Phase Diagram for Ideal Diblock-Copolymer Micelles Compared to Polymerization-Induced Self Assembly


Abstract
: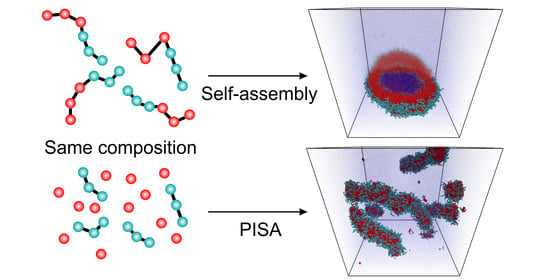
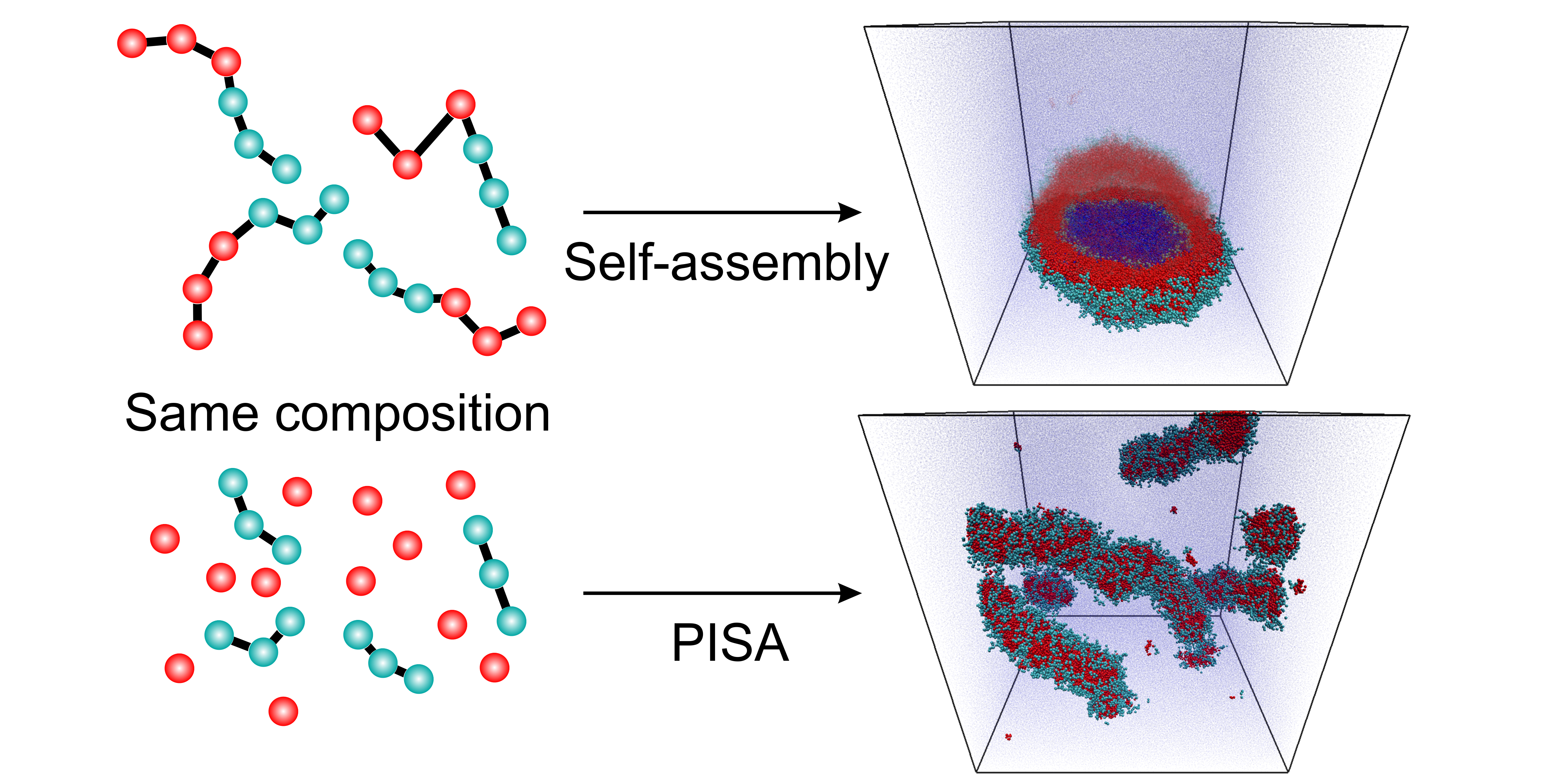{kind=link}
{kind=link}
{kind=link}
{kind=link}
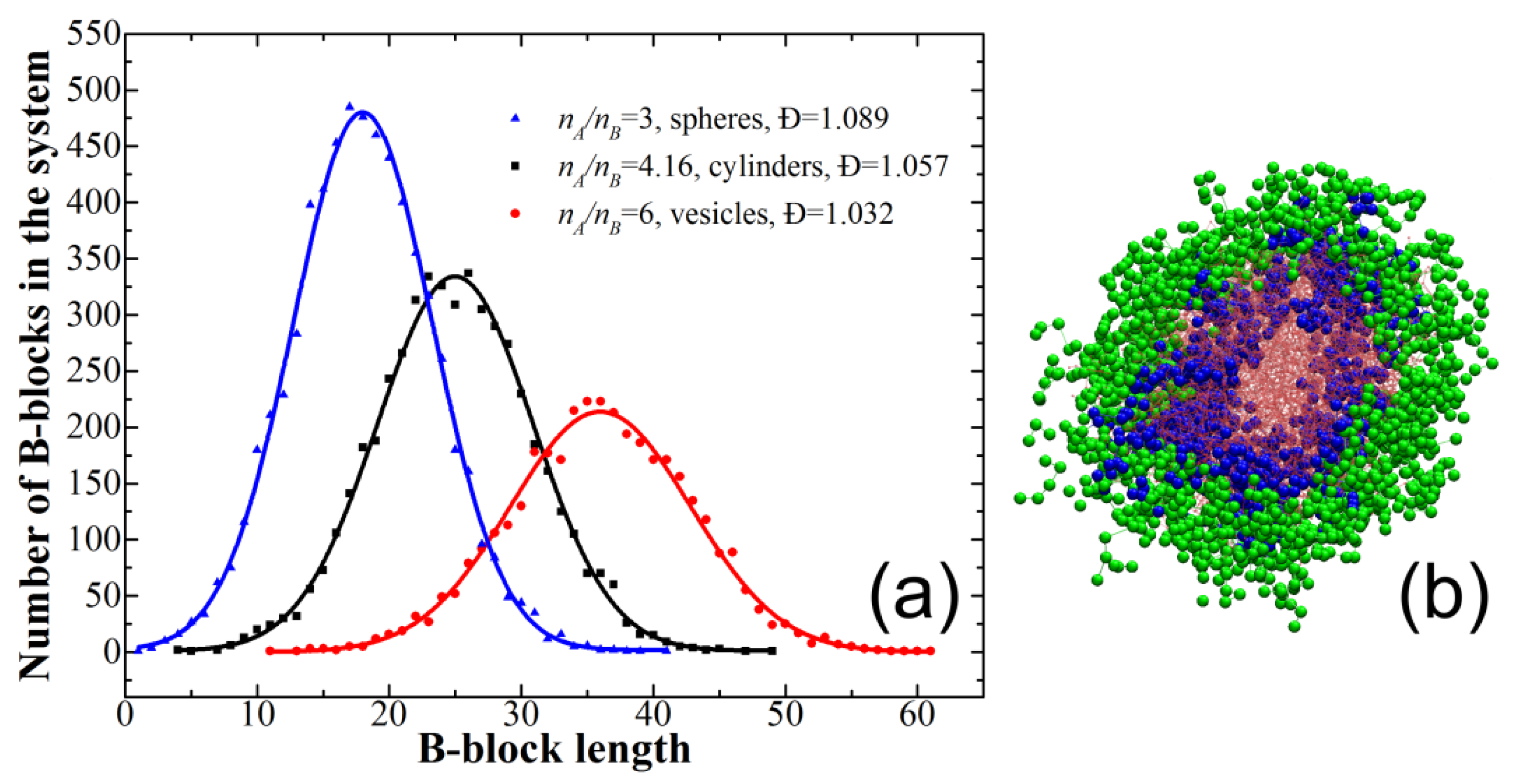{kind=link}
1. Introduction
2. Method and Model
2.1. Method
2.2. Reaction Scheme
- A growing chain end i is selected at random;
- The list of all free monomer beads located closer than the reaction radius Rchem from the bead i is created. The closest bead j is determined, and a bond between the beads i and j is created with the probability pp. If the bond is not created, the procedure is repeated with the next closest bead from the list until a bond is created or there are no more unchecked monomers.
2.3. Model and System
3. Results and Discussions
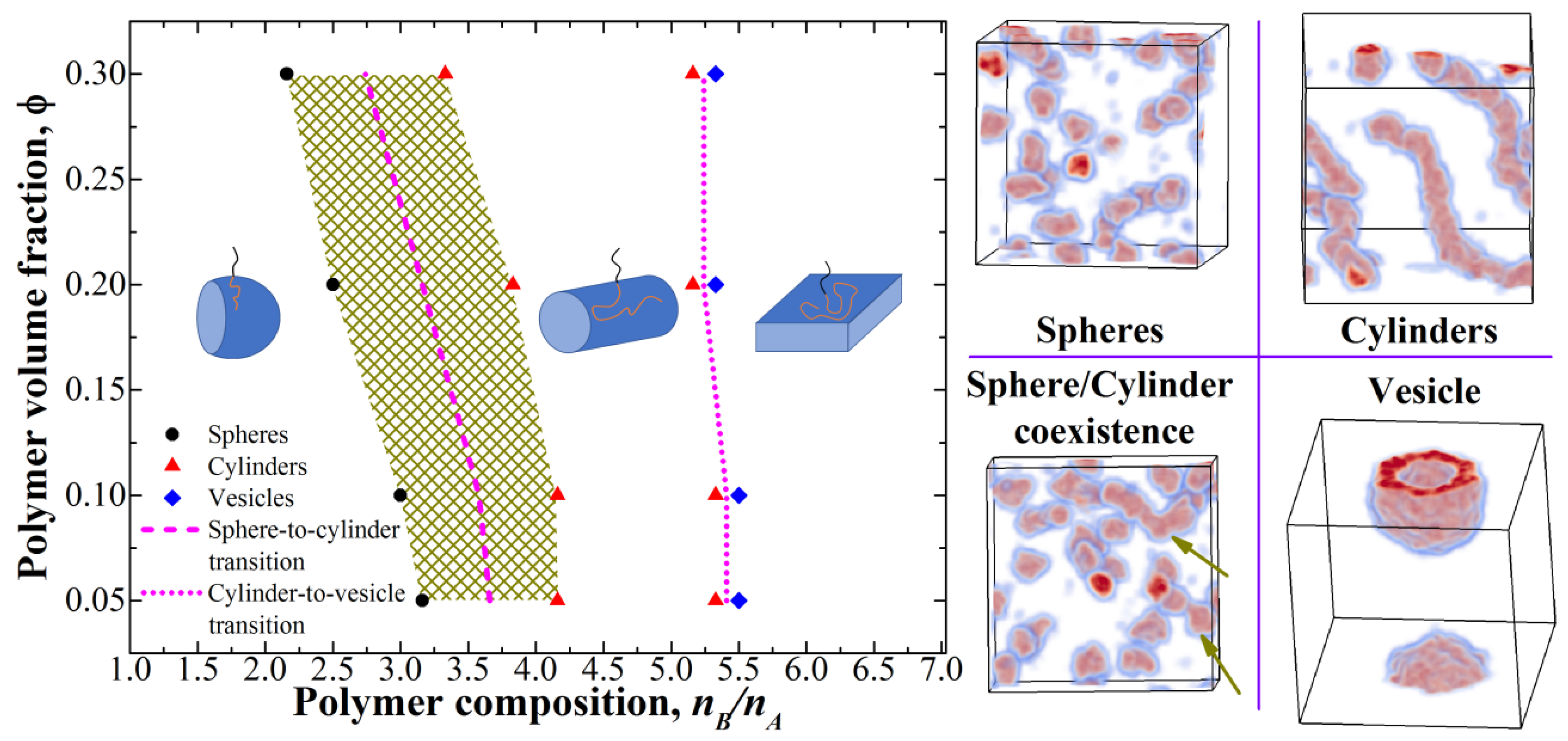
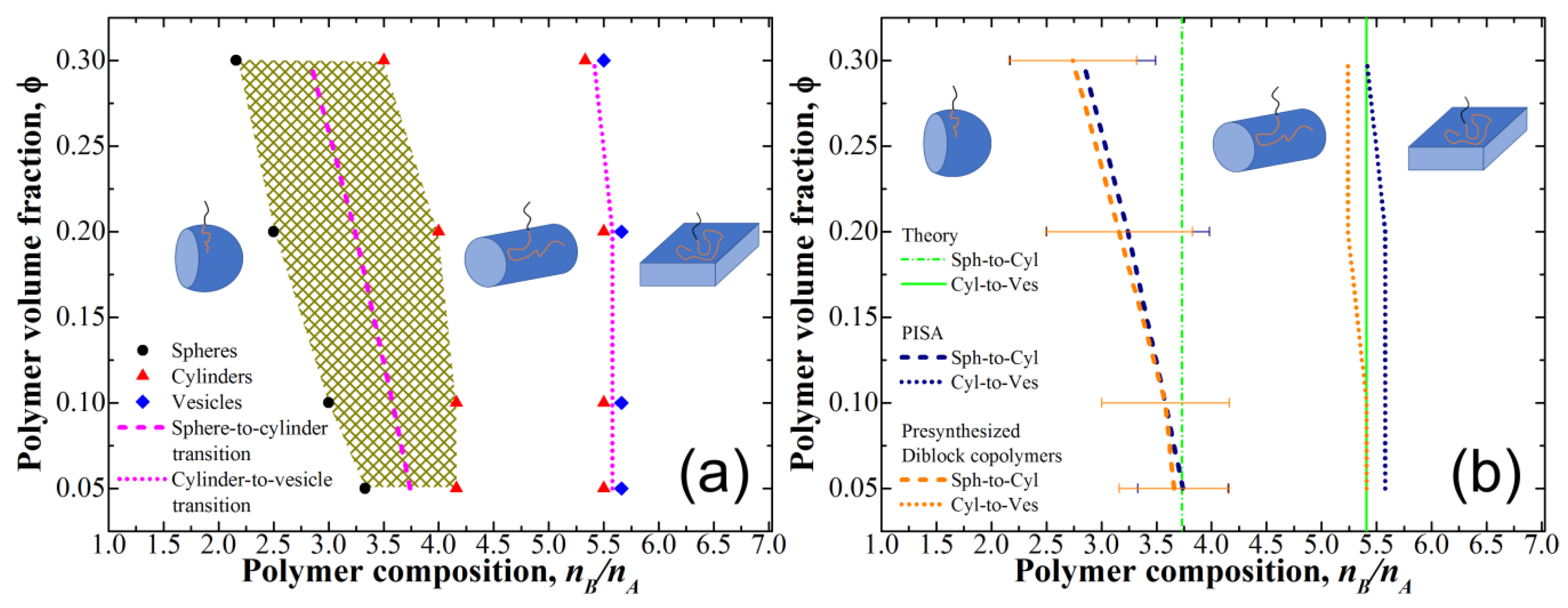
3.1. Micelle Formation in Solution of Presynthesized Monodisperse Diblock-Copolymers
3.2. Phase Diagram for PISA and Comparison to the Theoretical Predictions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tritschler, U.; Pearce, S.; Gwyther, J.; Whittell, G.R.; Manners, I. 50th Anniversary Perspective: Functional Nanoparticles from the Solution Self-Assembly of Block Copolymers. Macromolecules 2017, 50, 3439–3463. [Google Scholar] [CrossRef] [Green Version]
- Delaittre, G.; Nicolas, J.; Lefay, C.; Save, M.; Charleux, B. Surfactant-free synthesis of amphiphilic diblock copolymer nanoparticles via nitroxide-mediated emulsion polymerization. Chem. Commun. 2005, 1, 614. [Google Scholar] [CrossRef] [PubMed]
- Delaittre, G.; Dire, C.; Rieger, J.; Putaux, J.-L.; Charleux, B. Formation of polymer vesicles by simultaneous chain growth and self-assembly of amphiphilic block copolymers. Chem. Commun. 2009, 2887. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.-M.; Hong, C.-Y.; Pan, C.-Y. One-pot synthesis of nanomaterials via RAFT polymerization induced self-assembly and morphology transition. Chem. Commun. 2009, 5883–5885. [Google Scholar] [CrossRef] [PubMed]
- Penfold, N.J.W.; Yeow, J.; Boyer, C.; Armes, S.P. Emerging Trends in Polymerization-Induced Self-Assembly. ACS Macro Lett. 2019, 8, 1029–1054. [Google Scholar] [CrossRef] [Green Version]
- Groison, E.; Brusseau, S.; D’Agosto, F.; Magnet, S.; Inoubli, R.; Couvreur, L.; Charleux, B. Well-Defined Amphiphilic Block Copolymer Nanoobjects via Nitroxide-Mediated Emulsion Polymerization. ACS Macro Lett. 2012, 1, 47–51. [Google Scholar] [CrossRef]
- Blanazs, A.; Ryan, A.J.; Armes, S.P. Predictive phase diagrams for RAFT aqueous dispersion polymerization: Effect of block copolymer composition, molecular weight, and copolymer concentration. Macromolecules 2012, 45, 5099–5107. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, N.; Han, J.; Yu, Q.; Guo, L.; Gao, P.; Lu, X.; Cai, Y. The direct synthesis of interface-decorated reactive block copolymer nanoparticles via polymerisation-induced self-assembly. Polym. Chem. 2015, 6, 4955–4965. [Google Scholar] [CrossRef]
- Yeow, J.; Sugita, O.R.; Boyer, C. Visible Light-Mediated Polymerization-Induced Self-Assembly in the Absence of External Catalyst or Initiator. ACS Macro Lett. 2016, 5, 558–564. [Google Scholar] [CrossRef]
- Mellot, G.; Beaunier, P.; Guigner, J.M.; Bouteiller, L.; Rieger, J.; Stoffelbach, F. Beyond Simple AB Diblock Copolymers: Application of Bifunctional and Trifunctional RAFT Agents to PISA in Water. Macromol. Rapid Commun. 2019, 40, 1800315. [Google Scholar] [CrossRef]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [CrossRef]
- Wan, W.-M.; Pan, C.-Y. Atom Transfer Radical Dispersion Polymerization in an Ethanol/Water Mixture. Macromolecules 2007, 40, 8897–8905. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Zhang, W. Synthesis of diblock copolymer nano-assemblies by PISA under dispersion polymerization: Comparison between ATRP and RAFT. Polym. Chem. 2017, 8, 6407–6415. [Google Scholar] [CrossRef]
- Wang, G.; Schmitt, M.; Wang, Z.; Lee, B.; Pan, X.; Fu, L.; Yan, J.; Li, S.; Xie, G.; Bockstaller, M.R.; et al. Polymerization-Induced Self-Assembly (PISA) Using ICAR ATRP at Low Catalyst Concentration. Macromolecules 2016, 49, 8605–8615. [Google Scholar] [CrossRef]
- Wang, J.; Cao, M.; Zhou, P.; Wang, G. Exploration of a Living Anionic Polymerization Mechanism into Polymerization-Induced Self-Assembly and Site-Specific Stabilization of the Formed Nano-Objects. Macromolecules 2020, 53, 3157–3165. [Google Scholar] [CrossRef]
- Le, D.; Keller, D.; Delaittre, G. Reactive and Functional Nanoobjects by Polymerization-Induced Self-Assembly. Macromol. Rapid Commun. 2019, 40, 1800551. [Google Scholar] [CrossRef]
- Liu, X.; Sun, M.; Sun, J.; Hu, J.; Wang, Z.; Guo, J.; Gao, W. Polymerization Induced Self-Assembly of a Site-Specific Interferon α-Block Copolymer Conjugate into Micelles with Remarkably Enhanced Pharmacology. J. Am. Chem. Soc. 2018, 140, 10435–10438. [Google Scholar] [CrossRef]
- Kaga, S.; Truong, N.P.; Esser, L.; Senyschyn, D.; Sanyal, A.; Sanyal, R.; Quinn, J.F.; Davis, T.P.; Kaminskas, L.M.; Whittaker, M.R. Influence of Size and Shape on the Biodistribution of Nanoparticles Prepared by Polymerization-Induced Self-Assembly. Biomacromolecules 2017, 18, 3963–3970. [Google Scholar] [CrossRef]
- Cheng, G.; Pérez-Mercader, J. Polymerization-Induced Self-Assembly for Artificial Biology: Opportunities and Challenges. Macromol. Rapid Commun. 2019, 40, 1800513. [Google Scholar] [CrossRef]
- D’Agosto, F.; Rieger, J.; Lansalot, M. RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem. Int. Ed. 2020, 59, 8368–8392. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Lv, Y.; Wang, L.; Xu, P.; Lin, J.; Lin, S. An insight into polymerization-induced self-assembly by dissipative particle dynamics simulation. Soft Matter 2016, 12, 6422–6429. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Yao, Q.; Sun, X.; Yan, Y.; Zhang, J. One-pot production of porous assemblies by PISA of star architecture copolymers: A simulation study. Phys. Chem. Chem. Phys. 2018, 20, 10069–10076. [Google Scholar] [CrossRef]
- Lv, Y.; Wang, L.; Liu, F.; Feng, W.; Wei, J.; Lin, S. Rod-coil block copolymer aggregates: Via polymerization-induced self-assembly. Soft Matter 2020, 16, 3466–3475. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.D.; Xue, Y.H.; Zhao, H.Y.; Liu, H.; Lu, Z.Y.; Gu, F.L. Insight into the Polymerization-Induced Self-Assembly via a Realistic Computer Simulation Strategy. Macromolecules 2019, 52, 6169–6180. [Google Scholar] [CrossRef]
- Borisov, O.V.; Zhulina, E.B. Morphology of micelles formed by diblock copolymer with a polyelectrolyte block. Macromolecules 2003, 36, 10029–10036. [Google Scholar] [CrossRef]
- Zhulina, E.B.; Adam, M.; LaRue, I.; Sheiko, S.S.; Rubinstein, M. Diblock Copolymer Micelles in a Dilute Solution. Macromolecules 2005, 38, 5330–5351. [Google Scholar] [CrossRef]
- Shusharina, N.P.; Zhulina, E.B.; Dobrynin, A.V.; Rubinstein, M. Scaling Theory of Diblock Polyampholyte Solutions. Macromolecules 2005, 38, 8870–8881. [Google Scholar] [CrossRef]
- Zhang, L.; Eisenberg, A. Multiple morphologies of “crew-cut” aggregates of polystyrene-b-poly(acrylic acid) block copolymers. Science 1995, 268, 1728–1731. [Google Scholar] [CrossRef] [Green Version]
- Zhulina, E.B.; Borisov, O.V. Theory of Block Polymer Micelles: Recent Advances and Current Challenges. Macromolecules 2012, 45, 4429–4440. [Google Scholar] [CrossRef]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics. Europhys. Lett. 1992, 19, 155–160. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Guseva, D.V.; Kudryavtsev, Y.V.; Khalatur, P.G.; Chertovich, A.V. Simulation of phase separation in melts of reacting multiblock copolymers. Polym. Sci. Ser. A 2011, 53, 1207–1216. [Google Scholar] [CrossRef]
- Glagolev, M.K.; Lazutin, A.A.; Vasilevskaya, V.V.; Khokhlov, A.R. Influence of cross-linking rate on the structure of hypercrosslinked networks: Multiscale computer simulation. Polymer 2016, 86, 168–175. [Google Scholar] [CrossRef]
- Yong, X.; Kuksenok, O.; Balazs, A.C. Modeling free radical polymerization using dissipative particle dynamics. Polymer 2015, 72, 217–225. [Google Scholar] [CrossRef]
- Genzer, J. In Silico Polymerization: Computer Simulation of Controlled Radical Polymerization in Bulk and on Flat Surfaces. Macromolecules 2006, 39, 7157–7169. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Chertovich, A.V. Copolymerization of Partly Incompatible Monomers: An Insight from Computer Simulations. Macromolecules 2017, 50, 4677–4685. [Google Scholar] [CrossRef] [Green Version]
- Kozhunova, E.Y.; Gavrilov, A.A.; Zaremski, M.Y.; Chertovich, A.V. Copolymerization on Selective Substrates: Experimental Test and Computer Simulations. Langmuir 2017, 33, 3548–3555. [Google Scholar] [CrossRef] [PubMed]
- Shupanov, R.; Chertovich, A.; Kos, P. Micellar polymerization: Computer simulations by dissipative particle dynamics. J. Comput. Chem. 2018, 39, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qian, H.-J.; Lu, Z.-Y. The influence of one block polydispersity on phase separation of diblock copolymers: The molecular mechanism for domain spacing expansion. Polymer 2013, 54, 3716–3722. [Google Scholar] [CrossRef]
- Markina, A.; Chertovich, A. Stable domain size and conformational segregation of short and long blocks during microphase separation in random block copolymers. Chem. Phys. Lett. 2015, 624, 74–77. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Kudryavtsev, Y.V.; Chertovich, A.V. Phase diagrams of block copolymer melts by dissipative particle dynamics simulations. J. Chem. Phys. 2013, 139, 224901. [Google Scholar] [CrossRef] [Green Version]
- Cooke, D.M.; Shi, A.-C. Effects of Polydispersity on Phase Behavior of Diblock Copolymers. Macromolecules 2006, 39, 6661–6671. [Google Scholar] [CrossRef]
- Lynd, N.A.; Hillmyer, M.A. Influence of Polydispersity on the Self-Assembly of Diblock Copolymers. Macromolecules 2005, 38, 8803–8810. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavrilov, A.A.; Shupanov, R.M.; Chertovich, A.V. Phase Diagram for Ideal Diblock-Copolymer Micelles Compared to Polymerization-Induced Self Assembly. Polymers 2020, 12, 2599. https://doi.org/10.3390/polym12112599
Gavrilov AA, Shupanov RM, Chertovich AV. Phase Diagram for Ideal Diblock-Copolymer Micelles Compared to Polymerization-Induced Self Assembly. Polymers. 2020; 12(11):2599. https://doi.org/10.3390/polym12112599
Chicago/Turabian StyleGavrilov, Alexey A., Ruslan M. Shupanov, and Alexander V. Chertovich. 2020. "Phase Diagram for Ideal Diblock-Copolymer Micelles Compared to Polymerization-Induced Self Assembly" Polymers 12, no. 11: 2599. https://doi.org/10.3390/polym12112599
APA StyleGavrilov, A. A., Shupanov, R. M., & Chertovich, A. V. (2020). Phase Diagram for Ideal Diblock-Copolymer Micelles Compared to Polymerization-Induced Self Assembly. Polymers, 12(11), 2599. https://doi.org/10.3390/polym12112599