1. Introduction
The colonization of marine species such as algae (soft fouling) and barnacles (hard fouling) on ship hulls or any other artificial submerged surfaces causes serious impacts on the marine environment [
1,
2]. It is noteworthy that such biofouling colonization on ship hulls increases their hydrodynamic drag leading to an increase in fuel consumption and greenhouse gases emission [
3,
4]. Following the international ban of tributyltin, almost 20 years ago, innovations in the field of antifouling coatings dealt with the development of coatings based on copper and co-biocides, targeting the largest biodiversity of micro- and macro-organisms as possible. During the past 10 years, more environmentally friendly antifouling coatings were developed, mainly by reducing the biocides content. The use of biocides remains the most efficient way to prevent the settlement of marine species [
5,
6,
7,
8,
9], however the regulations around the world tend to drastically reduce over the years the number of harmful chemical substances authorized in antifouling paints (e.g. the European Biocidal Product Regulation) [
7,
10].
Fouling Release Coatings (FRC) are antifouling coatings mainly based on a crosslinked poly(dimethylsiloxane) (PDMS) matrix. Contrary to the commonly used antifouling coatings, they do not release biocides and their antifouling performances are based on their specific properties (a low surface free energy, a low elastic modulus and a low surface roughness), which enable the release of fouling organisms in presence of hydrodynamic forces (e.g. during navigation) [
6,
7,
11]. Thus, their antifouling performances are strongly dependent on the ship velocity, and they tend to be fouled during idle periods. Moreover, FRC suffer from a low fouling release efficacy towards some microalgae such as
Amphora and
Navicula diatoms [
12,
13,
14]. To overcome these issues, many researchers are now focusing their work on the synthesis of novel additives or polymer matrixes for developing a new generation of FRC. A promising antifouling mechanism is to create inhospitable surfaces which weakens the strength of adhesion between the bioadhesive, secreted by the fouling organisms, and the submerged surface. Inhospitable surfaces can be obtained using surface-modifying compounds, such as blended or grafted amphiphilic copolymers [
15,
16,
17,
18,
19,
20,
21], zwitterionic polymers [
22,
23,
24,
25], or hydrogel-like polymers [
26,
27]. Specialty additives like fillers (sepiolite nanofibers, modified graphite, carbon nanotubes) and pigments (TiO
2, ZnO) are also known to enhance the fouling release properties once embedded in a PDMS matrix [
28,
29,
30,
31]. Besides, silicone oils used as additives in PDMS elastomers can also enhance the fouling release properties of FRC due to their appropriate critical surface tension, and to a leaching phenomenon, which leads to the detachment of fouling organisms by slipping [
32,
33,
34,
35].
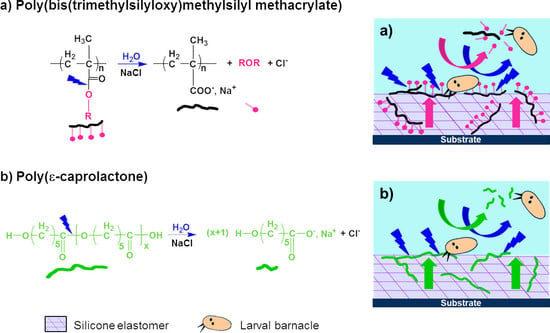
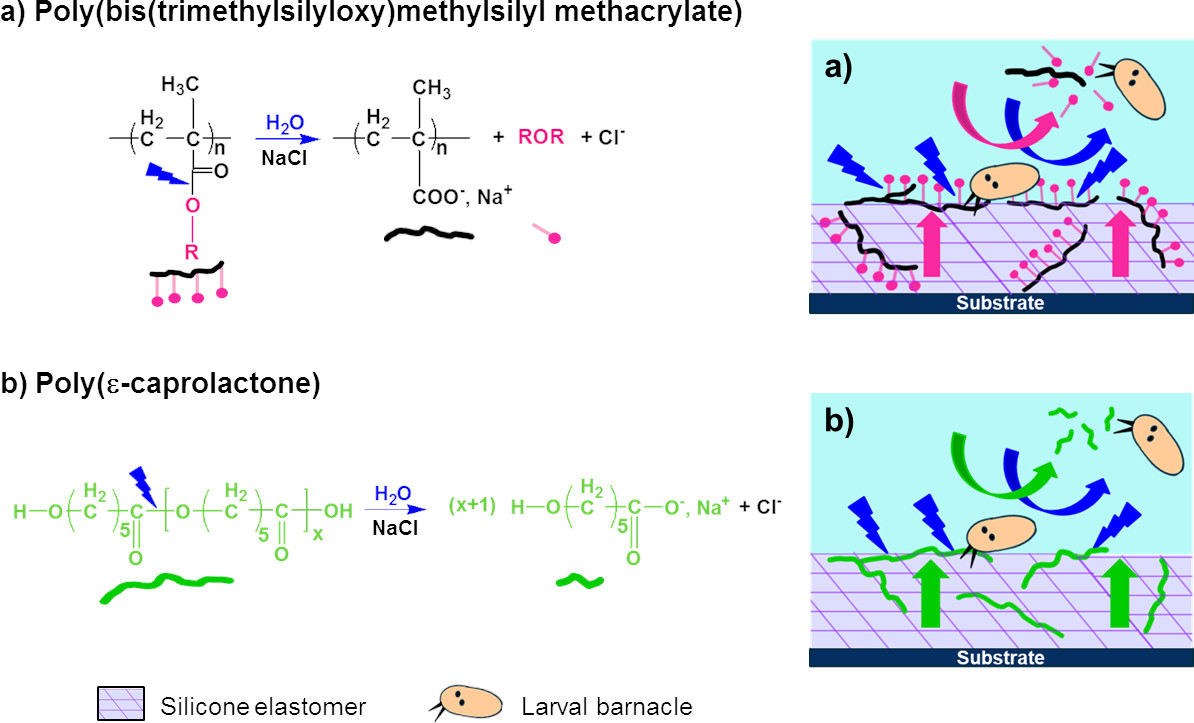
In this work, three hydrolyzable polymers were embedded in a silicone elastomer to combine the hydrophobic character of PDMS with the hydrophilicity of the hydrolyzed polymer additives. With these new silicone coatings, it is expected that in contact with water, the additive polymers will migrate more or less quickly towards the upper surface and create some hydrophilic and/or hydrolyzable regions among the hydrophobic PDMS phase. Thus, these coatings are expected to behave as evolving surfaces which could prevent the settlement of fouling organisms during static periods or limit their adhesion, without the use of biocides. Hydrolyzable polymers have already been used in the field of antifouling coatings, for the development of biocides-based erodible coatings. Bressy’s group synthesized poly(trialkylsilyl methacrylate)s to develop coatings with tunable erosion profiles [
36,
37,
38], Zhang’s and Réhel’s groups worked on degradable polyesters for antifouling applications [
39,
40,
41,
42,
43,
44,
45,
46]. Copolymers based on poly(ε-caprolactone), poly(δ-valerolactone), polybutylene succinate, poly(L-lactide), poly(ethylene adipate), and poly(2-methylene-1,3-dioxepane) were synthesized to promote the erosion of the polymer matrix and the controlled release of biocides.
In our study, three hydrolyzable polymers, with different kinetics of hydrolysis, were evaluated as polymer additives embedded in a silicone matrix. The aim was to select the most appropriate hydrolysis kinetics to improve the antifouling performances of PDMS-based coatings in static conditions. Poly(ε-caprolactone) (PCL) was chosen for its slow kinetics of hydrolysis which could lead to a more durable antifouling activity. PCL-
b-PDMS-
b-PCL, a triblock copolymer containing two PCL blocks and a central PDMS block, was also evaluated as its triblock structure should improve the miscibility of PCL segments in the silicone elastomer. The third hydrolyzable polymer, poly(bis(trimethylsilyloxy)methylsilyl methacrylate) (PMATM2), was selected due to its fast hydrolysis kinetics in opposition to the two previous ones [
38].
The coatings developed in this study were free of fillers and pigments in order to evaluate only the influence of the polymer additives on the properties of the PDMS elastomer matrix. The silicone-based coatings with hydrolyzable polymer additives were characterized in terms of surface free energy, wetting properties, roughness, elastic modulus, and hardness, since these parameters are the main drivers of antifouling and fouling release performances of PDMS elastomer-based coatings. A mass loss test was performed to estimate the degradation kinetics of the hydrolyzable polymers embedded in the silicone matrix. The characteristics of the coatings were correlated with their antifouling efficacy during immersion in the Mediterranean Sea (Toulon bay), in static conditions for 8 months. Finally, settlement bioassays using cyprids of Amphibalanus amphitrite gave further information on the antiadhesive properties of these silicone-based coatings with hydrolyzable polymer additives.
2. Materials and Methods
2.1. Materials
Poly(ε-caprolactone)-diol (PCL) (Mn,NMR = 3,000 g/mol) was supplied by Perstrop (Shanghai, China) and used without further purification. The polyester modified polysiloxane triblock copolymer PCL-b-PDMS-b-PCL (with Mn,NMR = 6,800 g/mol; Mn,NMR (PDMS block) ≈ 2,200 g/mol; total PCL end blocks ≈ 4,600 g/mol), and a polydiethoxysiloxane (PDES) crosslinking agent (Mn,NMR = 744 g/mol) were kindly supplied by Evonik (Essen, Germany). Dioctyltin dilaurate (DOTDL) catalyst was kindly supplied by TIB Chemicals (Mannheim, Germany). Bis-silanol polydimethylsiloxane oil (Mw = 24,000 g/mol) was kindly supplied by Dow Corning (Seneffe, Belgium). MATM2 monomer was kindly supplied by PPG (Amsterdam, The Netherlands) and was distilled under reduced pressure in presence of 2,6-di-tert-butyl-4-methylphenol before use. AIBN (2,2’-Azobis(2-methylpropionitrile)) was purchased from Sigma Aldrich (Saint-Quentin-Fallavier, France) and recrystallized in methanol before use. CPDB (2-Cyano-2-propyl dithiobenzoate) was purchased by Strem Chemicals (Bischheim, France) and used as received. Xylene reagent was purchased from Carlo Erba Reagents (Peypin, France) and distilled under reduced pressure before use. Diiodomethane (CH2I2, Sigma-Aldrich, Saint-Quentin-Fallavier, France) was used as received.
2.2. Synthesis of PMATM2
The PMATM2 homopolymer was prepared by a Reversible Addition-Fragmentation chain Transfer (RAFT) polymerization in xylene with AIBN as an initiator and CPDB as the transfer agent, as previously described by Lejars et al. [
38]. The reaction mixture was stirred at 75 °C for 20 h under nitrogen atmosphere. The synthesized polymer was precipitated in a great excess of cold methanol to provide a very sticky pink polymer. The reaction was followed by
1H NMR analysis to assess the conversion of MATM2 monomer by measuring the disappearance of the vinylic protons at 5.8 ppm [
38]. Once the conversion no longer evolved, the reaction was stopped. The number average molar mass (
Mn, NMR) of the purified PMATM2 was assessed by
1H NMR analysis assuming that there is one CPDB molecule per polymer chain using Equation (1).
1H NMR (PMATM2) (400 MHz, CDCl
3, δ, ppm): 7.9 (2H, m, aromatic –CH–), 7.5 (1H, m, aromatic –CH–), 7.3 (2H, m, aromatic –CH–), 1.7-2.5 (2H, wide signal, –CH
2–), 0.8-1.5 (3H, wide signal, –CH
3), 0.2–0.3 (3H, s, –SiCH
3), 0.02-0.2 (18H, m, (–[SiCH
3]
3)
2).
corresponds to the intensity of the signal at (0.3–0.02) ppm (21H, m, –SiCH3). corresponds to the intensity of the signal at 7.9 ppm (2H, m, aromatic –CH–) corresponding to the CPDB moiety. Mrepeat unit corresponds to the molar mass of the repeat unit (305 g/mol). MCPDB corresponds to the molar mass of the RAFT agent moiety (221.34 g/mol).
2.3. Characterization Techniques of the Polymer Additives
2.3.1. Nuclear Magnetic Resonance Spectroscopy (NMR)
1H NMR spectra were recorded on a Brüker Advance 400 (400 MHz) spectrometer, in deuterium chloroform (CDCl3), at room temperature.
2.3.2. Size Exclusion Chromatography (SEC)
The number average molar mass (
Mn,TD-SEC) and dispersity (Đ
M,TD-SEC) were measured on a Triple Detection Size Exclusion Chromatography (TD-SEC) with OmniSEC software (Viscotek, Vénissieux, France). The instrument is composed of a GPC Max (comprising a degazer, a pump and an autosampler) with TDA-302 (RI detector, right and low angle light scattering detector at 670 nm and viscometer) and UV detector (Knauer, Berlin, Germany). The following Viscotek columns were used: a HHR-H precolumn, a GMHHR-H and a GMHHR-L ViscoGel columns. THF was used as the eluent with a flow rate of 1.0 mL/min at 30 °C. Purified polymers were dissolved in THF at ca. 10 mg/mL and filtered on a 0.2 µm PTFE filter. Molar masses were calculated using the value of the refractive index increment d
n/d
c for PMATM2 (d
n/d
c= 0.044 mL/g in THF) and PCL (d
n/d
c= 0.071 mL/g in THF) using the OmniSEC software [
38,
47]. The analysis of the PCL-
b-PDMS-
b-PCL triblock copolymer was not possible by TD-SEC in THF due to its refractive index similar to
nTHF, resulting in no signal with the RI and light scattering detectors. This triblock copolymer was thus analyzed on a SEC apparatus (Waters, Milford, CT, USA) equipped with a refractive index detector, with toluene as eluent. The columns used were Styragel HR1, HR4, and HR5. Toluene was used as the eluent at a flow rate of 1.0 mL/min. Polystyrene standards (
Mn values varying from 400 to 20,000 g/mol) were used to generate a conventional calibration curve. Data were analyzed using Breeze software (Waters, Milford, CT, USA).
2.3.3. Differential Scanning Calorimetry (DSC)
The thermal behavior of polymers was analyzed on a Differential Scanning Calorimetry equipment (TA Instruments, New Castle, DE, USA) under a nitrogen flow of 50 mL/min. To determine the crystallinity (Xc) of PCL and PCL-b-PDMS-b-PCL, samples (10–15 mg) were first heated at 100 °C for 2 min, then cooled down to −90 °C at a rate of 1 °C/min, kept at −90 °C for 2 min, and then reheated to 100 °C at a rate of 1 °C/min. All values were recorded from the second heating run. To determine the glass transition temperature of the amorphous PMATM2, the sample was cooled down to −90 °C at a rate of 20 °C/min, kept at −90 °C for 2 min, and then reheated to 30 °C at a rate of 20 °C/min. To determine the glass transition temperature of the semi-crystalline PCL, modulated DSC was performed, the sample was first heated at 100 °C for 2 min then cooled down to −90 °C at a rate of 5 °C/min, kept at −90 °C for 2 min, and then reheated to 100 °C at a rate of 2 °C/min (conventional DSC experiment did not allow the visualization of the glass transition, regardless of the program choice).
2.4. Preparation of Polymer Additive Films
To assess the wettability properties of the polymer additives alone, each polymer was dissolved in chloroform (50 wt %) and consecutively applied with a bar coater (100 µm wet) on a thin sandblasted poly(vinylchloride) (PVC) substrate (2 × 4 cm2) previously cleaned with ethanol. The samples were dried for 48 h at room temperature before contact angles analysis.
To determine the mass loss kinetics of the three hydrolyzable polymers, the chloroform-based solutions were also casted on glass slides (76 × 26 mm2) previously cleaned with acetone. They were immersed in deionized water for a certain amount of time until the polymer films onto the glass slides cracked into pieces and could no longer be weighed.
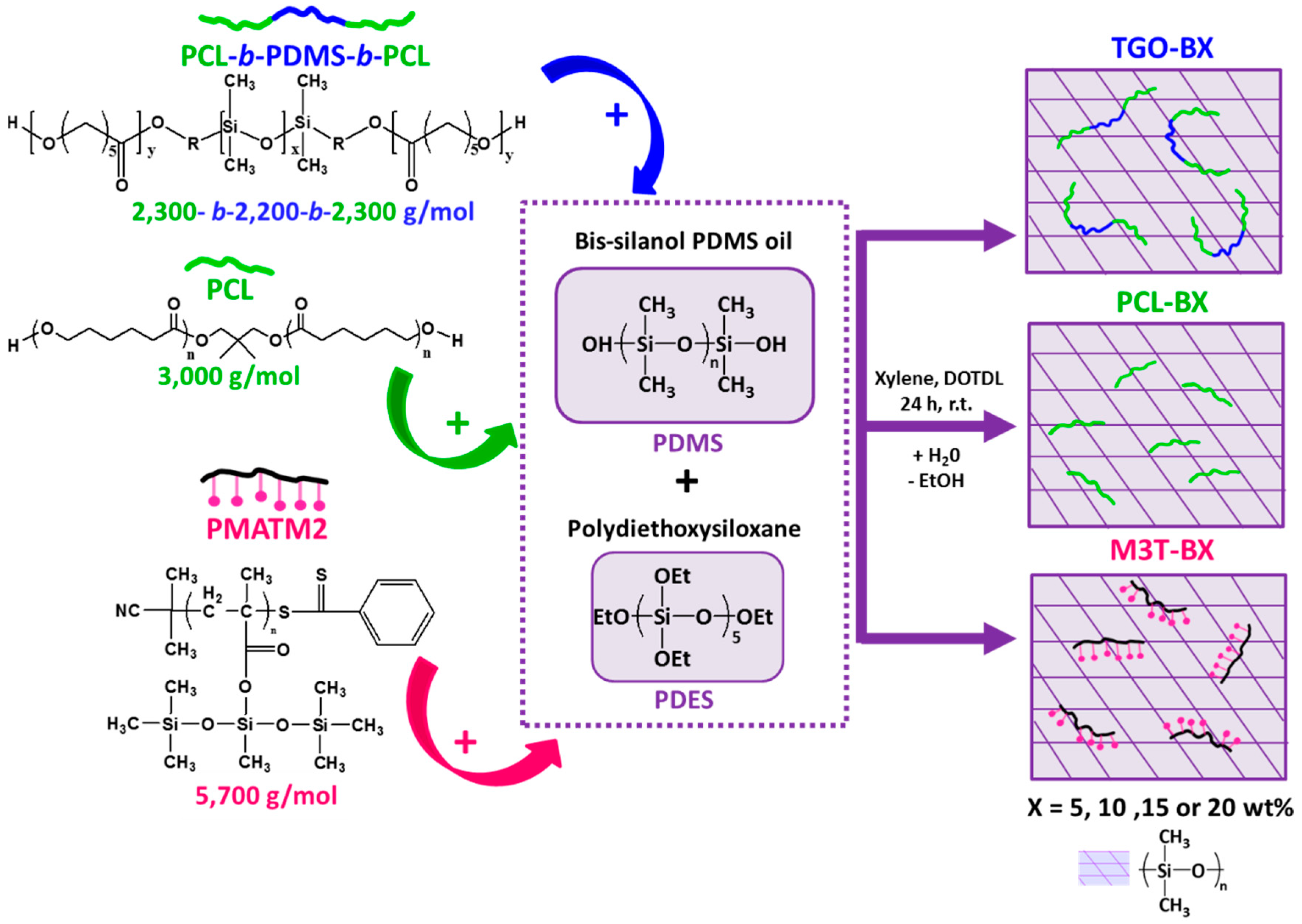
2.5. Preparation of PDMS-Based Coatings
The hydrolyzable additives-based silicone coatings were prepared as described in
Figure 1. The hydrolyzable polymers were dissolved in xylene (60–80 wt %) for 15 min of stirring. The xylene-based solutions were consecutively dispersed within the bis silanol-terminated PDMS at 1500 rpm for 20 min with a Dispermat® apparatus. The DOTDL catalyst was added using a micropipette). The PDES crosslinking agent was further added into the solution at 1500 rpm for 10 min. The massic ratio of PDMS:PDES:DOTDL was 100 : 4.4 : 0.1 for all the mixtures. The solvent amount (21–26)%was adjusted to have a similar viscosity for all formulations (full description in
Table 1). The coatings were prepared immediately after mixing (to avoid phase separation), by casting the formulation onto various substrates. The coatings were cured at room temperature for 24 h with a relative humidity of 38%–48%, measured by a hygrometer. For contact angle measurements, roughness measurements and the hydrolytic degradation tests, samples were prepared by casting ca. 2 g of the formulations onto cleaned glass microscope slides (76 × 24 mm
2). Samples for dynamic mechanical analyses (DMA) investigations were 1 mm-thick free standing films obtained after casting formulations onto 5 × 5 cm
2 cleaned smooth PVC panels, and gently detached from the substrate with a plastic tweezer. The samples for DMA analyses were also used for the hardness measurements by stacking 6 specimens. Additional applications over 10 × 10 cm
2 sandblasted and cleaned PVC panels were performed with a bar coater (wet thickness of 300 µm) for field tests and biological assays. The nomenclature of the coatings is YYY-BX, where YYY is the type of hydrolyzable polymer additives (PCL for the PCL homopolymer, TGO for the triblock PCL-
b-PDMS-
b-PCL and M3T for the PMATM2 homopolymer), B stands for blend, and X corresponds to the mass fraction of the hydrolyzable polymer within the dry coating.
Due to the limited miscibility of PCL within the silicone matrix, the maximum amount of PCL was up to 15 wt %. Above this quantity, the coatings with PCL-based additives displayed a macroscopic phase segregation, while coatings with 20 wt % of PMATM2 could be achieved without incompatibility issues at the macroscale level.
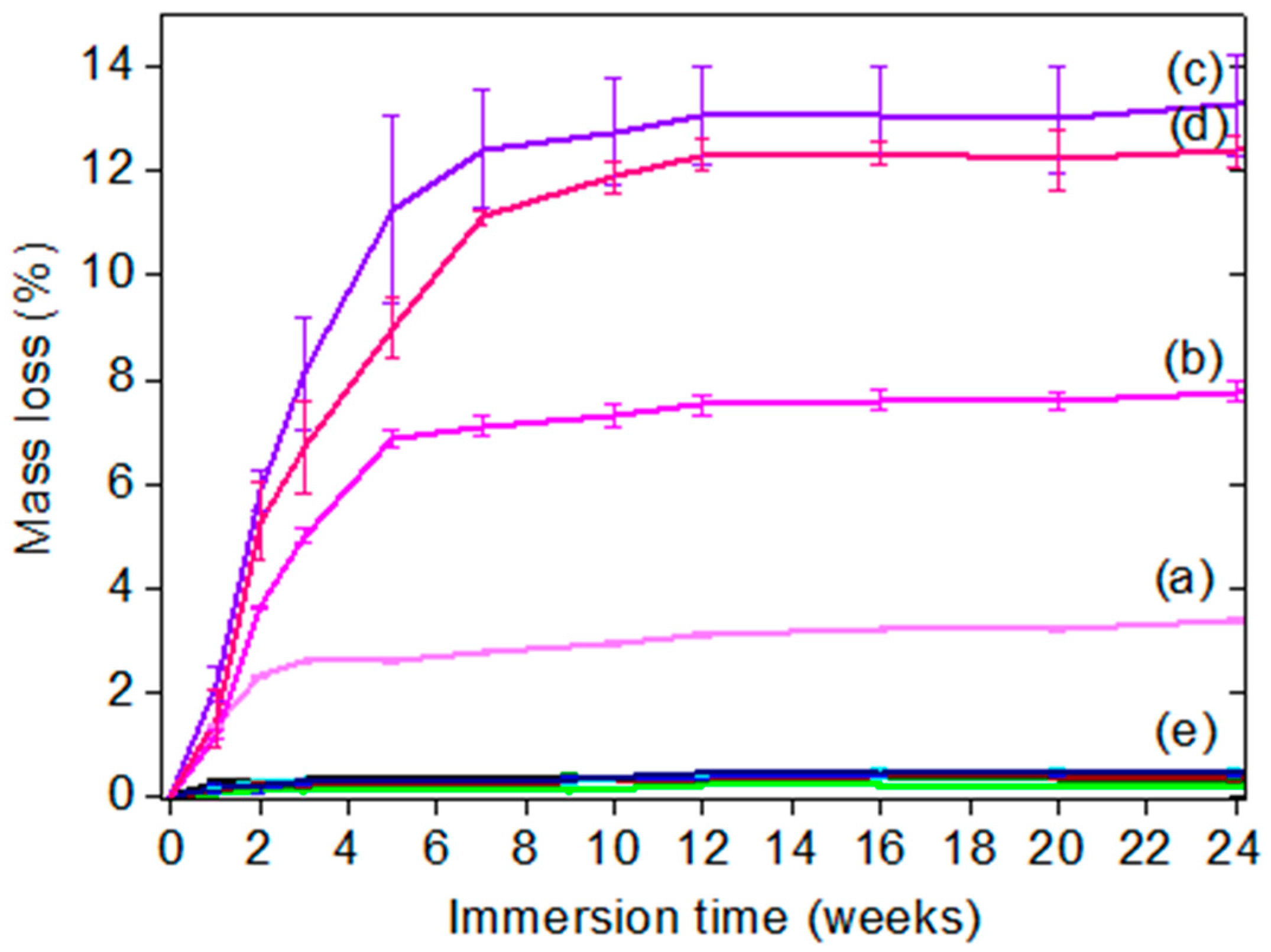
2.6. Hydrolytic Degradation of Coatings
The mass loss of hydrolyzable additives-based silicone coatings was carried out by immersing coated glass slides for 24 weeks in deionized water. Before gravimetric measurements, the coatings were gently rinsed with deionized water and dried at room temperature for 6 h. The reported mass loss results are the mean value of three replicates, weighted on an analytic balance (Denver Instrument) with a precision of 0.1 mg and calibrated prior to each measurement. The mass loss (wt %) was calculated using Equation (2).
where
Wo is the initial mass of the coated glass slide (g),
Wt is the mass of the coated glass slide after an immersion time
t (g) and Ws is the mass of the non-coated glass slide (g).
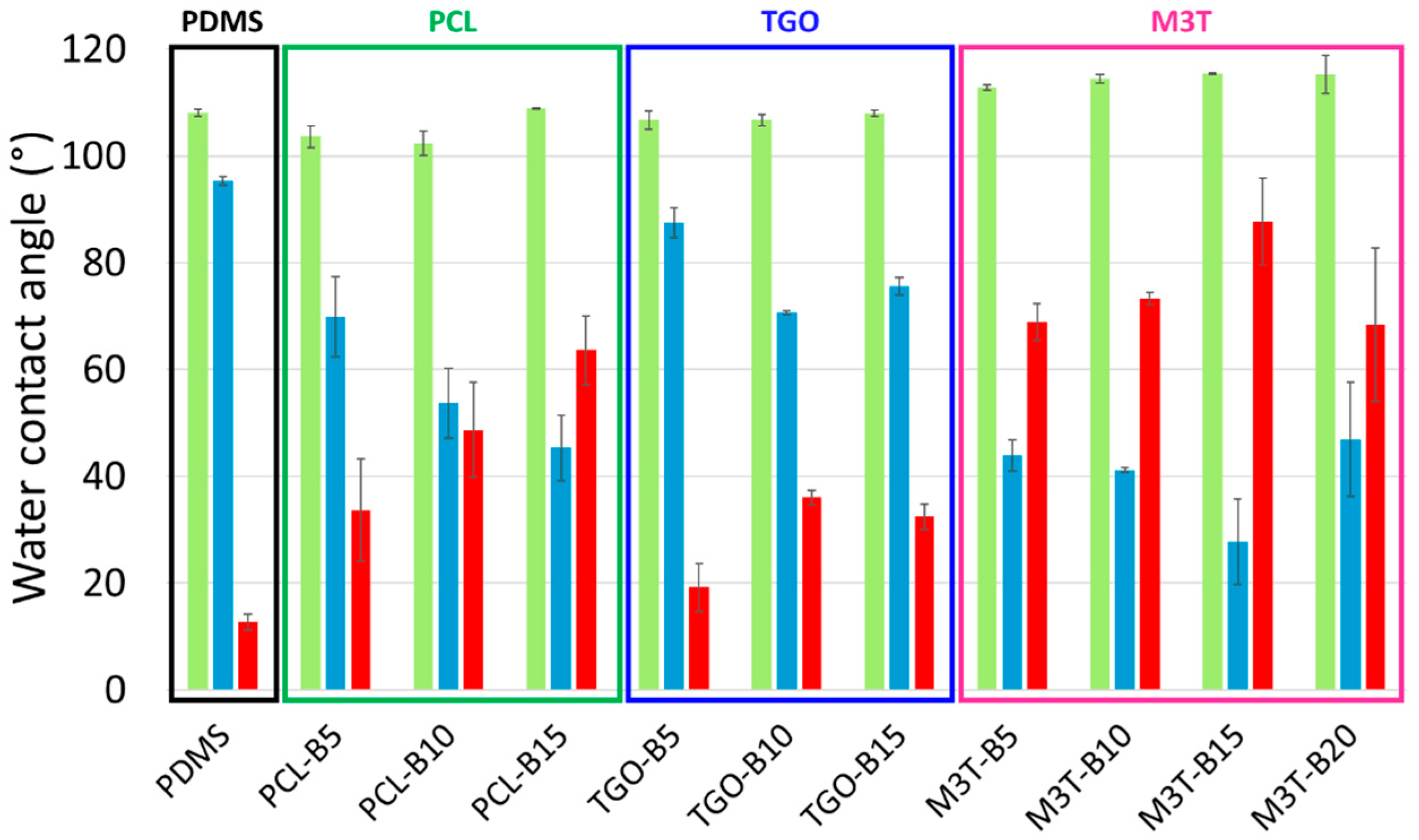
2.7. Contact Angle Measurement and Surface Free Energy of Coatings
Static contact angle measurements were performed using a DSA 30 apparatus (Krüss, Hambourg, Germany) by the sessile drop technique under ambient conditions. Five contact angle measurements were carried out with 2 µL-droplets of deionized water (θ
w) and diiodomethane (θ
diiodo), after 4 s of stabilization. The surface free energy of the coatings (
) was determined by the Krüss Advance software using the Owens Wendt method [
6]. Both the dispersive (
) and the polar (
) components of the surface free energy were assessed (
). Measurements were performed on pristine samples and samples immersed in deionized water for 5 weeks.
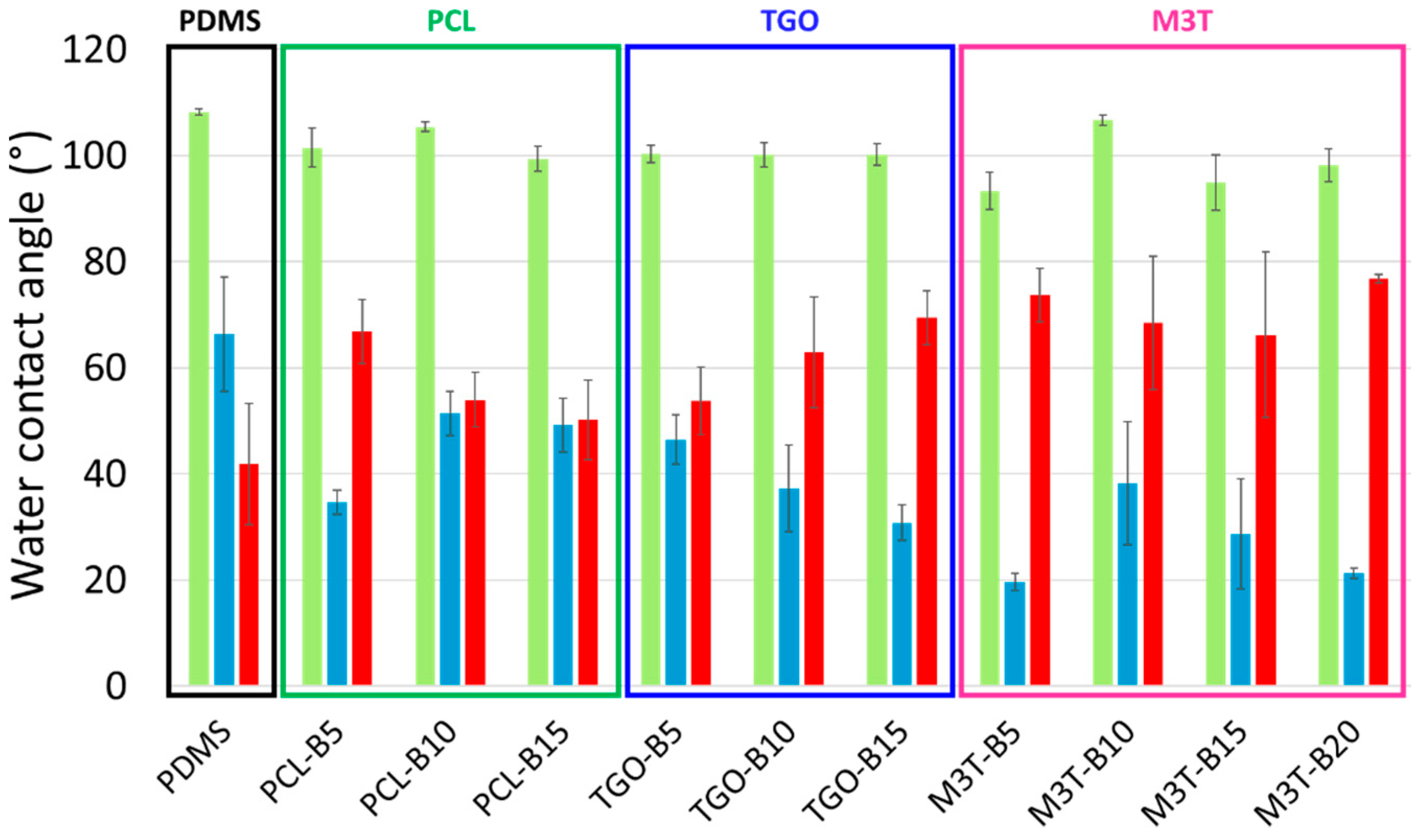
2.8. Dynamic Contact Angle Measurements of Coatings (DCA)
DCA experiments were carried out by the advancing-receding drop method using the DSA 30 apparatus (Krüss, Hambourg, Germany) under ambient conditions. A 4 µL-deionized water drop was first placed onto the coating with the syringe tip still immersed within the droplet, than the droplet is grown at a rate of 0.75 µL/s until a final volume of 25 µL for the measurement of the advancing contact angle (θw,adv). The receding contact angle (θw,rec) was measured by withdrawing the liquid at the same rate. For each coating, the reported θw,adv and θw,rec were the average values obtained from 1 cycle of advancing-receding on 3 deionized water droplets.
2.9. Surface Roughness Measurements of Coatings
Surface roughness profiles were measured by a contact type stylus profiler (Taylor Hobson, Leicester, UK) using a 2 µm radius tip and a 0.1 µm radius diamond tip, with a minimum applicable 1 mN stylus load. The stylus moved across 15 mm length of the coating surface, at a constant velocity of 0.50 mm/s to obtain surface height variations. Ra values were assessed from the average of three measurements. According to ISO 4288-1996, the selected cut-off wavelength (low-pass filter) was λc = 0.8mm since Ra < 2 µm. The high-pass filter (λs) was fixed at 0.0025 mm for all the samples.
2.10. Hardness Measurements
The hardness of the coatings were determined with a Shore A Durometer (Hildebrand, Oberboihingen, Germany) which indicates the relative resistance of a material to indentation with a load applied to the indenter. The reported hardness values were the average of 5 measurements on 6 mm-thick stacked specimens after 15 s of stabilization according to the standard ISO 868.
2.11. Dynamic Mechanical Analysis of Coatings
The dynamic mechanical analyses were performed using free standing films of 10–14 mm length, 8–10 mm width and 0.6–1.0 mm thick. The storage modulus E’ of samples was determined at 25 °C with a dynamic mechanical analyzer DMA (TA Instrument Q800, New Castle, DE, USA) operating in tensile strain-sweep mode. A frequency of 1 Hz, a preload of 0.01 N, and amplitudes from 5 to 50 µm (within the linear viscoelastic region) were used. The results are the average of at least 5 measurements on 3 different samples.
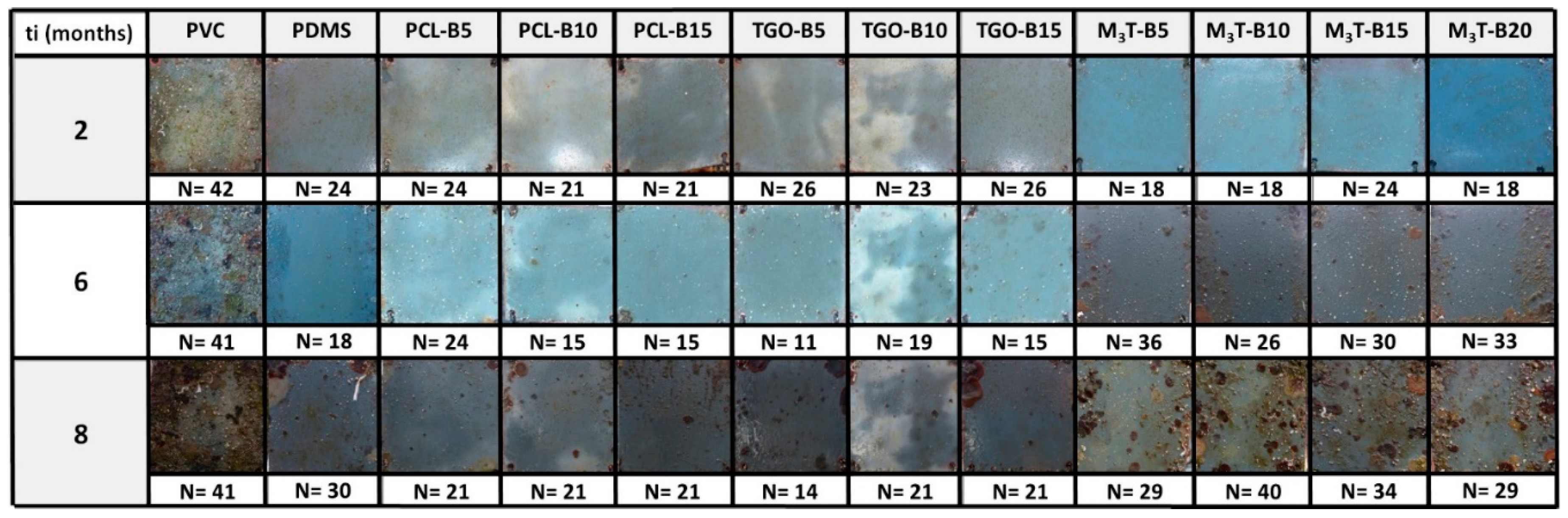
2.12. Marine Field Test
The coated panels (10 × 10 cm
2) were fully immersed in a vertical position in the Mediterranean Sea, in the Toulon Bay (43°06′25″N; 5°55′41″E), at a relatively shallow depth of water (1 m). The Antifouling (AF) and Fouling Release (FR) performances in static conditions were evaluated every month, over a period of 8 months (from June 2017 to January 2018). Two replicates of each coating were investigated, from which one was partially-cleaned with a sponge, each month, for assessing the FR properties. A sand-blasted uncoated PVC panel was also immersed as a negative control. A French practice adapted from the French NF T 34-552 standard was used to assess the AF efficacy of the coatings. This standard requires to report: (i) the type of macrofoulers attached to the surface, and (ii) the estimated percentage of the surface covered by each type of macrofoulers (intensity factor, i.e. IF, see
Table 2). The inspection was performed 1 cm from the edges of the panel. An efficacy factor N was defined as follows in Equation (3).
where SF is defined as a severity factor (see
Table 3), which takes into account the frictional drag penalty of ship hulls attributable to increased surface roughness due to foulers [
48,
49]. The best antifouling efficacy is assigned to coatings with a N value of 5, which corresponds to a surface fully covered by a biofilm. The worse antifouling efficacy is attributed to the negative control with a N value which tends to ca. 40.
The FR performance, ranging from 0 to 2 (0 = worst, 2 = best), was evaluated by cleaning the coating with a sponge on the lower half of the coated panel and assessing the level of detachment of the marine organisms.
These two inspection procedures provide meaningful information on both the AF/FR activity of the coatings and their long-term durability in an aggressive, real-world environment. However static immersion tests may be susceptible to the seasonal diversity and abundance of fouling, at the test site [
50]. The conclusions on the field test will thus consider the seasonal fouling activity (provided in
Figures S5, S6, and S7).
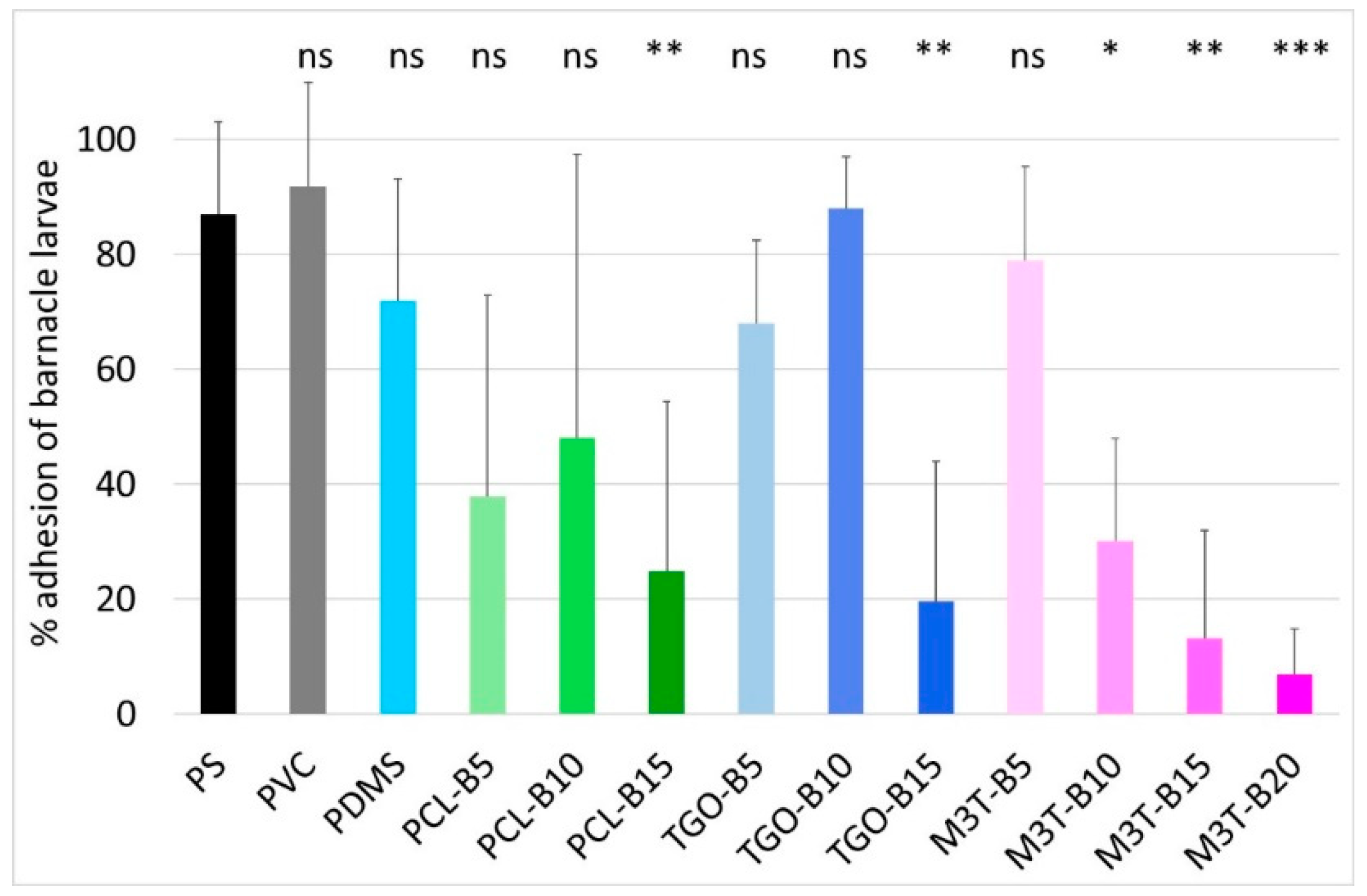
2.13. Larval Barnacle Culture and Settlement Assay of A. Amphitrite Cyprids
Adult and larval barnacle cultures were performed with
Amphibalanus (=
Balanus)
amphitrite as described by Othmani et al. [
51]. For anti-settlement assays, 2 mL of filtered sea water (FSW) were pipetted onto four different locations of each coating. Active cyprids were pipetted from the stock solution (~100 cyprids/mL) together with 200 μL of FSW. Thus, around 10 to 20 cyprids were allocated into each drop while adding 200 μL to the 2 mL drop of FSW. Incubation of cyprids on coating samples was carried out, at 22 °C, for 7 days, in a humid chamber to avoid excessive evaporation of water in the dark. This procedure allowed efficient settlement and transformation of cyprids, after which, time attached or metamorphosed individuals were counted under a binocular microscope. A poly(styrene) (PS) plate was used as control to evaluate the ability of the larvae to settle. A one-way ANOVA (Analysis of Variance) followed by post-hoc tests (Tuckey) was performed to determine which of the coatings showed a significant difference compared to the control for cyprid adhesion. The adhesion percentage is defined as the number of alive fixed cyprids divided by the total number of tested cyprids. Barnacle larvae are prone to settle on the inert PS control surface with adhesion of 80%–100% of the larvae. Further details of the bioassay are supplied in
Table S1.
4. Conclusions
We have explored the possibility to use water-hydrolyzable PCL, PCL-b-PDMS-b-PCL copolymer, and PMATM2 as polymer additives in a silicone matrix for developing AF/FR coatings. All the additive-based PDMS elastomer coatings showed a low surface free energy, a low elastic modulus as well as a surface chemical reorganization during water immersion which make them suitable for marine applications. Particularly, PMATM2-based coatings provided an interesting evolving surface chemistry thanks to a fast hydrolysis rate of the pendant ester group and a very dynamic surface chemistry reflected by a water contact angle hysteresis up to 60°. The antifouling efficacy of PMATM2-based elastomers gave slightly better results than the additive-free PDMS coating for the first 2 months of field immersion due to the fast hydrolysis of pMATM2. Nevertheless, after 6 months of field immersion the antifouling behavior of these coatings decreased which indicates that the hydrolysis did not last enough to provide durable antifouling properties.
Finally, the importance of hydrolysis kinetics has been highlighted in this article. For antifouling applications, PMATM2 is suitable for coatings with a short-term efficacy and good antiadhesive properties towards barnacle larvae. Although PCL and PCL-b-PDMS-b-PCL could not hydrolyze in the silicone elastomer after 24 weeks of water immersion, PCL-BX and TGO-BX coatings exhibited a longer antifouling efficacy in field than the additive-free PDMS coating, and they also showed good A. amphitrite antiadhesive properties with 15 wt % of hydrolyzable polymer content.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}