Cellulose Fibers Hydrophobization via a Hybrid Chemical Modification


Abstract
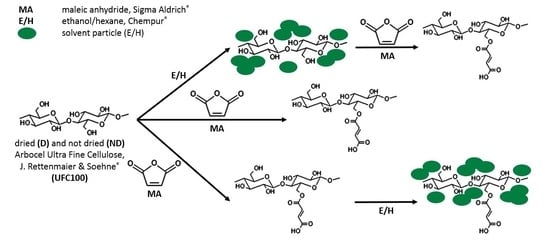
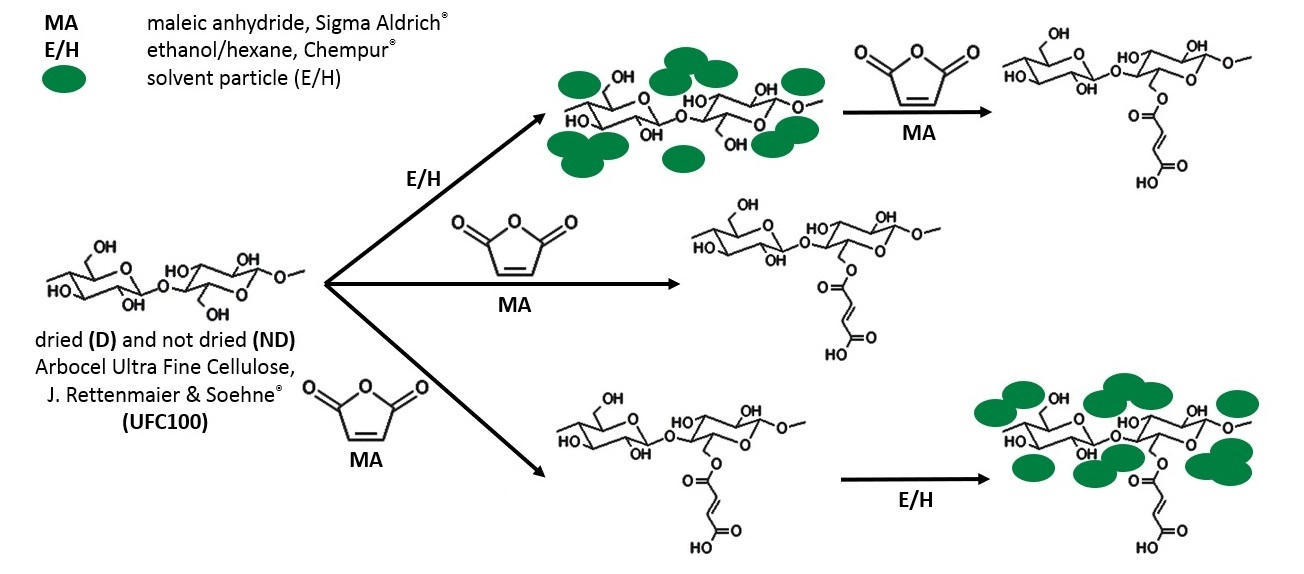
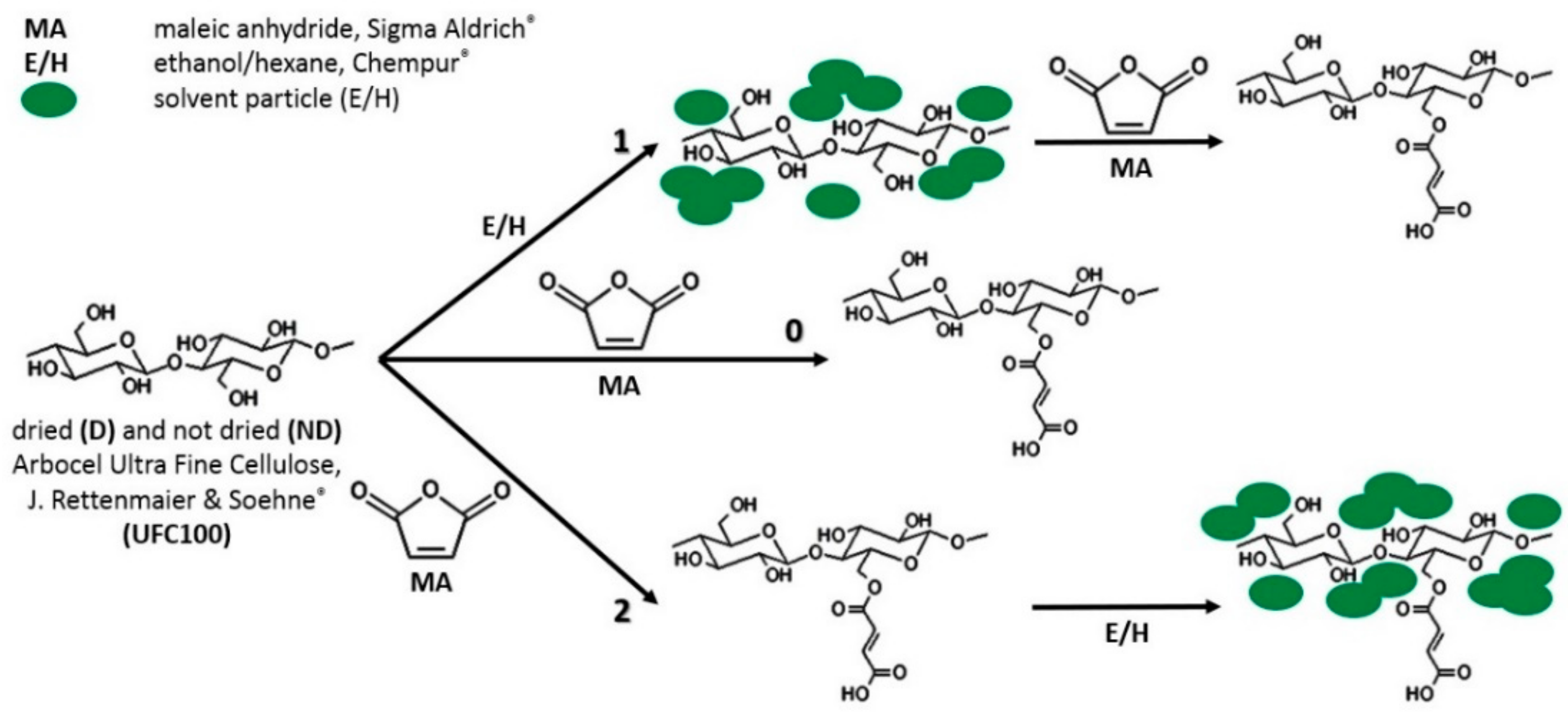
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cellulose Fibers Hybrid Chemical Modification
2.3. Fourier Transform Infrared Spectroscopy (FT-IR)
2.4. Near Infrared Spectroscopy (NIR)
2.5. Thermogravimetric Analysis (TGA)
2.6. Differential Scanning Calorimetry (DSC)
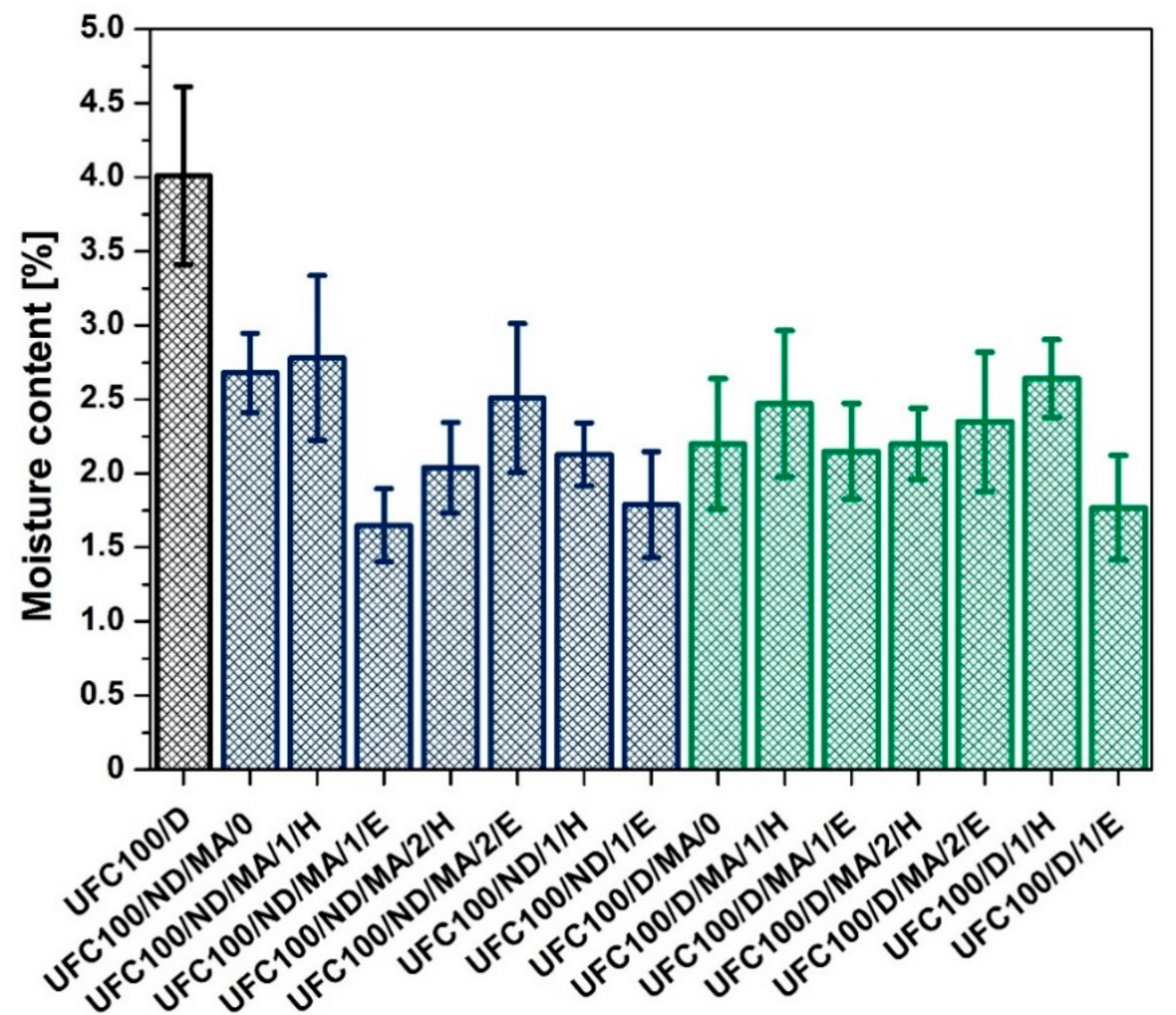
2.7. Fischer Titration
3. Results and Discussion
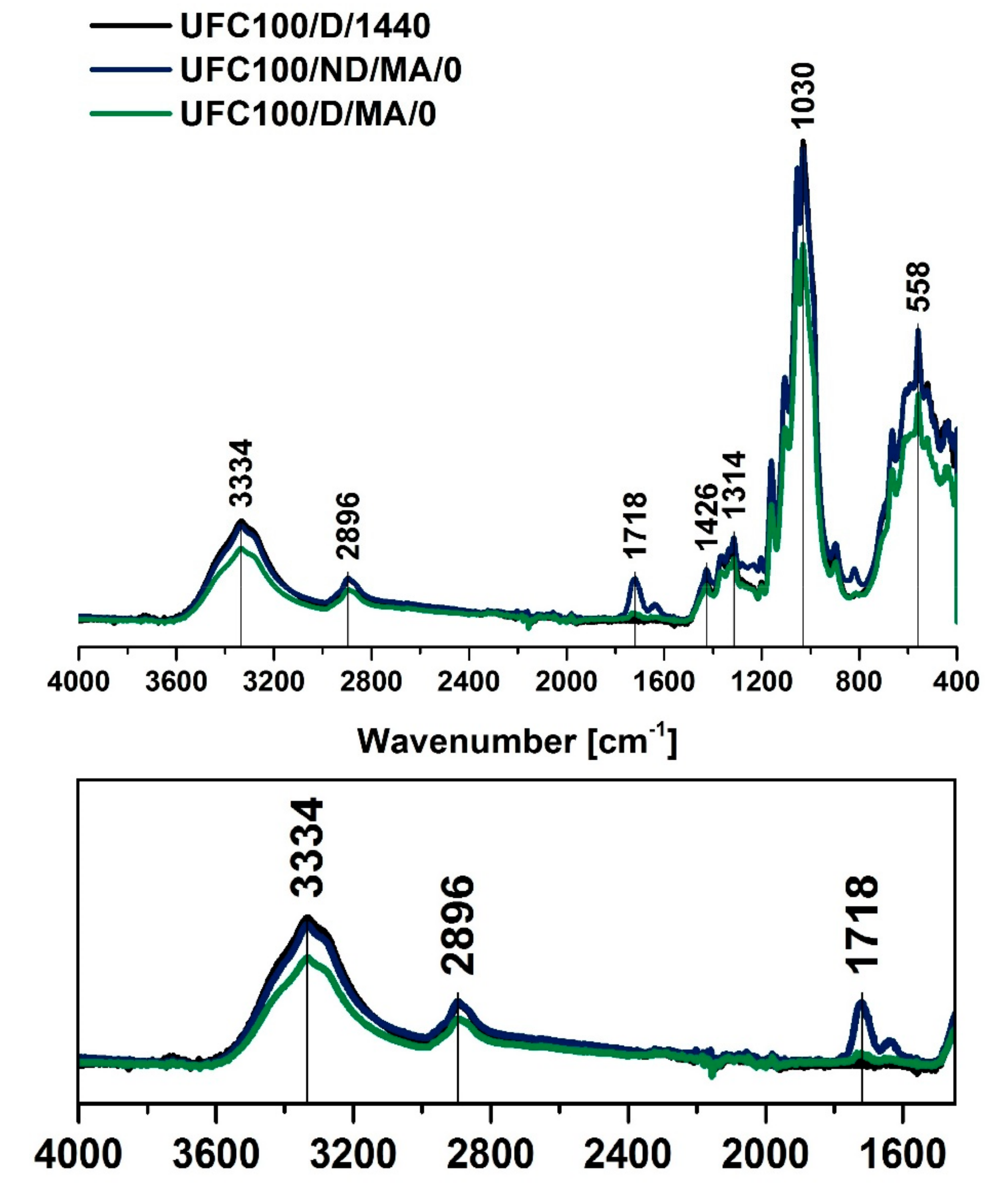
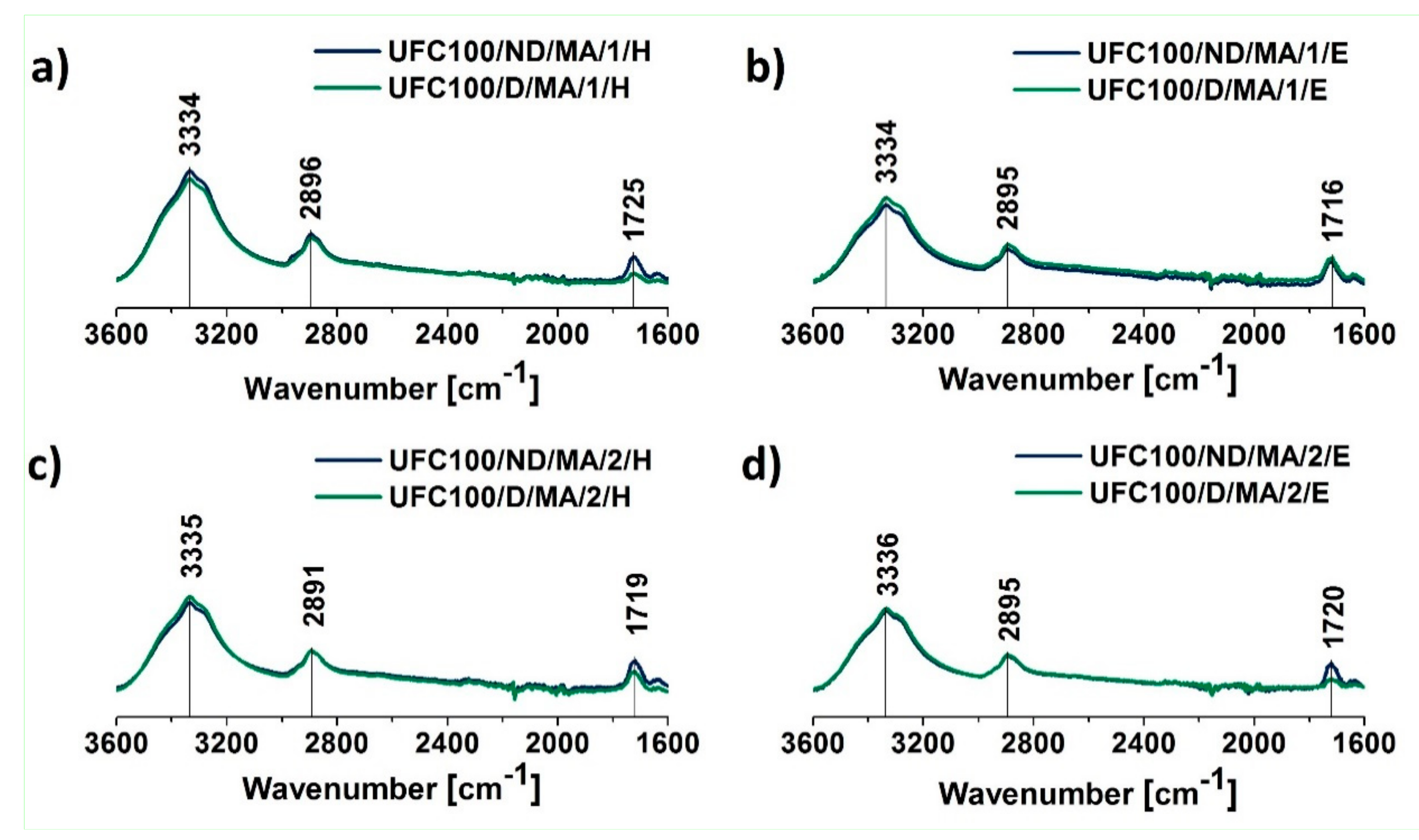
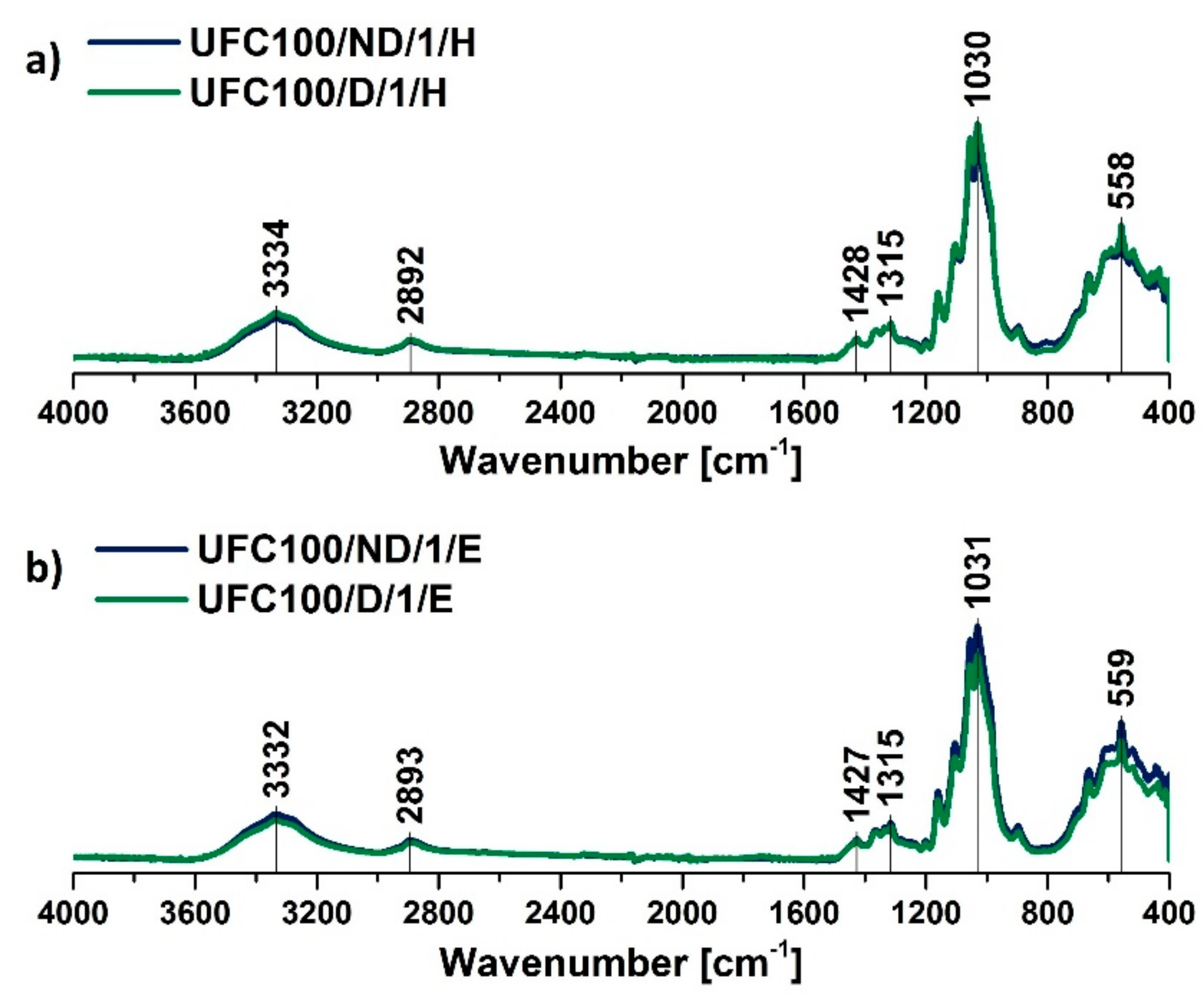
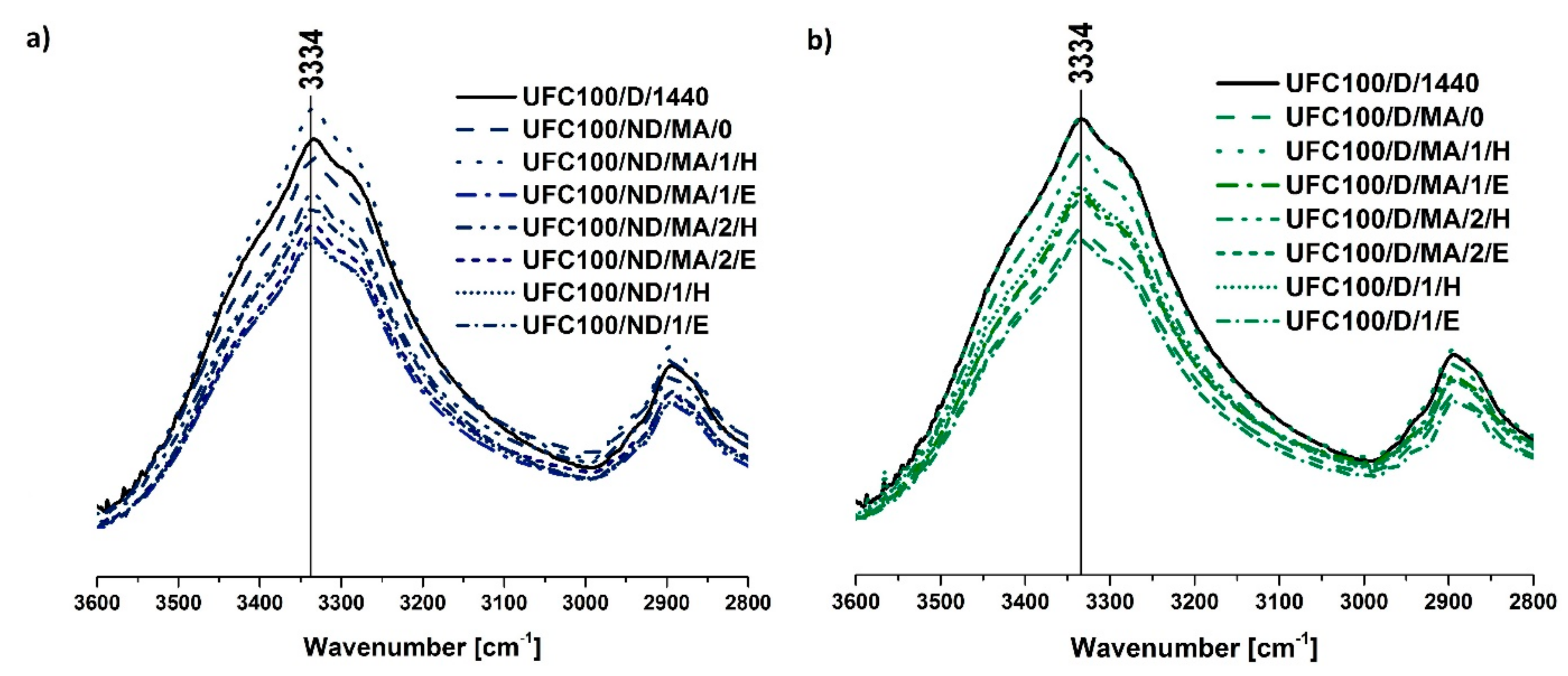
3.1. FT-IR Investigation
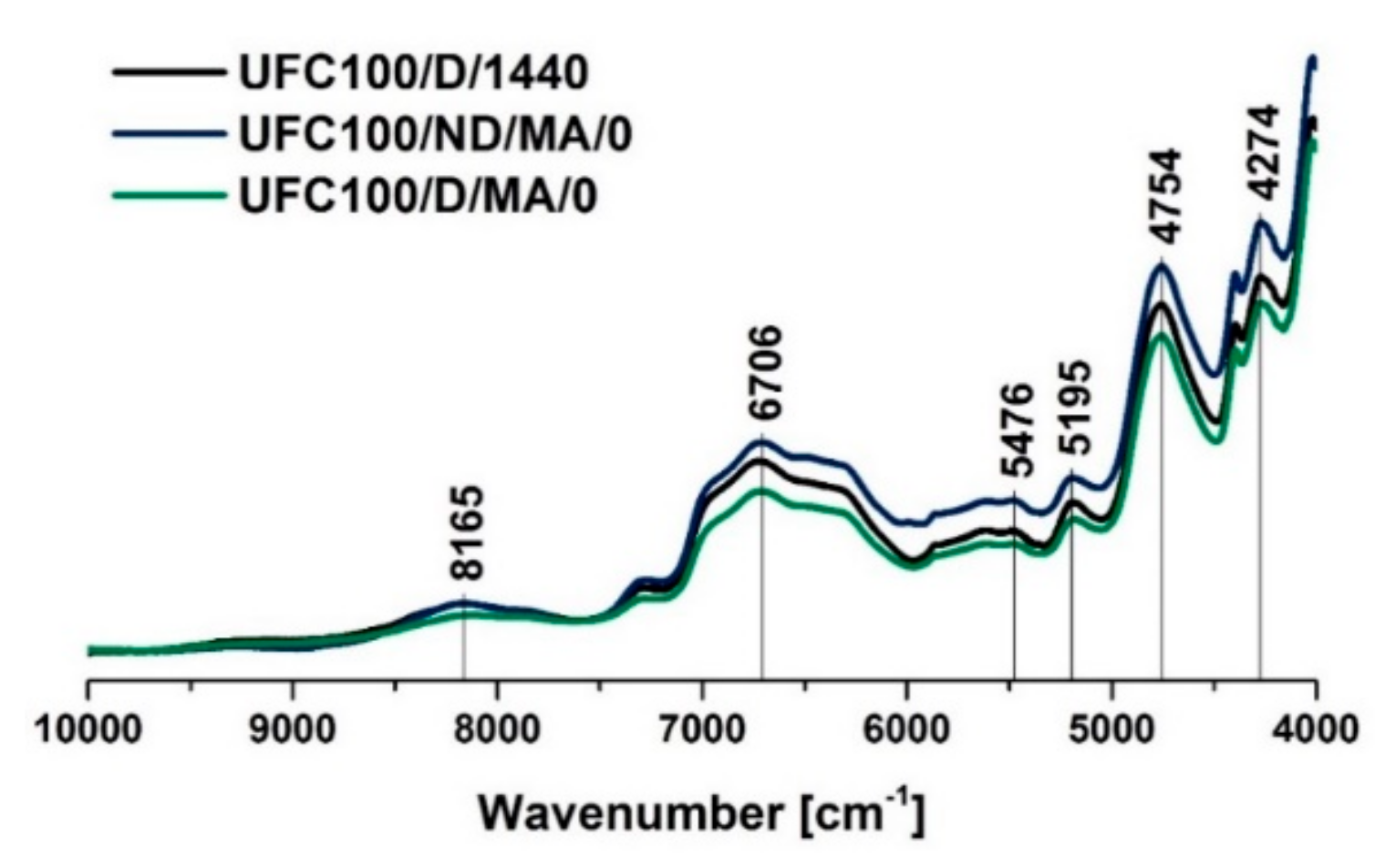
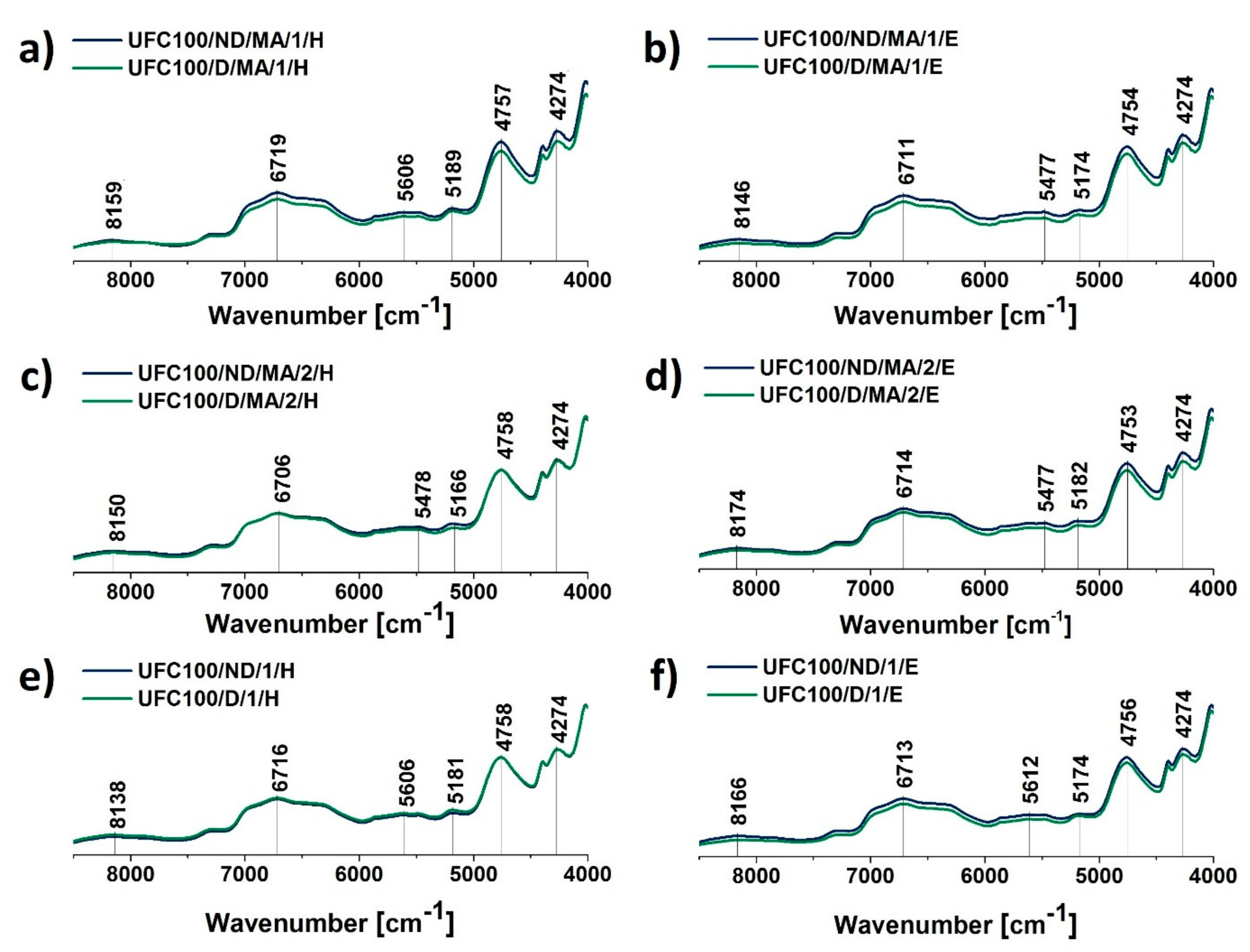
3.2. NIR Analysis
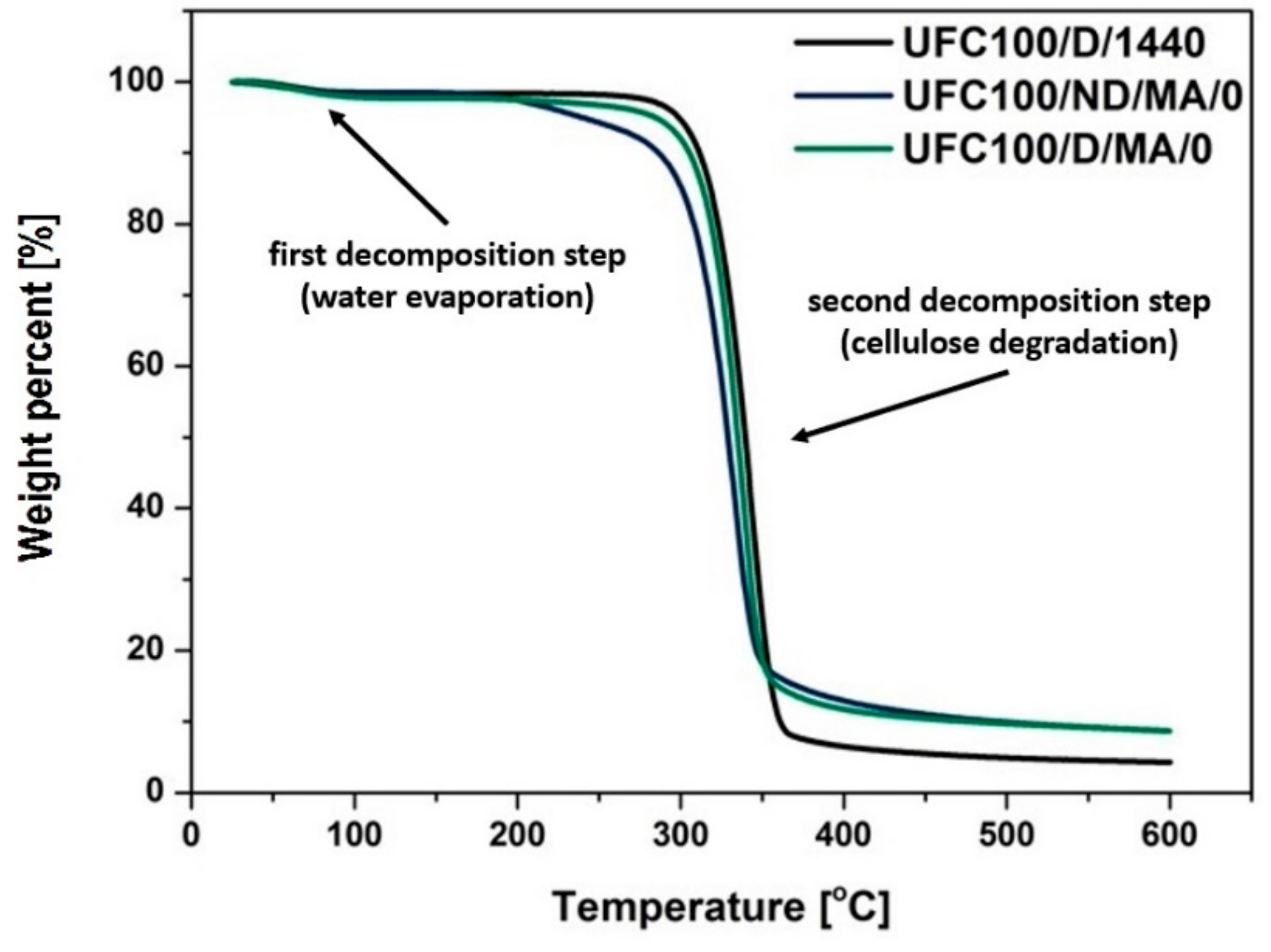
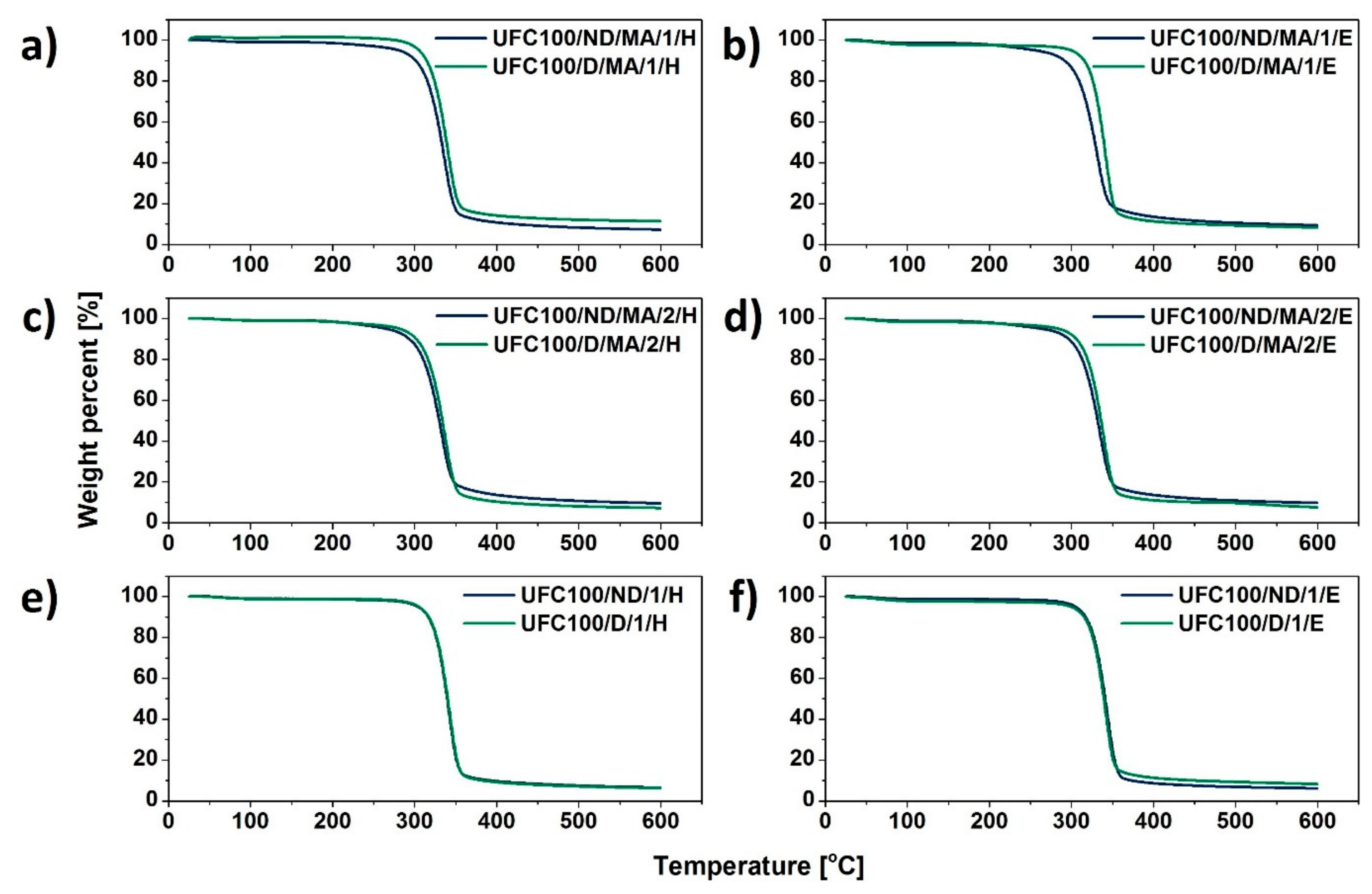
3.3. TGA Investigation
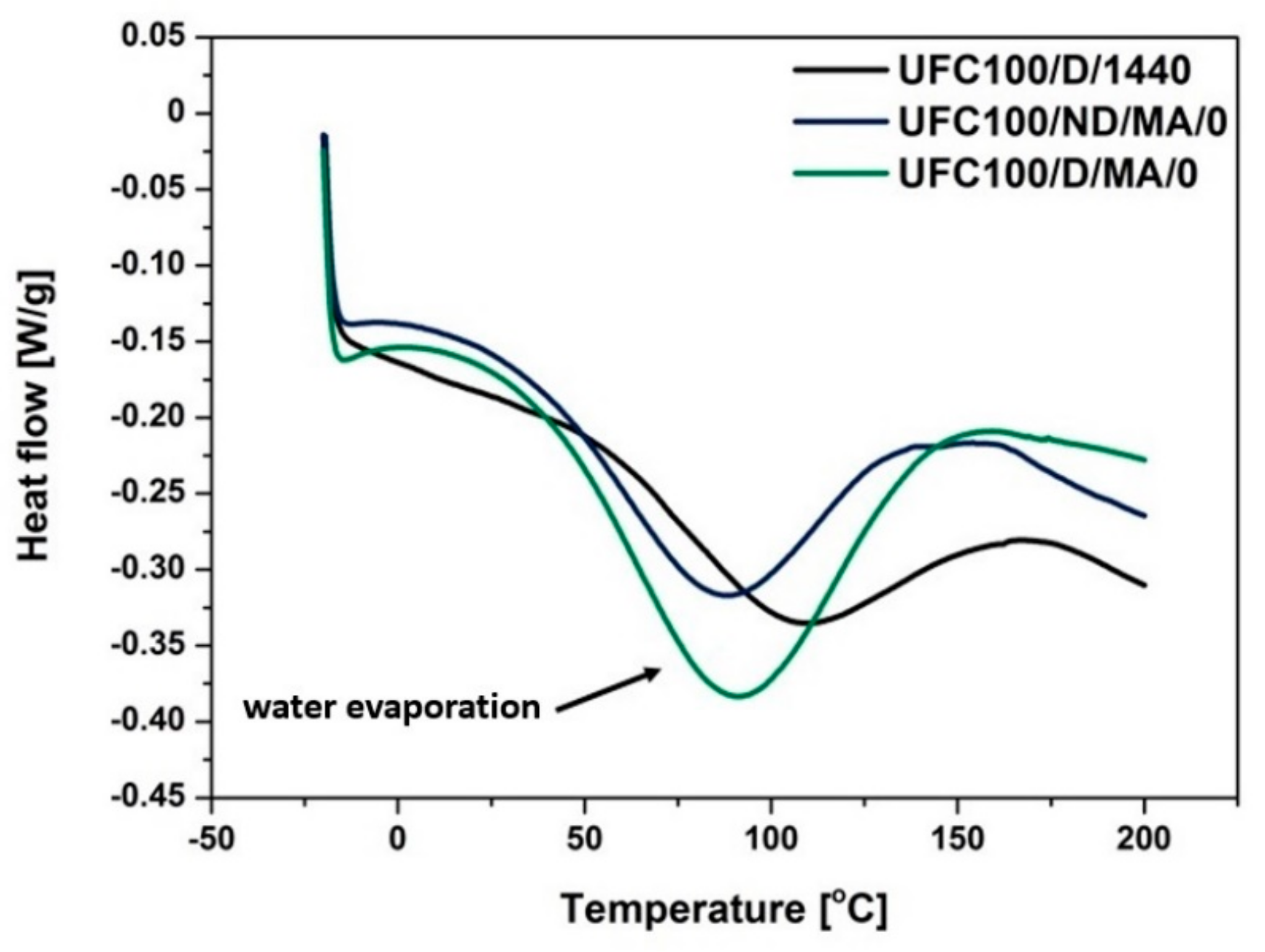
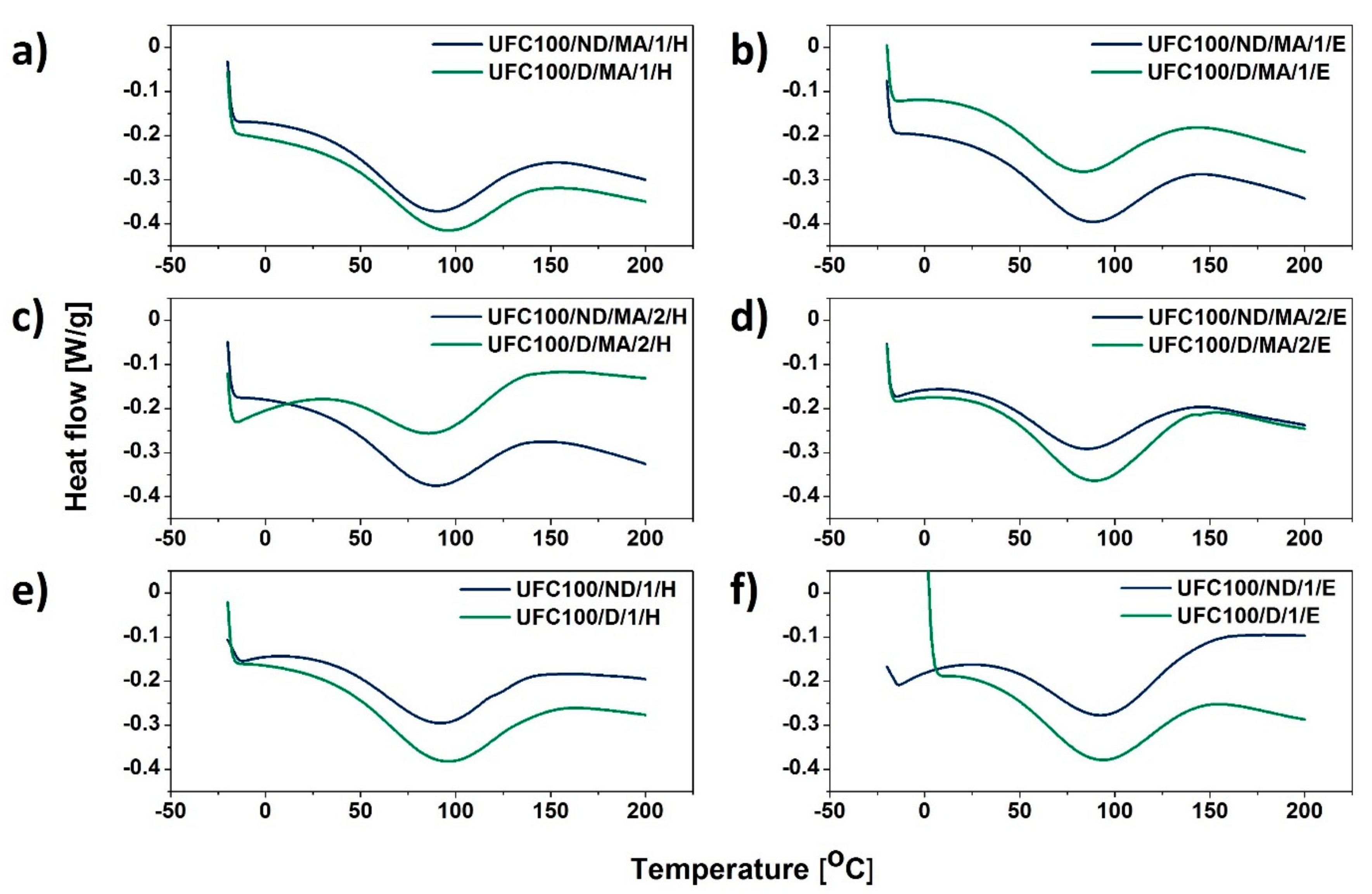
3.4. DSC Analysis
3.5. Fischer Titration Investigation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sharma, P.R.; Varma, A.J. Thermal stability of cellulose and their nanoparticles: Effect of incremental increases in carboxyl and aldehyde groups. Carbohydr. Polym. 2014, 114, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.R.; Joshi, R.; Sharma, S.K.; Hsiao, B.S. A Simple Approach to Prepare Carboxycellulose Nanofibers from Untreated Biomass. Biomacromolecules 2017, 18, 2333–2342. [Google Scholar] [CrossRef] [PubMed]
- Ngwabebhoh, F.A.; Yildiz, U. Nature-derived fibrous nanomaterial toward biomedicine and environmental remediation: Today’s state and future prospects. J. Appl. Polym. Sci. 2019, 1, 1–21. [Google Scholar] [CrossRef]
- Zhu, H.; Luo, W.; Ciesielski, P.N.; Fang, Z.; Zhu, J.Y.; Henriksson, G.; Himmel, M.E.; Hu, L. Wood-Derived Materials for Green Electronics, Biological Devices, and Energy Applications. Chem. Rev. 2016, 116, 9305–9374. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.R.; Chattopadhyay, A.; Sharma, S.K.; Hsiao, B.S. Efficient Removal of UO22+ from Water Using Carboxycellulose Nanofibers Prepared by the Nitro-Oxidation Method. Ind. Eng. Chem. Res. 2017, 56, 13885–13893. [Google Scholar] [CrossRef]
- Sharma, P.R.; Chattopadhyay, A.; Sharma, S.K.; Geng, L.; Amiralian, N.; Martin, D.; Hsiao, B.S. Nanocellulose from Spinifex as an Effective Adsorbent to Remove Cadmium(II) from Water. ACS Sustain. Chem. Eng. 2018, 6, 3279–3290. [Google Scholar] [CrossRef]
- Sharma, P.R.; Zheng, B.; Sharma, S.K.; Zhan, C.; Wang, R.; Bhatia, S.R.; Hsiao, B.S. High Aspect Ratio Carboxycellulose Nanofibers Prepared by Nitro-Oxidation Method and Their Nanopaper Properties; ACS Publications: Washington, DC, USA, 2018; Volume 1, ISBN 1631632779. [Google Scholar]
- Sharma, P.R.; Chattopadhyay, A.; Zhan, C.; Sharma, S.K.; Geng, L.; Hsiao, B.S. Lead removal from water using carboxycellulose nanofibers prepared by nitro-oxidation method. Cellulose 2018, 25, 1961–1973. [Google Scholar] [CrossRef]
- Zhan, C.B.; Sharma, P.R.; Geng, L.H.; Sharma, S.K.; Wang, R.F.; Joshi, R.; Hsiao, B.S. Structural characterization of carboxylcellulose nanofibers extracted from underutilized sources. Sci. China Technol. Sci. 2019, 20–23. [Google Scholar]
- Sharma, P.R.; Sharma, S.K.; Antoine, R.; Hsiao, B.S. Efficient Removal of Arsenic Using Zinc Oxide Nanocrystal-Decorated Regenerated Microfibrillated Cellulose Scaffolds. ACS Sustain. Chem. Eng. 2019, 7, 6140–6151. [Google Scholar] [CrossRef]
- Klemm, D.; Cranston, E.D.; Fischer, D.; Gama, M.; Kedzior, S.A.; Kralisch, D.; Kramer, F.; Kondo, T.; Lindström, T.; Nietzsche, S.; et al. Nanocellulose as a natural source for groundbreaking applications in materials science: Today’s state. Mater. Today 2018, 21, 720–748. [Google Scholar] [CrossRef]
- Foster, E.J.; Moon, R.J.; Agarwal, U.P.; Bortner, M.J.; Bras, J.; Camarero-Espinosa, S.; Chan, K.J.; Clift, M.J.D.; Cranston, E.D.; Eichhorn, S.J.; et al. Current characterization methods for cellulose nanomaterials. Chem. Soc. Rev. 2018, 47, 2609–2679. [Google Scholar] [CrossRef] [PubMed]
- Mihranyan, A.; Llagostera, A.P.; Karmhag, R.; Strømme, M.; Ek, R. Moisture sorption by cellulose powders of varying crystallinity. Int. J. Pharm. 2004, 269, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Espert, A.; Vilaplana, F.; Karlsson, S. Comparison of water absorption in natural cellulosic fibres from wood and one-year crops in polypropylene composites and its influence on their mechanical properties. Compos. Part A Appl. Sci. Manuf. 2004, 35, 1267–1276. [Google Scholar] [CrossRef]
- Chen, H.; Miao, M.; Ding, X. Influence of moisture absorption on the interfacial strength of bamboo/vinyl ester composites. Compos. Part A Appl. Sci. Manuf. 2009, 40, 2013–2019. [Google Scholar] [CrossRef]
- Sombatsompop, N.; Chaochanchaikul, K. Effect of moisture content on mechanical properties, thermal and structural stability and extrudate texture of poly(vinyl chloride)/wood sawdust composites. Polym. Int. 2004, 53, 1210–1218. [Google Scholar] [CrossRef]
- Scheirs, J.; Camino, G.; Tumiatti, W. Overview of water evolution during the thermal degradation of cellulose. Eur. Polym. J. 2001, 37, 933–942. [Google Scholar] [CrossRef]
- Lu, P.; Hsieh, Y. Lo Preparation and properties of cellulose nanocrystals: Rods, spheres, and network. Carbohydr. Polym. 2010, 82, 329–336. [Google Scholar] [CrossRef]
- Worasuwannarak, N.; Sonobe, T.; Tanthapanichakoon, W. Pyrolysis behaviors of rice straw, rice husk, and corncob by TG-MS technique. J. Anal. Appl. Pyrolysis 2007, 78, 265–271. [Google Scholar] [CrossRef]
- Van de Velden, M.; Baeyens, J.; Brems, A.; Janssens, B.; Dewil, R. Fundamentals, kinetics and endothermicity of the biomass pyrolysis reaction. Renew. Energy 2010, 35, 232–242. [Google Scholar] [CrossRef]
- Banyasz, J.L.; Li, S.; Lyons-Hart, J.; Shafer, K.H. Gas evolution and the mechanism of cellulose pyrolysis. Fuel 2001, 80, 1757–1763. [Google Scholar] [CrossRef]
- Collard, F.X.; Blin, J. A review on pyrolysis of biomass constituents: Mechanisms and composition of the products obtained from the conversion of cellulose, hemicelluloses and lignin. Renew. Sustain. Energy Rev. 2014, 38, 594–608. [Google Scholar] [CrossRef]
- Wang, Z.; McDonald, A.G.; Westerhof, R.J.M.; Kersten, S.R.A.; Cuba-Torres, C.M.; Ha, S.; Pecha, B.; Garcia-Perez, M. Effect of cellulose crystallinity on the formation of a liquid intermediate and on product distribution during pyrolysis. J. Anal. Appl. Pyrolysis 2013, 100, 56–66. [Google Scholar] [CrossRef]
- Liu, D.; Yu, Y.; Wu, H. Differences in water-soluble intermediates from slow pyrolysis of amorphous and crystalline cellulose. Energy Fuels 2013, 27, 1371–1380. [Google Scholar] [CrossRef]
- Demirbas, A. Biomass resource facilities and biomass conversion processing for fuels and chemicals. Energy Convers. Manag. 2001, 42, 1357–1378. [Google Scholar] [CrossRef]
- Azeez, A.M.; Meier, D.; Odermatt, J. Temperature dependence of fast pyrolysis volatile products from European and African biomasses. J. Anal. Appl. Pyrolysis 2011, 90, 81–92. [Google Scholar] [CrossRef]
- Heywood, R.J.; Xiao, X.; Emsley, A.M.; Ali, M. Degradation of cellulosic insulation in power transformers. Part 2: Formation of furan products in insulating oil. IEE Proc. Sci. Meas. Technol. 2002, 147, 110–114. [Google Scholar]
- Heywood, R.J.; Xiao, X.; Ali, M.; Emsley, A.M. Degradation of cellulosic insulation in power transformers. Part 3: Effects of oxygen and water on ageing in oil. IEE Proc. Sci. Meas. Technol. 2002, 147, 115–119. [Google Scholar]
- Poletto, M.; Zattera, A.J.; Forte, M.M.C.; Santana, R.M.C. Thermal decomposition of wood: Influence of wood components and cellulose crystallite size. Bioresour. Technol. 2012, 109, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Ornaghi Júnior, H.L.; Zattera, A.J. Native cellulose: Structure, characterization and thermal properties. Materials 2014, 7, 6105–6119. [Google Scholar] [CrossRef]
- Cichosz, S.; Masek, A.; Wolski, K.; Zaborski, M. Universal approach of cellulose fibres chemical modification result analysis via commonly used techniques. Polym. Bull. 2018, 76, 2147–2162. [Google Scholar] [CrossRef]
- Cichosz, S.; Masek, A.; Wolski, K. Innovative cellulose fibres reinforced ethylene-norbornene copolymer composites of an increased degradation potential. Polym. Degrad. Stab. 2019, 159, 174–183. [Google Scholar] [CrossRef]
- Leszczyńska, A.; Radzik, P.; Szefer, E.; Mičušík, M.; Omastová, M.; Pielichowski, K. Surface Modification of Cellulose Nanocrystals with Succinic Anhydride. Polymers 2019, 11, 866. [Google Scholar] [CrossRef] [PubMed]
- Manaf, M.E.A.; Tsuji, M.; Nobukawa, S.; Yamaguchi, M. Effect of moisture on the orientation birefringence of cellulose esters. Polymers 2011, 3, 955–966. [Google Scholar] [CrossRef]
- Mao, H.; Wei, C.; Gong, Y.; Wang, S.; Ding, W. Mechanical and Water-Resistant Properties of Eco-Friendly Chitosan Membrane Reinforced with Cellulose Nanocrystals. Polymers 2019, 11, 166. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Fei, M.-E.; Ban, Y.; Jia, A.; Qiu, R. Preparation and evaluation of green composites from microcrystalline cellulose and a soybean-oil derivative. Polymers 2017, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Vuoti, S.; Laatikainen, E.; Heikkinen, H.; Johansson, L.S.; Saharinen, E.; Retulainen, E. Chemical modification of cellulosic fibers for better convertibility in packaging applications. Carbohydr. Polym. 2013, 96, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Cichosz, S.; Masek, A. Cellulose structure and property changes indicated via wetting-drying cycles. Polym. Degrad. Stab. 2019, 167, 33–43. [Google Scholar] [CrossRef]
- Kittle, J.D.; Du, X.; Jiang, F.; Qian, C.; Heinze, T.; Roman, M.; Esker, A.R. Equilibrium water contents of cellulose films determined via solvent exchange and quartz crystal microbalance with dissipation monitoring. Biomacromolecules 2011, 12, 2881–2887. [Google Scholar] [CrossRef]
- Vuoti, S.; Talja, R.; Johansson, L.S.; Heikkinen, H.; Tammelin, T. Solvent impact on esterification and film formation ability of nanofibrillated cellulose. Cellulose 2013, 20, 2359–2370. [Google Scholar] [CrossRef]
- Iwamoto, S.; Endo, T. 3 Nm Thick Lignocellulose Nano Fibers Obtained From Esterified Wood With Maleic Anhydride. ACS Macro Lett. 2015, 4, 80–83. [Google Scholar] [CrossRef]
- Broido, A. A simple, sensitive graphical method of treating thermogravimetric analysis data. J. Polym. Sci. Part A-2 Polym. Phys. 1969, 7, 1761–1773. [Google Scholar] [CrossRef]
- Łojewska, J.; Miśkowiec, P.; Łojewski, T.; Proniewicz, L.M. Cellulose oxidative and hydrolytic degradation: In situ FTIR approach. Polym. Degrad. Stab. 2005, 88, 512–520. [Google Scholar] [CrossRef]
- Gulmine, J.; Janissek, P.; Heise, H.; Akcelrud, L. Polyethylene characterization by FTIR. Polym. Test. 2002, 21, 557–563. [Google Scholar] [CrossRef]
- Morán, J.I.; Alvarez, V.A.; Cyras, V.P.; Vázquez, A. Extraction of cellulose and preparation of nanocellulose from sisal fibers. Cellulose 2008, 15, 149–159. [Google Scholar] [CrossRef]
- Patel, H.A.; Somani, R.S.; Bajaj, H.C.; Jasra, R.V. Preparation and characterization of phosphonium montmorillonite with enhanced thermal stability. Appl. Clay Sci. 2007, 35, 194–200. [Google Scholar] [CrossRef]
- Stenstad, P.; Andresen, M.; Tanem, B.S.; Stenius, P. Chemical surface modifications of microfibrillated cellulose. Cellulose 2008, 15, 35–45. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Salmén, L.; Bergström, E. Cellulose structural arrangement in relation to spectral changes in tensile loading FTIR. Cellulose 2009, 16, 975–982. [Google Scholar] [CrossRef]
- Ołdak, D.; Kaczmarek, H.; Buffeteau, T.; Sourisseau, C. Photo- and bio-degradation processes in polyethylene, cellulose and their blends studied by ATR-FTIR and raman spectroscopies. J. Mater. Sci. 2005, 40, 4189–4198. [Google Scholar] [CrossRef]
- Colom, X.; Carrillo, F.; Nogués, F.; Garriga, P. Structural analysis of photodegraded wood by means of FTIR spectroscopy. Polym. Degrad. Stab. 2003, 80, 543–549. [Google Scholar] [CrossRef]
- Colom, X.; Carrasco, F.; Pagès, P.; Canavate, J. Effects of different treatments on the interface of HDPE/lignocellulosic fiber composites. Compos. Sci. Technol. 2003, 63, 161–169. [Google Scholar] [CrossRef]
- Ibrahim, M.; Osman, O.; Mahmoud, A.A. Spectroscopic analyses of cellulose and chitosan: FTIR and modeling approach. J. Comput. Theor. Nanosci. 2011, 8, 117–123. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Seo, G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar] [CrossRef]
- Goussé, C.; Chanzy, H.; Excoffier, G.; Soubeyrand, L.; Fleury, E. Stable suspensions of partially silylated cellulose whiskers dispersed in organic solvents. Polymer 2002, 43, 2645–2651. [Google Scholar] [CrossRef]
- Cao, Y.; Tan, H. Structural characterization of cellulose with enzymatic treatment. J. Mol. Struct. 2004, 705, 189–193. [Google Scholar] [CrossRef]
- Sato, A.; Kabusaki, D.; Okumura, H.; Nakatani, T.; Nakatsubo, F.; Yano, H. Surface modification of cellulose nanofibers with alkenyl succinic anhydride for high-density polyethylene reinforcement. Compos. Part A Appl. Sci. Manuf. 2016, 83, 72–79. [Google Scholar] [CrossRef]
- Célino, A.; Gonçalves, O.; Jacquemin, F.; Fréour, S. Qualitative and quantitative assessment of water sorption in natural fibres using ATR-FTIR spectroscopy. Carbohydr. Polym. 2014, 101, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Schwanninger, M.; Rodrigues, J.C.; Fackler, K. A review of band assignments in near infrared spectra of wood and wood components. J. Near Infrared Spectrosc. 2011, 19, 287–308. [Google Scholar] [CrossRef]
- Ali, M.; Emsley, A.M.; Herman, H.; Heywood, R.J. Spectroscopic studies of the ageing of cellulosic paper. Polymer 2001, 42, 2893–2900. [Google Scholar] [CrossRef]
- Inagaki, T.; Siesler, H.W.; Mitsui, K.; Tsuchikawa, S. Difference of the crystal structure of cellulose in wood after hydrothermal and aging degradation: A NIR spectroscopy and XRD study. Biomacromolecules 2010, 11, 2300–2305. [Google Scholar] [CrossRef]
- Tsuchikawa, S.; Siesler, H.W. Near-Infrared Spectroscopic Monitoring of the Diffusion Process of Deuterium-Labeled Molecules in Wood. Part I: Softwood. Appl. Spectrosc. 2003, 57, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Rantanen, J.; Räsänen, E.; Tenhunen, J.; Känsäkoski, M.; Mannermaa, J.P.; Yliruusi, J. In-line moisture measurement during granulation with a four-wavelength near infrared sensor: An evaluation of particle size and binder effects. Eur. J. Pharm. Biopharm. 2000, 50, 271–276. [Google Scholar] [CrossRef]
- Ma, S.; Yu, S.J.; Wang, Z.H.; Zheng, X.L. Ultrasound-Assisted Modification of Beet Pulp Cellulose With Phthalic Anhydride in Ionic Liquid. Cellul. Chem. Technol. 2013, 47, 527–533. [Google Scholar]
- Wyasu, G.; Gimba, C.E.; Agbaji, E.B.; Ndukwe, G.I. Thermo-gravimetry(TGA) and DSC of thermal analysis techniques in production of active carbon from lignocellulosic materials. Pelagia Res. Libr. Adv. Appl. Sci. Res. 2016, 7, 109–115. [Google Scholar]
- Li, Y.; Xiao, H.; Chen, M.; Song, Z.; Zhao, Y. Absorbents based on maleic anhydride-modified cellulose fibers/diatomite for dye removal. J. Mater. Sci. 2014, 49, 6696–6704. [Google Scholar] [CrossRef]
- Das, K.; Ray, D.; Bandyopadhyay, N.R.; Sengupta, S. Study of the Properties of Microcrystalline Cellulose Particles from Different Renewable Resources by XRD, FTIR, Nanoindentation, TGA and SEM. J. Polym. Environ. 2010, 18, 355–363. [Google Scholar] [CrossRef]
- Völker, S.; Rieckmann, T. Thermokinetic investigation of cellulose pyrolysis—Impact of initial and final mass on kinetic results. J. Anal. Appl. Pyrolysis 2002, 62, 165–177. [Google Scholar] [CrossRef]
- Park, S.; Venditti, R.A.; Jameel, H.; Pawlak, J.J. Hard to remove water in cellulose fibers characterized by high resolution thermogravimetric analysis—Methods development. Cellulose 2006, 13, 23–30. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Habibi, Y.; Rojas, O.J.; Pawlak, J.J. The effect of chemical composition on microfibrillar cellulose films from wood pulps: Mechanical processing and physical properties. Bioresour. Technol. 2010, 101, 5961–5968. [Google Scholar] [CrossRef]
- Shen, D.K.; Gu, S. The mechanism for thermal decomposition of cellulose and its main products. Bioresour. Technol. 2009, 100, 6496–6504. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, X.C.; Dong, C.Q.; Zhang, Z.F.; Zhang, X.M.; Zhu, X.F. Influence of pyrolysis temperature and time on the cellulose fast pyrolysis products: Analytical Py-GC/MS study. J. Anal. Appl. Pyrolysis 2011, 92, 430–438. [Google Scholar] [CrossRef]
- Huang, F.Y. Thermal properties and thermal degradation of cellulose tri-stearate (CTs). Polymers 2012, 4, 1012–1024. [Google Scholar] [CrossRef]
- Ouajai, S.; Shanks, R.A. Composition, structure and thermal degradation of hemp cellulose after chemical treatments. Polym. Degrad. Stab. 2005, 89, 327–335. [Google Scholar] [CrossRef]
- Fernandes Diniz, J.M.B.; Gil, M.H.; Castro, J.A.A.M. Hornification—Its origin and interpretation in wood pulps. Wood Sci. Technol. 2004, 37, 489–494. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, F.; Keer, L.M. Influence of surface modification on the microstructure and thermo-mechanical properties of bamboo fibers. Materials 2015, 8, 6597–6608. [Google Scholar] [CrossRef] [PubMed]
- Ciolacu, D.; Ciolacu, F.; Popa, V.I. Amorphous cellulose—Structure and characterization. Cellul. Chem. Technol. 2011, 45, 13–21. [Google Scholar]
- Szcześniak, L.; Rachocki, A.; Tritt-Goc, J. Glass transition temperature and thermal decomposition of cellulose powder. Cellulose 2008, 15, 445–451. [Google Scholar] [CrossRef]
- Kestens, V.; Conneely, P.; Bernreuther, A. Vaporisation coulometric Karl Fischer titration: A perfect tool for water content determination of difficult matrix reference materials. Food Chem. 2008, 106, 1454–1459. [Google Scholar] [CrossRef]
- Ronkart, S.N.; Paquot, M.; Fougnies, C.; Deroanne, C.; Van Herck, J.C.; Blecker, C. Determination of total water content in inulin using the volumetric Karl Fischer titration. Talanta 2006, 70, 1006–1010. [Google Scholar] [CrossRef]
- Arbelaiz, A.; Fernández, B.; Ramos, J.A.; Retegi, A.; Llano-Ponte, R.; Mondragon, I. Mechanical properties of short flax fibre bundle/polypropylene composites: Influence of matrix/fibre modification, fibre content, water uptake and recycling. Compos. Sci. Technol. 2005, 65, 1582–1592. [Google Scholar] [CrossRef]
- Ul-Islam, M.; Khan, T.; Park, J.K. Water holding and release properties of bacterial cellulose obtained by in situ and ex situ modification. Carbohydr. Polym. 2012, 88, 596–603. [Google Scholar] [CrossRef]
- Tomšič, B.; Simončič, B.; Orel, B.; Vilčnik, A.; Spreizer, H. Biodegradability of cellulose fabric modified by imidazolidinone. Carbohydr. Polym. 2007, 69, 478–488. [Google Scholar] [CrossRef]
- Ishii, D.; Tatsumi, D.; Matsumoto, T. Effect of solvent exchange on the solid structure and dissolution behavior of cellulose. Biomacromolecules 2003, 4, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Ben Ohoud, M.; Obrecht, F.; Gatineua, L.; Levitz, P.; Van Damme, H. Surface area, mass fractal dimension, and apparent density of powders. J. Colloid Interface Sci. 1988, 124, 156–161. [Google Scholar] [CrossRef]
- Park, S.; Venditti, R.A.; Jameel, H.; Pawlak, J.J. Changes in pore size distribution during the drying of cellulose fibers as measured by differential scanning calorimetry. Carbohydr. Polym. 2006, 66, 97–103. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Acetone | Ethanol (99.9%) | Hexane |
|---|---|---|---|
| Melting point (°C) | −95 | −117 | −95 |
| Boiling point (°C) | 55–57 | 78 | 68 |
| Viscosity at 20 °C (mPas) | 0.330 | 1.078 | 0.310 |
| Density at 20 °C (g/cm3) | 0.791 | 0.790 | 0.660 |
| Vapor pressure at 20 °C (mbar) | 233 | 60 | 160 |
| Solubility in water (g/cm3) | yes | yes | 0.1 |
| Solubility in organic solvents | yes | yes | yes |
| Sample | UFC100 Dried before Modification (D) | UFC100 Not Dried before Modification (ND) | Solvent Exchange | MA Treated | |||
|---|---|---|---|---|---|---|---|
| Path 1 | Path 2 | ||||||
| H | E | H | E | ||||
| UFC100/ND/MA/0 | ------ | ✔ | ------ | ------ | ------ | ------ | ✔ |
| UFC100/ND/MA/1/H | ------ | ✔ | ✔ | ------ | ------ | ------ | ✔ |
| UFC100/ND/MA/1/E | ------ | ✔ | ------ | ✔ | ------ | ------ | ✔ |
| UFC100/ND/MA/2/H | ------ | ✔ | ------ | ------ | ✔ | ------ | ✔ |
| UFC100/ND/MA/2/E | ------ | ✔ | ------ | ------ | ------ | ✔ | ✔ |
| UFC100/ND/1/H | ------ | ✔ | ✔ | ------ | ------ | ------ | ------ |
| UFC100/ND/1/E | ------ | ✔ | ------ | ✔ | ------ | ------ | ------ |
| UFC100/D/MA/0 | ✔ | ------ | ------ | ------ | ------ | ------ | ✔ |
| UFC100/D/MA/1/H | ✔ | ------ | ✔ | ------ | ------ | ------ | ✔ |
| UFC100/D/MA/1/E | ✔ | ------ | ------ | ✔ | ------ | ------ | ✔ |
| UFC100/D/MA/2/H | ✔ | ------ | ------ | ------ | ✔ | ------ | ✔ |
| UFC100/D/MA/2/E | ✔ | ------ | ------ | ------ | ------ | ✔ | ✔ |
| UFC100/D/1/H | ✔ | ------ | ✔ | ------ | ------ | ------ | ------ |
| UFC100/D/1/E | ✔ | ------ | ------ | ✔ | ------ | ------ | ------ |
| Wavenumber (cm−1) | Chemical Group | Ref. |
|---|---|---|
| 558 | C–OH out-of-plane bending, C–C | [48] |
| 1200–900 | –OH, –COO | [46] |
| 1100–1000 | CO–O–CO | [49] |
| 1030 | C–O stretching vibration | [50] |
| 1100 | –OH | [44] |
| 1158 | C–O stretching vibration, C–O–C bridge | [51] |
| 1245 | –CH3 | [52] |
| 1314 | C–O, C=O, C=C, COOH | [44] |
| 1426 | C–H bending of CH2 | [53] |
| 1635 | –OH bending of adsorbed water, C=C | [54] |
| 1718 | C=O | [47] |
| 2896 | CH stretching vibration | [45] |
| 3334 | –OH, water | [43] |
| Sample | I1720 (–) |
|---|---|
| UFC100/ND/MA/0 | 0.035 |
| UFC100/ND/MA/1/H | 0.020 |
| UFC100/ND/MA/1/E | 0.016 |
| UFC100/ND/MA/2/H | 0.024 |
| UFC100/ND/MA/2/E | 0.022 |
| UFC100/D/MA/0 | 0.007 |
| UFC100/D/MA/1/H | 0.007 |
| UFC100/D/MA/1/E | 0.019 |
| UFC100/D/MA/2/H | 0.016 |
| UFC100/D/MA/2/E | 0.010 |
| Wavenumber (cm−1) | Chemical Group | Ref. |
|---|---|---|
| 4754 | –OH, C=O | [60] |
| 5220–5150 | –OH, water | [59] |
| 6706 | –OH, water, hydrogen bonds | [61] |
| 4724 | C=C | [62] |
| 6772 | –OH | [63] |
| 7290 | C–H | [59] |
| 8165 | C–H | [59] |
| Sample | T05% (°C) | T10% (°C) | T15% (°C) | T20% (°C) | T50% (°C) | T80% (°C) | T90% (°C) |
|---|---|---|---|---|---|---|---|
| UFC100/ND/MA/0 | 240 | 286 | 300 | 309 | 328 | 347 | 496 |
| UFC100/ND/MA/1/H | 280 | 301 | 310 | 315 | 333 | 343 | 418 |
| UFC100/ND/MA/1/E | 254 | 292 | 302 | 309 | 328 | 346 | 538 |
| UFC100/ND/MA/2/H | 265 | 294 | 304 | 310 | 330 | 348 | 546 |
| UFC100/ND/MA/2/E | 263 | 296 | 307 | 313 | 332 | 349 | 562 |
| UFC100/ND/1/H | 304 | 316 | 322 | 326 | 340 | 351 | 392 |
| UFC100/ND/1/E | 304 | 317 | 323 | 327 | 341 | 352 | 372 |
| UFC100/D/MA/0 | 284 | 305 | 314 | 318 | 335 | 349 | 478 |
| UFC100/D/MA/1/H | 305 | 314 | 320 | 324 | 339 | 354 | 560 |
| UFC100/D/MA/1/E | 299 | 316 | 322 | 326 | 340 | 351 | 449 |
| UFC100/D/MA/2/H | 281 | 302 | 310 | 316 | 334 | 348 | 406 |
| UFC100/D/MA/2/E | 284 | 306 | 314 | 320 | 336 | 350 | 452 |
| UFC100/D/1/H | 306 | 317 | 323 | 327 | 340 | 352 | 383 |
| UFC100/D/1/E | 300 | 316 | 322 | 326 | 340 | 351 | 449 |
| Sample | (kJ/mol) | (kJ/mol) |
|---|---|---|
| UFC100/ND/MA/0 | 73 ± 3 | 76 ± 1 |
| UFC100/ND/MA/1/H | 73 ± 2 | 103 ± 2 |
| UFC100/ND/MA/1/E | 69 ± 2 | 85 ± 1 |
| UFC100/ND/MA/2/H | 72 ± 2 | 85 ± 1 |
| UFC100/ND/MA/2/E | 66 ± 2 | 87 ± 1 |
| UFC100/ND/1/H | 77 ± 2 | 112 ± 1 |
| UFC100/ND/1/E | 73 ± 2 | 125 ± 3 |
| UFC100/D/MA/0 | 54 ± 1 | 111 ± 2 |
| UFC100/D/MA/1/H | 75 ± 3 | 115 ± 2 |
| UFC100/D/MA/1/E | 74 ± 2 | 127 ± 3 |
| UFC100/D/MA/2/H | 45 ± 1 | 105 ± 2 |
| UFC100/D/MA/2/E | 73 ± 3 | 110 ± 2 |
| UFC100/D/1/H | 76 ± 2 | 125 ± 3 |
| UFC100/D/1/E | 74 ± 2 | 127 ± 3 |
| Sample | Tpeak (°C) | ΔH (J/g) |
|---|---|---|
| UFC100/ND/MA/0 | 86 | 45.67 |
| UFC100/ND/MA/1/H | 88 | 56.68 |
| UFC100/ND/MA/1/E | 86 | 52.99 |
| UFC100/ND/MA/2/H | 87 | 52.04 |
| UFC100/ND/MA/2/E | 84 | 39.95 |
| UFC100/ND/1/H | 94 | 54.69 |
| UFC100/ND/1/E | 94 | 54.96 |
| UFC100/D/MA/0 | 90 | 76.86 |
| UFC100/D/MA/1/H | 92 | 51.75 |
| UFC100/D/MA/1/E | 82 | 45.98 |
| UFC100/D/MA/2/H | 88 | 35.52 |
| UFC100/D/MA/2/E | 88 | 58.66 |
| UFC100/D/1/H | 94 | 64.44 |
| UFC100/D/1/E | 92 | 56.46 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cichosz, S.; Masek, A. Cellulose Fibers Hydrophobization via a Hybrid Chemical Modification. Polymers 2019, 11, 1174. https://doi.org/10.3390/polym11071174
Cichosz S, Masek A. Cellulose Fibers Hydrophobization via a Hybrid Chemical Modification. Polymers. 2019; 11(7):1174. https://doi.org/10.3390/polym11071174
Chicago/Turabian StyleCichosz, Stefan, and Anna Masek. 2019. "Cellulose Fibers Hydrophobization via a Hybrid Chemical Modification" Polymers 11, no. 7: 1174. https://doi.org/10.3390/polym11071174
APA StyleCichosz, S., & Masek, A. (2019). Cellulose Fibers Hydrophobization via a Hybrid Chemical Modification. Polymers, 11(7), 1174. https://doi.org/10.3390/polym11071174