Drying of the Natural Fibers as A Solvent-Free Way to Improve the Cellulose-Filled Polymer Composite Performance



Abstract
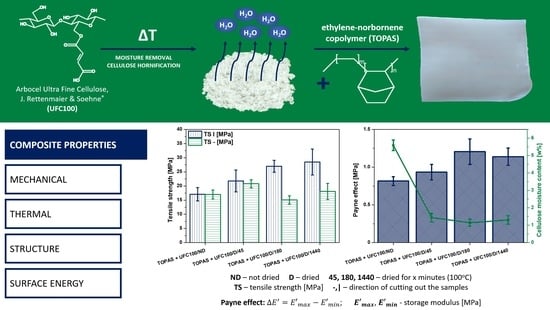
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cellulose Fibers Samples of Varied Moisture Contents Preparation
2.3. Polymer Composite Samples Preparation
2.4. Cellulose Fibers Properties Characterization
2.4.1. Fourier Transform Infrared Spectroscopy (FT-IR)
2.4.2. Thermogravimetric Analysis (TGA)
- —specimen mass at the time t [g];
- —specimen mass at the beginning of considered decomposition step [g];
- —specimen mass at the end of considered decomposition step [g];
- —temperature [K];
- —gas constant [8.31 J/(mol·K)].
2.4.3. Fischer Titration
2.4.4. Detection of Mass Change during Moisture Adsorption/Desorption
- —specimen mass at the time t [g];
- —specimen mass at the beginning of considered decomposition step [g];
- —sample mass after 1440 min of experiment [g].
2.5. Polymer Composite Samples Characterization
2.5.1. Static Mechanical Analysis
- —tensile strength of the samples cut out, respectively, vertically and horizontally [MPa];
- —elongation at break of the samples cut out, respectively, vertically and horizontally [%].
2.5.2. Dynamic Mechanical Analysis (DMA)
- —storage modulus maximum and minimum value [MPa].
- —storage modulus of filled polymer composite [MPa]
- —storage modulus of neat polymer matrix [MPa]
- —filler volume fraction [-]
3. Results and Discussion
3.1. Cellulose Fibers of Various Moisture Contents Influence on Polymer Composite Properties
3.1.1. Cellulose Fibers Characterization
FT-IR Investigation
Fischer Titration and Mass Changes Analysis
TGA Investigation
3.1.2. Polymer Composite Properties
Static Mechanical Characterization
DMA Investigation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kemp, R.; Loorbach, D.; Rotmans, J. Transition management as a model for managing processes of co-evolution towards sustainable development. Int. J. Sust. Dev. World 2007, 14, 37–41. [Google Scholar] [CrossRef]
- Datta, J.; Kopczyńska, P. From polymer waste to potential main industrial products: Actual state of recycling and recovering. Crit. Rev. Environ. Sci. Technol. 2016, 46, 905–946. [Google Scholar] [CrossRef]
- Do Sul, J.A.I.; Costa, M.F. The present and future of microplastic pollution in the marine environment. Environ. Pollut. 2014, 185, 352–364. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, P.; Wang, M.; Zhao, H.; Yang, J.; Xu, F. Sustainable, reusable, and superhydrophobic aerogels from microfibrillated cellulose for highly effective oil/water separation. ACS Sustain. Chem. Eng. 2016, 4, 6409–6416. [Google Scholar] [CrossRef]
- Liu, W.; Fei, M.; Ban, Y.; Jia, A.; Qiu, R. Preparation and evaluation of green composites from microcrystalline cellulose and a soybean-oil derivative. Polymers 2017, 9, 541. [Google Scholar] [CrossRef]
- Huang, F.Y. Thermal properties and thermal degradation of cellulose tri-stearate (CTs). Polymers 2012, 4, 1012–1024. [Google Scholar] [CrossRef]
- Manaf, M.E.A.; Tsuji, M.; Nobukawa, S.; Yamaguchi, M. Effect of moisture on the orientation birefringence of cellulose esters. Polymers 2011, 3, 955–966. [Google Scholar] [CrossRef]
- Li, G.C.; Anastas, P. Green chemistry themed issue green chemistry: Present and future. Chem. Soc. Rev. 2012, 41, 1413–1414. [Google Scholar] [CrossRef]
- Thakur, M.K.; Gupta, R.K.; Thakur, V.K. Surface modification of cellulose using silane coupling agent. Carbohydr. Polym. 2014, 111, 849–855. [Google Scholar] [CrossRef]
- Yadav, M.; Liu, Y.-K.; Chiu, F.-C. Fabrication of cellulose nanocrystal/silver/alginate bionanocomposite films with enhanced mechanical and barrier properties for food packaging application. Nanomaterials 2019, 9, 1523. [Google Scholar] [CrossRef]
- Yadav, M.; Chiu, F.C. Cellulose nanocrystals reinforced κ-carrageenan based UV resistant transparent bionanocomposite films for sustainable packaging applications. Carbohydr. Polym. 2019, 211, 181–194. [Google Scholar] [CrossRef]
- Simon, M.O.; Li, C.J. Green chemistry oriented organic synthesis in water. Chem. Soc. Rev. 2012, 41, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.T.; Hengge, N.N.; Sims, R.C.; Miller, C.D. Acetone, butanol, and ethanol production from wastewater algae. Bioresour. Technol. 2012, 111, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Clausen, L.R.; Houbak, N.; Elmegaard, B. Technoeconomic analysis of a methanol plant based on gasification of biomass and electrolysis of water. Energy 2010, 35, 2338–2347. [Google Scholar] [CrossRef]
- Zabed, H.; Sahu, J.N.; Boyce, A.N.; Faruq, G. Fuel ethanol production from lignocellulosic biomass: An overview on feedstocks and technological approaches. Renew. Sustain. Energy Rev. 2016, 66, 751–774. [Google Scholar] [CrossRef]
- Green, E.M. Fermentative production of butanol-the industrial perspective. Curr. Opin. Biotechnol. 2011, 22, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.; François, J.M. Microbial production of propanol. Biotechnol. Adv. 2016, 34, 984–996. [Google Scholar] [CrossRef]
- Löbs, A.K.; Lin, J.L.; Cook, M.; Wheeldon, I. High throughput, colorimetric screening of microbial ester biosynthesis reveals high ethyl acetate production from Kluyveromyces marxianus on C5, C6, and C12 carbon sources. Biotechnol. J. 2016, 11, 1274–1281. [Google Scholar] [CrossRef]
- Khoo, H.H.; Wong, L.L.; Tan, J.; Isoni, V.; Sharratt, P. Synthesis of 2-methyl tetrahydrofuran from various lignocellulosic feedstocks: Sustainability assessment via LCA. Resour. Conserv. Recycl. 2015, 95, 174–182. [Google Scholar] [CrossRef]
- Dunn, P.J. The importance of green chemistry in process research and development. Chem. Soc. Rev. 2012, 41, 1452–1461. [Google Scholar] [CrossRef]
- Prat, D.; Hayler, J.; Wells, A. A survey of solvent selection guides. Green Chem. 2014, 16, 4546–4551. [Google Scholar] [CrossRef]
- Ghernaout, D.; Ghernaout, B.; Naceur, M.W. Embodying the chemical water treatment in the green chemistry—A review. Desalination 2011, 271, 1–10. [Google Scholar] [CrossRef]
- Funari, C.S.; Carneiro, R.L.; Khandagale, M.M.; Cavalheiro, A.J.; Hilder, E.F. Acetone as a greener alternative to acetonitrile in liquid chromatographic fingerprinting. J. Sep. Sci. 2015, 38, 1458–1465. [Google Scholar] [CrossRef]
- Huang, X.; Korányi, T.I.; Boot, M.D.; Hensen, E.J.M. Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics. Green Chem. 2015, 17, 4941–4950. [Google Scholar] [CrossRef]
- Tamura, M.; Noro, K.; Honda, M.; Nakagawa, Y.; Tomishige, K. Highly efficient synthesis of cyclic ureas from CO2 and diamines by a pure CeO2 catalyst using a 2-propanol solvent. Green Chem. 2013, 15, 1567–1577. [Google Scholar] [CrossRef]
- Jessop, P.G.; Jessop, D.A.; Fu, D.; Phan, L. Solvatochromic parameters for solvents of interest in green chemistry. Green Chem. 2012, 14, 1245–1259. [Google Scholar] [CrossRef]
- Barta, K.; Matson, T.D.; Fettig, M.L.; Scott, S.L.; Iretskii, A.V.; Ford, P.C. Catalytic disassembly of an organosolv lignin via hydrogen transfer from supercritical methanol. Green Chem. 2010, 12, 1640–1647. [Google Scholar] [CrossRef]
- Uyttebroek, M.; Van Hecke, W.; Vanbroekhoven, K. Sustainability metrics of 1-butanol. Catal. Today 2015, 239, 7–10. [Google Scholar] [CrossRef]
- Yeung, D.K.J.; Gao, T.; Huang, J.; Sun, S.; Guo, H.; Wang, J. Organocatalytic 1,3-dipolar cycloaddition reactions of ketones and azides with water as a solvent. Green Chem. 2013, 15, 2384–2388. [Google Scholar] [CrossRef]
- Yang, D.; Fu, H. A simple and practical copper-catalyzed approach to substituted phenols from aryl halides by using water as the solvent. Chem. A Eur. J. 2010, 16, 2366–2370. [Google Scholar] [CrossRef]
- Adschiri, T.; Lee, Y.W.; Goto, M.; Takami, S. Green materials synthesis with supercritical water. Green Chem. 2011, 13, 1380–1390. [Google Scholar] [CrossRef]
- Ibarra, I.A.; Bayliss, P.A.; Pérez, E.; Yang, S.; Blake, A.J.; Nowell, H.; Allan, D.R.; Poliakoff, M.; Schröder, M. Near-critical water, a cleaner solvent for the synthesis of a metal-organic framework. Green Chem. 2012, 14, 117–122. [Google Scholar] [CrossRef]
- Butler, R.N.; Coyne, A.G. Water: Nature’s reaction enforcer-comparative effects for organic synthesis “in-water” and “on-water”. Chem. Rev. 2010, 110, 6302–6337. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.; Doan, H.A.; Pursell, C.J.; Grabow, L.C.; Chandler, B.D. The critical role of water at the gold-titania interface in catalytic CO oxidation. Science 2014, 345, 1599–1602. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, F.; Nau, W.M.; Schneider, H.J. The hydrophobic effect revisited—Studies with supramolecular complexes imply high-energy water as a noncovalent driving force. Angew. Chem. Int. Ed. 2014, 53, 11158–11171. [Google Scholar] [CrossRef] [PubMed]
- Butler, R.N.; Coyne, A.G. Organic synthesis reactions on-water at the organic-liquid water interface. Org. Biomol. Chem. 2016, 14, 9945–9960. [Google Scholar] [CrossRef]
- Cao, J.J.; Zhou, F.; Zhou, J. Improving the atom efficiency of the wittig reaction by a “waste as catalyst/Co-catalyst” strategy. Angew. Chem. Int. Ed. 2010, 49, 4976–4980. [Google Scholar] [CrossRef]
- Sheldon, R.A. Fundamentals of green chemistry: Efficiency in reaction design. Chem. Soc. Rev. 2012, 41, 1437–1451. [Google Scholar] [CrossRef]
- Dicks, A.P. Don’t forget the workup. J. Chem. Educ. 2015, 92, 405. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Ashori, A.; Babaee, M.; Jonoobi, M.; Hamzeh, Y. Solvent-free acetylation of cellulose nanofibers for improving compatibility and dispersion. Carbohydr. Polym. 2014, 102, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Chen, S.; Xu, Q.; Wang, H. Solvent-free acetylation of bacterial cellulose under moderate conditions. Carbohydr. Polym. 2011, 83, 1575–1581. [Google Scholar] [CrossRef]
- Tian, D.; Zhang, X.; Lu, C.; Yuan, G.; Zhang, W.; Zhou, Z. Solvent-free synthesis of carboxylate-functionalized cellulose from waste cotton fabrics for the removal of cationic dyes from aqueous solutions. Cellulose 2014, 21, 473–484. [Google Scholar] [CrossRef]
- Soliman, E.M.; Ahmed, S.A.; Fadl, A.A. Removal of calcium ions from aqueous solutions by sugar cane bagasse modified with carboxylic acids using microwave-assisted solvent-free synthesis. Desalination 2011, 278, 18–25. [Google Scholar] [CrossRef]
- Baidya, A.; Ganayee, M.A.; Ravindran, S.J.; Tam, K.C.; Das, S.K.; Ras, R.H.A.; Pradeep, T. Organic solvent-free fabrication of durable and multifunctional superhydrophobic paper from waterborne fluorinated cellulose nanofiber building blocks. ACS Nano 2017, 11, 11091–11099. [Google Scholar] [CrossRef]
- El Boustani, M.; Brouillette, F.; Lebrun, G.; Belfkira, A. Solvent-free acetylation of lignocellulosic fibers at room temperature: Effect on fiber structure and surface properties. J. Appl. Polym. Sci. 2015, 132, 1–8. [Google Scholar] [CrossRef]
- Monteil-Rivera, F.; Paquet, L. Solvent-free catalyst-free microwave-assisted acylation of lignin. Ind. Crops Prod. 2015, 65, 446–453. [Google Scholar] [CrossRef]
- Ballesteros, J.E.M.; dos Santos, V.; Mármol, G.; Frías, M.; Fiorelli, J. Potential of the hornification treatment on eucalyptus and pine fibers for fiber-cement applications. Cellulose 2017, 24, 2275–2286. [Google Scholar] [CrossRef]
- Minor, J.L. Hornification—Its origin and meaning. Progress Pap. Recycl. 1994, 3, 93–95. [Google Scholar]
- Diniz, J.M.B.F.; Gil, M.H.; Castro, J.A.A.M. Hornification—Its origin and interpretation in wood pulps. Wood Sci. Technol. 2004, 37, 489–494. [Google Scholar] [CrossRef]
- Zimmermann, R.; Nitschke, C.; Potthast, A.; Werner, C.; Freudenberg, U.; Scherr, G.; Müller, Y.; Eichhorn, K.-J.; Tot, I.; Rosenau, T. The impact of esterification reactions on physical properties of cellulose thin films. Soft Matter 2010, 6, 3680–3684. [Google Scholar]
- Mao, H.; Wei, C.; Gong, Y.; Wang, S.; Ding, W. Mechanical and water-resistant properties of eco-friendly chitosan membrane reinforced with cellulose nanocrystals. Polymers 2019, 11, 166. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Venditti, R.A.; Jameel, H.; Pawlak, J.J. Changes in pore size distribution during the drying of cellulose fibers as measured by differential scanning calorimetry. Carbohydr. Polym. 2006, 66, 97–103. [Google Scholar] [CrossRef]
- Kuciel, S.; Jakubowska, P.; Kuźniar, P. A study on the mechanical properties and the influence of water uptake and temperature on biocomposites based on polyethylene from renewable sources. Compos. Part B Eng. 2014, 64, 72–77. [Google Scholar] [CrossRef]
- Haddad, H.; Al Kobaisi, M. Influence of moisture content on the thermal and mechanical properties and curing behavior of polymeric matrix and polymer concrete composite. Mater. Des. 2013, 49, 850–856. [Google Scholar] [CrossRef]
- Al-Oqla, F.M.; Salit, M.S.; Ishak, M.R.; Aziz, N.A. A novel evaluation tool for enhancing the selection of natural fibers for polymeric composites based on fiber moisture content criterion. BioResources 2015, 10, 299–312. [Google Scholar] [CrossRef]
- Broido, A. A simple, sensitive graphical method of treating thermogravimetric analysis data. J. Polym. Sci. Part A 2 Polym. Phys. 1969, 7, 1761–1773. [Google Scholar] [CrossRef]
- De Caro, C.A.; Aichert, A.; Walter, C.M. Efficient, precise and fast water determination by the Karl Fischer titration. Food Control 2001, 12, 431–436. [Google Scholar] [CrossRef]
- Merabia, S.; Sotta, P.; Long, D. A microscopic model for the reinforcement and the nonlinear behavior of filled elastomers and thermoplastic elastomers (Payne and Mullins effects). Macromolecules 2008, 41, 8252–8266. [Google Scholar] [CrossRef]
- Jyoti, J.; Singh, B.P.; Arya, A.K.; Dhakate, S.R. Dynamic mechanical properties of multiwall carbon nanotube reinforced ABS composites and their correlation with entanglement density, adhesion, reinforcement and C factor. RSC Adv. 2016, 6, 3997–4006. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D., II; Shin, Y.; Seo, G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Łojewska, J.; Miśkowiec, P.; Łojewski, T.; Proniewicz, L.M. Cellulose oxidative and hydrolytic degradation: In situ FTIR approach. Polym. Degrad. Stab. 2005, 88, 512–520. [Google Scholar] [CrossRef]
- Gulmine, J.; Janissek, P.; Heise, H.; Akcelrud, L. Polyethylene characterization by FTIR. Polym. Test. 2002, 21, 557–563. [Google Scholar] [CrossRef]
- Morán, J.I.; Alvarez, V.A.; Cyras, V.P.; Vázquez, A. Extraction of cellulose and preparation of nanocellulose from sisal fibers. Cellulose 2008, 15, 149–159. [Google Scholar] [CrossRef]
- Patel, H.A.; Somani, R.S.; Bajaj, H.C.; Jasra, R.V. Preparation and characterization of phosphonium montmorillonite with enhanced thermal stability. Appl. Clay Sci. 2007, 35, 194–200. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Salmén, L.; Bergström, E. Cellulose structural arrangement in relation to spectral changes in tensile loading FTIR. Cellulose 2009, 16, 975–982. [Google Scholar] [CrossRef]
- Ołdak, D.; Kaczmarek, H.; Buffeteau, T.; Sourisseau, C. Photo- and bio-degradation processes in polyethylene, cellulose and their blends studied by ATR-FTIR and raman spectroscopies. J. Mater. Sci. 2005, 40, 4189–4198. [Google Scholar] [CrossRef]
- Colom, X.; Carrillo, F.; Nogués, F.; Garriga, P. Structural analysis of photodegraded wood by means of FTIR spectroscopy. Polym. Degrad. Stab. 2003, 80, 543–549. [Google Scholar] [CrossRef]
- Colom, X.; Carrasco, F.; Pagès, P.; Canavate, J. Effects of different treatments on the interface of HDPE/lignocellulosic fiber composites. Compos. Sci. Technol. 2003, 63, 161–169. [Google Scholar] [CrossRef]
- Ibrahim, M.; Osman, O.; Mahmoud, A.A. Spectroscopic analyses of cellulose and chitosan: FTIR and modeling approach. J. Comput. Theor. Nanosci. 2011, 8, 117–123. [Google Scholar] [CrossRef]
- Célino, A.; Gonçalves, O.; Jacquemin, F.; Fréour, S. Qualitative and quantitative assessment of water sorption in natural fibers using ATR-FTIR spectroscopy. Carbohydr. Polym. 2014, 101, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Kachrimanis, K.; Noisternig, M.F.; Griesser, U.J.; Malamataris, S. Dynamic moisture sorption and desorption of standard and silicified microcrystalline cellulose. Eur. J. Pharm. Biopharm. 2006, 64, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Okubayashi, S.; Griesser, U.J.; Bechtold, T. A kinetic study of moisture sorption and desorption on lyocell fibers. Carbohydr. Polym. 2004, 58, 293–299. [Google Scholar] [CrossRef]
- Sun, C.C. Mechanism of moisture induced variations in true density and compaction properties of microcrystalline cellulose. Int. J. Pharm. 2008, 346, 93–101. [Google Scholar] [CrossRef]
- Hofstetter, K.; Hinterstoisser, B.; Salmén, L. Moisture uptake in native cellulose—The roles of different hydrogen bonds: A dynamic FT-IR study using Deuterium exchange. Cellulose 2006, 13, 131–145. [Google Scholar] [CrossRef]
- Ma, S.; Yu, S.J.; Wang, Z.H.; Zheng, X.L. Ultrasound-assisted modification of beet pulp cellulose with phthalic anhydride in ionic liquid. Cell. Chem. Technol. 2013, 47, 527–533. [Google Scholar]
- Wyasu, G.; Gimba, C.E.; Agbaji, E.B.; Ndukwe, G.I. Thermo-gravimetry (TGA) and DSC of thermal analysis techniques in production of active carbon from lignocellulosic materials. Pelagia Res. Libr. Adv. Appl. Sci. Res. 2016, 7, 109–115. [Google Scholar]
- Li, Y.; Xiao, H.; Chen, M.; Song, Z.; Zhao, Y. Absorbents based on maleic anhydride-modified cellulose fibers/diatomite for dye removal. J. Mater. Sci. 2014, 49, 6696–6704. [Google Scholar] [CrossRef]
- Ouajai, S.; Shanks, R.A. Composition, structure and thermal degradation of hemp cellulose after chemical treatments. Polym. Degrad. Stab. 2005, 89, 327–335. [Google Scholar] [CrossRef]
- Luo, X.L.; Zhu, J.Y.; Gleisner, R.; Zhan, H.Y. Effects of wet-pressing-induced fiber hornification on enzymatic saccharification of lignocelluloses. Cellulose 2011, 18, 1055–1062. [Google Scholar] [CrossRef]
- Ruffini, G. Improvement in bonding of wood pulps by the presence of acidic groups. Sven. Papprstidn 1966, 69, 72–76. [Google Scholar]
- Cabalova, I.; Kacik, F.; Geffert, A.; Kacikov, D. The effects of paper recycling and its environmental impact. Environ. Manag. Pract. 2011, 17, 329–350. [Google Scholar]
- Huber, T.; Müssig, J.; Curnow, O.; Pang, S.; Bickerton, S.; Staiger, M.P. A critical review of all-cellulose composites. J. Mater. Sci. 2012, 47, 1171–1186. [Google Scholar] [CrossRef]
- Alamri, H.; Low, I.M. Mechanical properties and water adsorption behavior of recycled cellulose fiber reinforced epoxy composites. Polym. Test. 2012, 31, 620–628. [Google Scholar] [CrossRef]
- Nishino, T.; Hirao, K.; Kotera, M.; Nakamae, K.; Inagaki, H. Kenaf reinforced biodegradable composite. Compos. Sci. Technol. 2003, 63, 1281–1286. [Google Scholar] [CrossRef]
- Bendahou, A.; Kaddami, H.; Dufresne, A. Investigation on the effect of cellulosic nanoparticles’ morphology on the properties of natural rubber based nanocomposites. Eur. Polym. J. 2010, 46, 609–620. [Google Scholar] [CrossRef]
- Cho, M.J.; Park, B.D. Tensile and thermal properties of nanocellulose-reinforced poly(vinyl alcohol) nanocomposites. J. Ind. Eng. Chem. 2011, 17, 36–40. [Google Scholar] [CrossRef]
- Gao, H.; Xie, Y.; Ou, R.; Wang, Q. Grafting effects of polypropylene/polyethylene blends with maleic anhydride on the properties of the resulting wood-plastic composites. Compos. Part A Appl. Sci. Manuf. 2012, 43, 150–157. [Google Scholar] [CrossRef]
- Georgopoulos, S.T.; Tarantili, P.A.; Avgerinos, E.; Andreopoulos, A.G.; Koukios, E.G. Thermoplastic polymers reinforced with fibrous agricultural residues. Polym. Degrad. Stab. 2005, 90, 303–312. [Google Scholar] [CrossRef]
- Samerzadeh, S.H. Fiber matrix interactions in jute reinforced polyester resin. Polym. Compos. 1986, 7, 23–25. [Google Scholar] [CrossRef]
- Lee, B.H.; Kim, H.J.; Yu, W.R. Fabrication of long and discontinuous natural fiber reinforced polypropylene biocomposites and their mechanical properties. Fibers Polym. 2009, 10, 83–90. [Google Scholar] [CrossRef]
- Essabir, H.; Elkhaoulani, A.; Benmoussa, K.; Bouhfid, R.; Arrakhiz, F.Z.; Qaiss, A. Dynamic mechanical thermal behavior analysis of doum fibers reinforced polypropylene composites. Mater. Des. 2013, 51, 780–788. [Google Scholar] [CrossRef]
- Essabir, H.; Nekhlaoui, S.; Malha, M.; Bensalah, M.O.; Arrakhiz, F.Z.; Qaiss, A.; Bouhfid, R. Bio-composites based on polypropylene reinforced with Almond Shells particles: Mechanical and thermal properties. Mater. Des. 2013, 51, 225–230. [Google Scholar] [CrossRef]
- Saba, N.; Safwan, A.; Sanyang, M.L.; Mohammad, F.; Pervaiz, M.; Jawaid, M.; Alothman, O.Y.; Sain, M. Thermal and dynamic mechanical properties of cellulose nanofibers reinforced epoxy composites. Int. J. Biol. Macromol. 2017, 102, 822–828. [Google Scholar] [CrossRef]
- Barczewski, M.; Mysiukiewicz, O.; Szulc, J.; Kloziński, A. Poly(lactic acid) green composites filled with linseed cake as an agricultural waste filler. Influence of oil content within the filler on the rheological behavior. J. Appl. Polym. Sci. 2019, 136, 1–12. [Google Scholar] [CrossRef]
- Lion, A.; Kardelky, C.; Haupt, P. On the frequency and amplitude dependence of the Payne effect: Theory and experiments. Rubber Chem. Technol. 2011, 76, 533–547. [Google Scholar] [CrossRef]
- Scagliusi, S.R.; Cardoso, E.C.L.; Parra, D.F.; Lima, L.F.C.P.; Lugão, A.B. Evaluation of “Payne effect” in radiation-induced modification of chlorobutyl rubber. Radiat. Phys. Chem. 2013, 84, 42–46. [Google Scholar] [CrossRef]
- Fröhlich, J.; Niedermeier, W.; Luginsland, H.D. The effect of filler-filler and filler-elastomer interaction on rubber reinforcement. Compos. Part A Appl. Sci. Manuf. 2005, 36, 449–460. [Google Scholar] [CrossRef]
- Robertson, C.G.; Wang, X. Isoenergetic jamming transition in particle-filled systems. Phys. Rev. Lett. 2005, 95, 1–4. [Google Scholar] [CrossRef]
- Peng, Y.; Gardner, D.J.; Han, Y. Drying cellulose nanofibrils: In search of a suitable method. Cellulose 2012, 19, 91–102. [Google Scholar] [CrossRef]
- Štefelová, J.; Slovák, V.; Siqueira, G.; Olsson, R.T.; Tingaut, P.; Zimmermann, T.; Sehaqui, H. Drying and pyrolysis of cellulose nanofibers from wood, bacteria, and algae for char application in oil adsorption and dye adsorption. ACS Sustain. Chem. Eng. 2017, 5, 2679–2692. [Google Scholar] [CrossRef]
- Kontturi, E.; Vuorinen, T. Indirect evidence of supramolecular changes within cellulose microfibrils of chemical pulp fibers upon drying. Cellulose 2009, 16, 65–74. [Google Scholar] [CrossRef]
- Nazhad, M. Fundamentals of strength loss in recycled paper. TAPPI J. 1994, 77, 171–179. [Google Scholar]
- Spence, K.L.; Venditti, R.A.; Habibi, Y.; Rojas, O.J.; Pawlak, J.J. The effect of chemical composition on microfibrillar cellulose films from wood pulps: Mechanical processing and physical properties. Bioresour. Technol. 2010, 101, 5961–5968. [Google Scholar] [CrossRef]
- Luo, X.; Zhu, J.Y. Effects of drying-induced fiber hornification on enzymatic saccharification of lignocelluloses. Enzym. Microb. Technol. 2010, 48, 92–99. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Reihmane, S.; Gassan, J. Properties and modification methods for vegetable fibers for natural fiber composites. J. Appl. Polym. Sci. 1996, 59, 1329–1336. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Faruk, O.; Huque, M. Physico-mechanical studies of wood fiber reinforced composites. Polym. Plast. Technol. Eng. 2002, 41, 435–451. [Google Scholar] [CrossRef]
- Gassan, J.; Bledzki, A.K. Effect of moisture content on the properties of silanized jute-epoxy composites. Polym. Compos. 1997, 18, 179–184. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Drying Time [min] |
|---|---|
| UFC100/ND | – |
| UFC100/D/45 | 45 |
| UFC100/D/90 | 90 |
| UFC100/D/180 | 180 |
| UFC100/D/1440 | 1440 |
| Wavenumber [cm−1] | Chemical Group | Ref. |
|---|---|---|
| 557 | C–OH out-of-plane bending, C–C | [66] |
| 1200–900 | –OH, –COO | [65] |
| 1100–1000 | CO–O–CO | [67] |
| 1058 | C–O stretching vibration | [68] |
| 1100 | –OH | [63] |
| 1158 | C–O stretching vibration, C–O–C bridge | [69] |
| 1245 | –CH3 | [70] |
| 1300–1100 | C–O, C=O, C=C, COOH | [63] |
| 1463 | C–H bending of CH2 | [71] |
| 1653 | OH bending of adsorbed water, C=C | [61] |
| 2900–2800 | CH stretching vibration | [64] |
| 3331 | –OH, water | [62] |
| Sample | T05% [°C] | T10% [°C] | T15% % [°C] | T20% [°C] | T50% [°C] | T80% [°C] | T90% [°C] |
|---|---|---|---|---|---|---|---|
| UFC100/ND | 172 | 311 | 322 | 328 | 343 | 356 | 365 |
| UFC100/D/1440 | 299 | 312 | 318 | 323 | 339 | 353 | 361 |
| Sample | ||
|---|---|---|
| UFC100/ND | 56 ± 2 | 116 ± 3 |
| UFC100/D/1440 | 66 ± 2 | 112 ± 3 |
| Sample | O [-] |
|---|---|
| TOPAS and UFC100/ND | 0.12 |
| TOPAS and UFC100/D/45 | 0.07 |
| TOPAS and UFC100/D/180 | 0.64 |
| TOPAS and UFC100/D/1440 | 0.55 |
| Sample | E′max [MPa] | E′min [MPa] | Payne Effect [MPa] |
|---|---|---|---|
| TOPAS and UFC100/ND | 0.8177 | 0.0011 | 0.82 |
| TOPAS and UFC100/D/45 | 0.9353 | 0.0014 | 0.93 |
| TOPAS and UFC100/D/180 | 1.2062 | 0.0010 | 1.21 |
| TOPAS and UFC100/D/1440 | 1.1396 | 0.0010 | 1.14 |
| Sample | E′max [MPa] | E″max [MPa] | r [-] |
|---|---|---|---|
| TOPAS | 1.26 | 0.32 | ---------- |
| TOPAS and UFC100/ND | 0.82 | 0.19 | −2.5 |
| TOPAS and UFC100/D/45 | 0.94 | 0.22 | −1.8 |
| TOPAS and UFC100/D/180 | 1.21 | 0.30 | −0.3 |
| TOPAS and UFC100/D/1440 | 1.14 | 0.29 | −0.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cichosz, S.; Masek, A. Drying of the Natural Fibers as A Solvent-Free Way to Improve the Cellulose-Filled Polymer Composite Performance. Polymers 2020, 12, 484. https://doi.org/10.3390/polym12020484
Cichosz S, Masek A. Drying of the Natural Fibers as A Solvent-Free Way to Improve the Cellulose-Filled Polymer Composite Performance. Polymers. 2020; 12(2):484. https://doi.org/10.3390/polym12020484
Chicago/Turabian StyleCichosz, Stefan, and Anna Masek. 2020. "Drying of the Natural Fibers as A Solvent-Free Way to Improve the Cellulose-Filled Polymer Composite Performance" Polymers 12, no. 2: 484. https://doi.org/10.3390/polym12020484
APA StyleCichosz, S., & Masek, A. (2020). Drying of the Natural Fibers as A Solvent-Free Way to Improve the Cellulose-Filled Polymer Composite Performance. Polymers, 12(2), 484. https://doi.org/10.3390/polym12020484