Zhao, Z.W.; Sun, Y.J.; Dong, F. Graphitic carbon nitride based nanocomposites: A review. Nanoscale 2015, 7, 15–37. [Google Scholar] [CrossRef]
- Ong, W.J.; Tan, L.L.; Ng, Y.H.; Yong, S.T.; Chai, S.P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer to Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Yetisen, A.K.; Butt, H.; Volpatti, L.R.; Pavlichenko, I.; Humar, M.; Kwok, S.J.J.; Koo, H.; Kim, K.S.; Naydenova, I.; Khademhosseini, A.; et al. Photonic hydrogel sensors. Biotechnol. Adv. 2016, 34, 250–271. [Google Scholar] [CrossRef]
- Saito, H.; Takeoka, Y.; Watanabe, M. Simple and precision design of porous gel as a visible indicator for ionic species and concentration. Chem. Commun. 2003, 2126–2127. [Google Scholar] [CrossRef]
- Kamenjicki, M.; Asher, S.A. Epoxide functionalized polymerized crystalline colloidal arrays. Sens. Actuators B 2005, 106, 373–377. [Google Scholar] [CrossRef]
- Holtz, J.H.; Asher, S.A. Polymerized colloidal crystal hydrogel films as intelligent chemical sensing materials. Nature 1997, 389, 829–832. [Google Scholar] [CrossRef]
- Ye, B.F.; Zhao, Y.J.; Cheng, Y.; Li, T.T.; Xie, Z.Y.; Zhao, X.W.; Gu, Z.Z. Colorimetric photonic hydrogel aptasensor for the screening of heavy metal ions. Nanoscale 2012, 4, 5998–6003. [Google Scholar] [CrossRef]
- Arunbabu, D.; Sannigrahi, A.; Jana, T. Photonic crystal hydrogel material for the sensing of toxic mercury ions (Hg2+) in water. Soft Matter 2011, 7, 2592–2599. [Google Scholar] [CrossRef]
- Lin, F.Y.; Yu, L.P. Thiourea functionalized hydrogel photonic crystal sensor for Cd2+ detection. Anal. Methods 2012, 4, 2838–2845. [Google Scholar] [CrossRef]
- Muscatello, M.M.W.; Stunja, L.E.; Asher, S.A. Polymerized Crystalline Colloidal Array Sensing of High Glucose Concentrations. Anal. Chem. 2009, 81, 4978–4986. [Google Scholar] [CrossRef]
- Zhang, C.J.; Cano, G.G.; Braun, P.V. Linear and Fast Hydrogel Glucose Sensor Materials Enabled by Volume Resetting Agents. Adv. Mater. 2014, 26, 5678–5683. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Wu, S.Z.; Sun, Z.W.; Xi, H.Y.; Li, R.F.; Hou, Z.L. Urea sensing materials via solidified crystalline colloidal arrays. Sens. Actuators B 2002, 81, 273–276. [Google Scholar] [CrossRef]
- Chen, C.; Zhu, Y.H.; Bao, H.; Shen, J.H.; Jiang, H.L.; Peng, L.M.; Yang, X.L.; Li, C.Z.; Chen, G.R. Ethanol-assisted multi-sensitive poly(vinyl alcohol) photonic crystal sensor. Chem. Commun. 2011, 47, 5530–5532. [Google Scholar] [CrossRef]
- Sharma, A.C.; Jana, T.; Kesavamoorthy, R.; Shi, L.J.; Virji, M.A.; Finegold, D.N.; Asher, S.A. A general photonic crystal sensing motif: Creatinine in bodily fluids. J. Am. Chem. Soc. 2004, 126, 2971–2977. [Google Scholar] [CrossRef] [PubMed]
- Kimble, K.W.; Walker, J.P.; Finegold, D.N.; Asher, S.A. Progress toward the development of a point-of-care photonic crystal ammonia sensor. Anal. Bioanal. Chem. 2006, 385, 678–685. [Google Scholar] [CrossRef]
- Alexeev, V.L.; Das, S.; Finegold, D.N.; Asher, S.A. Photonic crystal glucose-sensing material for noninvasive monitoring of glucose in tear fluid. Clin. Chem. 2004, 50, 2353–2360. [Google Scholar] [CrossRef]
- MacConaghy, K.I.; Chadly, D.M.; Stoykovich, M.P.; Kaar, J.L. Label-free detection of missense mutations and methylation differences in the p53 gene using optically diffracting hydrogels. Analyst 2015, 140, 6354–6362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Chao, X.; Liu, X.Y.; Asher, S.A. Two-dimensional array Debye ring diffraction protein recognition sensing. Chem. Commun. 2013, 49, 6337–6339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Cai, Z.Y.; Kwak, D.H.; Liu, X.Y.; Asher, S.A. Two-Dimensional Photonic Crystal Sensors for Visual Detection of Lectin Concanavalin A. Anal. Chem. 2014, 86, 9036–9041. [Google Scholar] [CrossRef] [PubMed]
- Amsden, B. Solute diffusion within hydrogels. Mechanisms and models. Macromolecules 1998, 31, 8382–8395. [Google Scholar] [CrossRef]
- Dembo, M.; Wang, Y.L. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys. J. 1999, 76, 2307–2316. [Google Scholar] [CrossRef]
- Colin-York, H.; Eggeling, C.; Fritzsche, M. Dissection of mechanical force in living cells by super-resolved traction force microscopy. Nat. Protocols 2017, 12, 783–796. [Google Scholar] [CrossRef]
- Harris, A.K.; Wild, P.; Stopak, D. Silicone rubber substrata: A new wrinkle in the study of cell locomotion. Science 1980, 208, 177–179. [Google Scholar] [CrossRef]
- Miller, J.S.; Shen, C.J.; Legant, W.R.; Baranski, J.D.; Blakely, B.L.; Chen, C.S. Bioactive hydrogels made from step-growth derived PEG-peptide macromers. Biomaterials 2010, 31, 3736–3743. [Google Scholar] [CrossRef] [PubMed]
- Legant, W.R.; Miller, J.S.; Blakely, B.L.; Cohen, D.M.; Genin, G.M.; Chen, C.S. Measurement of mechanical tractions exerted by cells in three-dimensional matrices. Nat. Methods 2010, 7, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Koch, T.M.; Munster, S.; Bonakdar, N.; Butler, J.P.; Fabry, B. 3D Traction Forces in Cancer Cell Invasion. PLoS ONE 2012, 7, e33476. [Google Scholar] [CrossRef] [PubMed]
- Colin-York, H.; Shrestha, D.; Felce, J.H.; Waithe, D.; Moeendarbary, E.; Davis, S.J.; Eggeling, C.; Fritzsche, M. Super-Resolved Traction Force Microscopy (STFM). Nano Lett. 2016, 16, 2633–2638. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.M.; Kyburz, K.A.; Anseth, K.S. Measuring dynamic cell-material interactions and remodeling during 3D human mesenchymal stem cell migration in hydrogels. Proc. Natl. Acad. Sci. USA 2015, 112, E3757–E3764. [Google Scholar] [CrossRef] [PubMed]
- Krajina, B.A.; Tropini, C.; Zhu, A.; DiGiacomo, P.; Sonnenburg, J.L.; Heilshorn, S.C.; Spakowitz, A.J. Dynamic Light Scattering Microrheology Reveals Multiscale Viscoelasticity of Polymer Gels and Precious Biological Materials. ACS Cent. Sci. 2017, 3, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Mason, T.G. Local collective motion analysis for multiprobe dynamic imaging and microrheology. J. Phys. Cond. Matter 2016, 28. [Google Scholar] [CrossRef]
- Squires, T.M.; Mason, T.G. Fluid Mechanics of Microrheology. Ann. Rev. Fluid Mech. 2010, 42, 413–438. [Google Scholar] [CrossRef]
- Crocker, J.C.; Valentine, M.T.; Weeks, E.R.; Gisler, T.; Kaplan, P.D.; Yodh, A.G.; Weitz, D.A. Two-point microrheology of inhomogeneous soft materials. Phys. Rev. Lett. 2000, 85, 888–891. [Google Scholar] [CrossRef]
- Yao, A.; Tassieri, M.; Padgett, M.; Cooper, J. Microrheology with optical tweezers. Lab Chip 2009, 9, 2568–2575. [Google Scholar] [CrossRef]
- Robertson-Anderson, R.M. Optical Tweezers Microrheology: From the Basics to Advanced Techniques and Applications. ACS Macro Lett. 2018, 7, 968–975. [Google Scholar] [CrossRef]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry(II). Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef] [PubMed]
- Caccavo, D.; Cascone, S.; Lamberti, G.; Barba, A. Hydrogels: Experimental characterization and mathematical modelling of their mechanical and diffusive behaviour. Chem. Soc. Rev. 2018, 47, 2357–2373. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
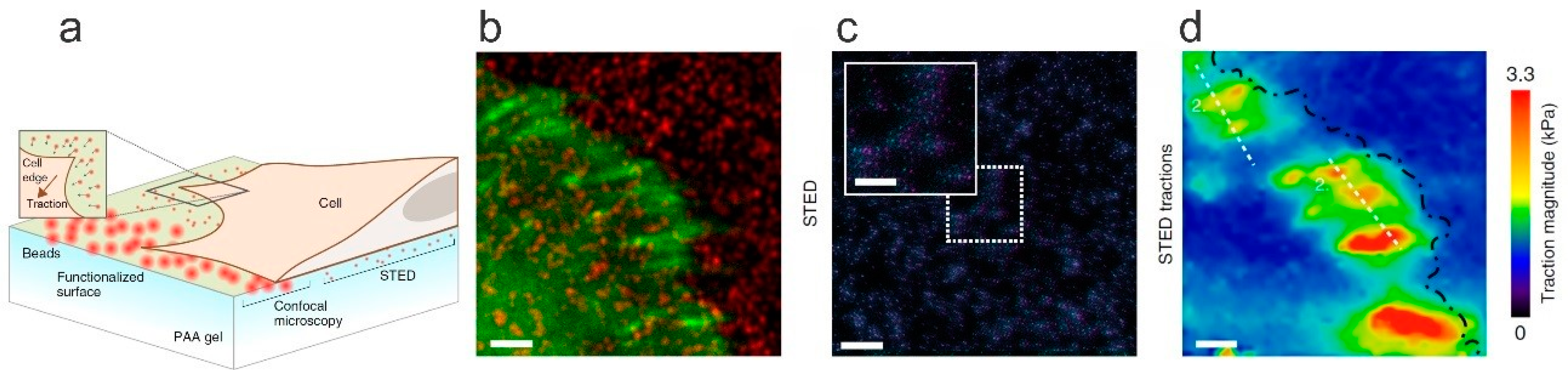{kind=link}
{kind=link}
| Polymer/Nanoparticle Materials | Self-Healing Test | Self-Healing Efficiency | Reference |
|---|---|---|---|
| Hydroxyapatite/calcium-containing silicate glass (bioactive glass) | Compression tests were performed using a Zwick Roell Z2.5 instrument | Complete recovery of mechanical properties | [126] |
| Poly(N,N-dimethylacrylamide) or poly(N-isopropylacrylamide)/Hectorite clay | Keeping surfaces in contact at ambient/elevated temperatures | Complete recovery of mechanical properties after 10 h | [110] |
| Bisphosphonated hyaluronan/calcium phosphate | Keeping surfaces in contact for short time (min 5 s) | Almost complete recovery of mechanical properties (98%) | [74] |
| Carboxybetaine methacrylamide and 2-hydroxyethyl methacrylate/Laponite clay | Keeping surfaces in contact for 5 min, ambient temperature | Mechanically stable and resistant to handling | [112] |
| Monomer acrylamide copolymerized with N-isopropylacrylamide/hectorite clay | Keeping surfaces in contact for 4 days at 20 °C or 4 h at 80 °C | Possessed ultrahigh extensibility and up to 90% strength recovery | [107] |
| Sodium polyacrylate polymer particles and hyperbranched bis-MPA polyester-64-hydroxyl/reduced graphene oxide | Contact with gentle pressure for 30 s | Nearly full restoration of the electrical conductivity | [127] |
| Poly(N,N-dimethylacrylamide)/graphene oxide-hectorite clay | Keeping the cut surfaces in contact and irradiated with a NIR laser under ambient condition | After healing for 2–3 min, strength recovery of ∼96% | [128] |
| Poly(acrylic acid)/graphene oxide | Contact at 45 °C for 48 h | Almost full recovery of mechanical properties | [118] |
| Poly(acrylamide)/exfoliated montmorillonite layers | Gels were dried and reswollen at room temperature, and then merged into a single bar | Fracturing did not occur at interface | [116] |
| Poly(acrylamide)/Laponite | Contact at 80 °C for 24 h | Healing efficiency to up to 50% | [121] |
| Poly(aspartamide)(GABA/DOPA/EA)/graphene oxide | Fractured gel pieced held in contact | Healing interface strong enough to be stretched without fracturing | [129] |
| 2-acrylamido-2-methyl propane sulfonic acid and acrylamide/zirconium hydroxide | Contact for 5 min-24 h at room temperature | healing efficiency of up to 86% (in strain efficiency) | [122] |
| Poly(ethylene glycol)/cellulose nanocrystals | Contact at 90 °C for a varying contact time under nitrogen | Up to 78% healing efficiency | [123] |
| Poly(acrylic acid)/iron ions and 2,2,6,6-tetramethylpiperidine-1-oxyl radical oxidized cellulose nanofibrils (additional cross-linker) | Contact immediately without applied stress for the prescribed contact time 25 °C | Healing interface strong enough to sustain self-supporting, bending, and lifting weights (350 g) | [111] |
| linear polyurethane chains/maleimide functionalized graphene oxide NPs | Contact at different temperatures for different times | Up to 99% healing efficiency, complete healing at 120 °C after 10 min | [29] |
| Poly(acrylamide)/Graphene oxide NPs | Contact for different times and different water content | Healing efficiency of up to 92.3% | [115] |
| Poly(N,N-dimethyl acrylamide)-gum arabic/TiO2 | Contact at different temperatures for different times | Healing interface could withstand various deformations such as bending, tying and subsequent elongation. Gels healed at room temperature broke easily at interface | [124] |
| Poly (2-acrylamido-2-methyl-1-propanesulfonic acid)/Laponite clay and N,N′-methylenebis (acrylamide) (additional cross-linker) | Contact at 50 °C for 24 h | Fracturing occurred at healing interface, the original network structure could not be completely recovered | [114] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dannert, C.; Stokke, B.T.; Dias, R.S. Nanoparticle-Hydrogel Composites: From Molecular Interactions to Macroscopic Behavior. Polymers 2019, 11, 275. https://doi.org/10.3390/polym11020275
Dannert C, Stokke BT, Dias RS. Nanoparticle-Hydrogel Composites: From Molecular Interactions to Macroscopic Behavior. Polymers. 2019; 11(2):275. https://doi.org/10.3390/polym11020275
Chicago/Turabian StyleDannert, Corinna, Bjørn Torger Stokke, and Rita S. Dias. 2019. "Nanoparticle-Hydrogel Composites: From Molecular Interactions to Macroscopic Behavior" Polymers 11, no. 2: 275. https://doi.org/10.3390/polym11020275
APA StyleDannert, C., Stokke, B. T., & Dias, R. S. (2019). Nanoparticle-Hydrogel Composites: From Molecular Interactions to Macroscopic Behavior. Polymers, 11(2), 275. https://doi.org/10.3390/polym11020275