The Competition of Termination and Shielding to Evaluate the Success of Surface-Initiated Reversible Deactivation Radical Polymerization


Abstract
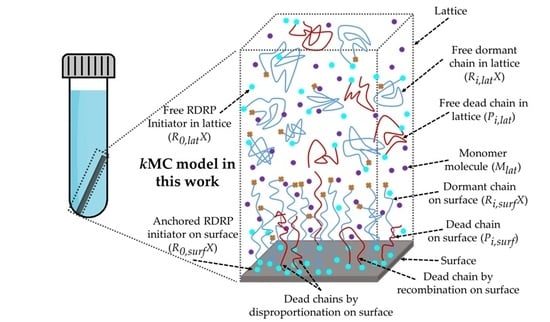
:
1. Introduction
2. Kinetic Modeling Methodology
2.1. Lattice Framework and Representation of Solution and Anchored Chemical Species
2.2. Matrix-Based Kinetic Monte Carlo Algorithm Implementation
2.2.1. Random Selection of Overall Reactions before Possible Confinement Correction
2.2.2. Initialization and Possible Correction for Confinement
3. Results and Discussion
3.1. From Conventional Molecular Properties to Molecular Height Distribution and Apparent Livingness
3.2. From Molecular Distributions to a 3D Vizualization of Tethered Polymer Chains
3.3. Effect of Average Initiator Surface Coverage on SI-RDRP Molecular Properties
3.4. Comparison of the Effect of the Translational and Segmental Diffusion Modes on SI-RDRP Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Edmondson, S.; Osborne, V.L.; Huck, W.T. Polymer brushes via surface-initiated polymerizations. Chem. Soc. Rev. 2004, 33, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Caykara, T. Polymer Brushes by Surface-Mediated RAFT Polymerization for Biological Functions. In Polymer Biopolymer Brushes; Azzaroni, O., Szleifer, I., Eds.; Wiley: Hoboken, NJ, USA, 2017; pp. 97–121. [Google Scholar]
- Lee, H.I.; Pietrasik, J.; Sheiko, S.S.; Matyjaszewski, K. Stimuli-responsive molecular brushes. Progress Polym. Sci. 2010, 35, 24–44. [Google Scholar] [CrossRef]
- Malinge, J.; Mousseau, F.; Zanchi, D.; Brun, G.; Tribet, C.; Marie, E. Tailored stimuli-responsive interaction between particles adjusted by straightforward adsorption of mixed layers of Poly(lysine)-g-PEG and Poly(lysine)-g-PNIPAM on anionic beads. J. Colloid Interface Sci. 2016, 461, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Xu, F.J. Biomolecule-functionalized polymer brushes. Chem. Soc. Rev. 2013, 42, 3394–3426. [Google Scholar] [CrossRef] [PubMed]
- Keating, J.J.T.; Imbrogno, J.; Belfort, G. Polymer Brushes for Membrane Separations: A Review. ACS Appl. Mater. Interfaces 2016, 8, 28383–28399. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Hakobyan, S.; Ramstedt, M.; Gautrot, J.E. Surface-initiated polymer brushes in the biomedical field: Applications in membrane science, biosensing, cell culture, regenerative medicine and antibacterial coatings. Chem. Rev. 2014, 114, 10976–11026. [Google Scholar] [CrossRef]
- Nagase, K.; Kobayashi, J.; Kikuchi, A.; Akiyama, Y.; Kanazawa, H.; Okano, T. Thermally-modulated on/off-adsorption materials for pharmaceutical protein purification. Biomaterials 2011, 32, 619–627. [Google Scholar] [CrossRef]
- Kurosawa, S.; Aizawa, H.; Talib, Z.A.; Atthoff, B.; Hilborn, J. Synthesis of tethered-polymer brush by atom transfer radical polymerization from a plasma-polymerized-film-coated quartz crystal microbalance and its application for immunosensors. Biosens. Bioelectron. 2004, 20, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Michl, T.D.; Giles, C.; Mocny, P.; Futrega, K.; Doran, M.R.; Klok, H.A.; Griesser, H.J.; Coad, B.R. Caspofungin on ARGET-ATRP grafted PHEMA polymers: Enhancement and selectivity of prevention of attachment of Candida albicans. Biointerphases 2017, 12, 05G602. [Google Scholar] [CrossRef] [PubMed]
- Divandari, M.; Morgese, G.; Trachsel, L.; Romio, M.; Dehghani, E.S.; Rosenboom, J.G.; Paradisi, C.; Zenobi-Wong, M.; Ramakrishna, S.N.; Benetti, E.M. Topology Effects on the Structural and Physicochemical Properties of Polymer Brushes. Macromolecules 2017, 50, 7760–7769. [Google Scholar] [CrossRef]
- Binder, K.; Milchev, A. Polymer brushes on flat and curved surfaces: How computer simulations can help to test theories and to interpret experiments. J. Polym. Sci. Part B Polym. Phys. 2012, 50, 1515–1555. [Google Scholar] [CrossRef]
- Barbey, R.; Lavanant, L.; Paripovic, D.; Schuwer, N.; Sugnaux, C.; Tugulu, S.; Klok, H.A. Polymer brushes via surface-initiated controlled radical polymerization: Synthesis, characterization, properties, and applications. Chem. Rev. 2009, 109, 5437–5527. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Ataman, N.C.; Mocny, P.; Wang, J.; Moraes, J.; Klok, H.A. Surface-Initiated Controlled Radical Polymerization: State-of-the-Art, Opportunities, and Challenges in Surface and Interface Engineering with Polymer Brushes. Chem. Rev. 2017, 117, 1105–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arraez, F.J.; Van Steenberge, P.H.M.; D’hooge, D.R. Conformational Distributions near and on the Substrate during Surface-Initiated Living Polymerization: A Lattice-Based Kinetic Monte Carlo Approach. Macromolecules 2020. [Google Scholar] [CrossRef]
- Giussi, J.M.; Cortez, M.L.; Marmisollé, W.A.; Azzaroni, O. Functionalization of Surfaces Using Polymer Brushes: An Overview of Techniques, Strategies, and Approaches. In Polymer and Biopolymer Brushes; Azzaroni, O., Szleifer, I., Eds.; Wiley: Hoboken, NJ, USA, 2017; pp. 1–27. [Google Scholar]
- Fristrup, C.J.; Jankova, K.; Hvilsted, S. Surface-initiated atom transfer radical polymerization—A technique to develop biofunctional coatings. Soft Matter 2009, 5. [Google Scholar] [CrossRef]
- Chen, T.; Amin, I.; Jordan, R. Patterned polymer brushes. Chem. Soc. Rev. 2012, 41, 3280–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coad, B.R.; Bilgic, T.; Klok, H.A. Polymer brush gradients grafted from plasma-polymerized surfaces. Langmuir 2014, 30, 8357–8365. [Google Scholar] [CrossRef]
- Karaiskos, E.; Bitsanis, I.A.; Anastasiadis, S.H. Monte Carlo Studies of Tethered Chains. J. Polym. Sci. Pol. Phys. 2009, 47, 2449–2461. [Google Scholar] [CrossRef]
- Iyer, K.S.; Zdyrko, B.; Malz, H.; Pionteck, J.; Luzinov, I. Polystyrene layers grafted to macromolecular anchoring layer. Macromolecules 2003, 36, 6519–6526. [Google Scholar] [CrossRef]
- Olivier, A.; Meyer, F.; Raquez, J.M.; Damman, P.; Dubois, P. Surface-initiated controlled polymerization as a convenient method for designing functional polymer brushes: From self-assembled monolayers to patterned surfaces. Progress Polym. Sci. 2012, 37, 157–181. [Google Scholar] [CrossRef]
- Mastan, E.; Li, X.H.; Zhu, S.P. Modeling and theoretical development in controlled radical polymerization. Progress Polym. Sci. 2015, 45, 71–101. [Google Scholar] [CrossRef]
- Xu, F.J.; Neoh, K.G.; Kang, E.T. Bioactive surfaces and biomaterials via atom transfer radical polymerization. Progress Polym. Sci. 2009, 34, 719–761. [Google Scholar] [CrossRef]
- Borner, H.G.; Duran, D.; Matyjaszewski, K.; da Silva, M.; Sheiko, S.S. Synthesis of molecular brushes with gradient in grafting density by atom transfer polymerization. Macromolecules 2002, 35, 3387–3394. [Google Scholar] [CrossRef]
- Li, C.; Benicewicz, B.C. Synthesis of Well-Defined Polymer Brushes Grafted onto Silica Nanoparticles via Surface Reversible Addition−Fragmentation Chain Transfer Polymerization. Macromolecules 2005, 38, 5929–5936. [Google Scholar] [CrossRef]
- Azzaroni, O.; Szleifer, I. Polymer and Biopolymer Brushes; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Krysiak, E.; Janasz, L.; Dupont, B.G.R.; Wypych-Puszkarz, A.; Matyjaszewski, K.; Ulanski, J. Poly(2-hydroxyethyl methacrylate) brushes synthesized by atom transfer radical polymerization from gold surface as a gate insulator in organic thin-film transistors. Thin Solid Films 2019, 669, 133–140. [Google Scholar] [CrossRef]
- Xie, G.; Khabibullin, A.; Pietrasik, J.; Yan, J.; Matyjaszewski, K. Polymer Brushes by Atom Transfer Radical Polymerization. In Polymer and Biopolymer Brushes; Azzaroni, O., Szleifer, I., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2018; pp. 29–78. [Google Scholar]
- Feng, W.; Brash, J.; Zhu, S.P. Atom-transfer radical grafting polymerization of 2-methacryloyloxyethyl phosphorylcholine from silicon wafer surfaces. J. Polym. Sci. Pol. Chem. 2004, 42, 2931–2942. [Google Scholar] [CrossRef]
- Feng, W.; Chen, R.X.; Brash, J.L.; Zhu, S.P. Surface-initiated atom transfer radical polymerization of oligo(ethylene glycol) methacrylate: Effect of solvent on graft density. Macromol. Rapid Commun. 2005, 26, 1383–1388. [Google Scholar] [CrossRef]
- Yamamoto, S.; Pietrasik, J.; Matyjaszewski, K. Temperature- and pH-responsive dense copolymer brushes prepared by ATRP. Macromolecules 2008, 41, 7013–7020. [Google Scholar] [CrossRef]
- Hui, C.M.; Pietrasik, J.; Schmitt, M.; Mahoney, C.; Choi, J.; Bockstaller, M.R.; Matyjaszewski, K. Surface-Initiated Polymerization as an Enabling Tool for Multifunctional (Nano-)Engineered Hybrid Materials. Chem. Mater. 2013, 26, 745–762. [Google Scholar] [CrossRef]
- Bartholome, C.; Beyou, E.; Bourgeat-Lami, E.; Chaumont, P.; Zydowicz, N. Nitroxide-Mediated Polymerizations from Silica Nanoparticle Surfaces: “Graft from” Polymerization of Styrene Using a Triethoxysilyl-Terminated Alkoxyamine Initiator. Macromolecules 2003, 36, 7946–7952. [Google Scholar] [CrossRef]
- Matsuno, R.; Yamamoto, K.; Otsuka, H.; Takahara, A. Polystyrene- and poly(3-vinylpyridine)-grafted magnetite nanoparticles prepared through surface-initiated nitroxide-mediated radical polymerization. Macromolecules 2004, 37, 2203–2209. [Google Scholar] [CrossRef]
- Bartholome, C.; Beyou, E.; Bourgeat-Lami, E.; Chaumont, P.; Lefebvre, F.; Zydowicz, N. Nitroxide-mediated polymerization of styrene initiated from the surface of silica nanoparticles. In situ generation and grafting of alkoxyamine initiators. Macromolecules 2005, 38, 1099–1106. [Google Scholar] [CrossRef]
- Moehrke, J.; Vana, P. The Kinetics of Surface-Initiated RAFT Polymerization of Butyl acrylate Mediated by Trithiocarbonates. Macromol. Chem. Phys. 2017, 218, 1600506. [Google Scholar] [CrossRef]
- Zengin, A.; Caykara, T. RAFT-mediated synthesis of poly[(oligoethylene glycol) methyl ether acrylate] brushes for biological functions. J. Polym. Sci. Pol. Chem. 2012, 50, 4443–4450. [Google Scholar] [CrossRef]
- Zengin, A.; Yildirim, E.; Caykara, T. RAFT-mediated synthesis and temperature-induced responsive properties of poly(2-(2-methoxyethoxy)ethyl methacrylate) brushes. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 954–962. [Google Scholar] [CrossRef]
- Zengin, A.; Karakose, G.; Caykara, T. Poly(2-(dimethylamino)ethyl methacrylate) brushes fabricated by surface-mediated RAFT polymerization and their response to pH. Eur. Polym. J. 2013, 49, 3350–3358. [Google Scholar] [CrossRef]
- Li, D.L.; Luo, Y.W.; Li, B.G.; Zhu, S.P. Effect of rate retardation in RAFT grafting polymerization from silicon wafer surface. J. Polym. Sci. Pol. Chem. 2008, 46, 970–978. [Google Scholar] [CrossRef]
- Turgman-Cohen, S.; Genzer, J. Computer Simulation of Concurrent Bulk- and Surface-Initiated Living Polymerization. Macromolecules 2012, 45, 2128–2137. [Google Scholar] [CrossRef]
- Bain, E.D.; Turgman-Cohen, S.; Genzer, J. Progress in Computer Simulation of Bulk, Confined, and Surface-initiated Polymerizations. Macromol. Theory Simul. 2013, 22, 8–30. [Google Scholar] [CrossRef]
- Turgman-Cohen, S.; Genzer, J. Simultaneous bulk- and surface-initiated controlled radical polymerization from planar substrates. J. Am. Chem. Soc. 2011, 133, 17567–17569. [Google Scholar] [CrossRef]
- Zhou, Y.-N.; Luo, Z.-H. State-of-the-Art and Progress in Method of Moments for the Model-Based Reversible-Deactivation Radical Polymerization. Macromol. React. Eng. 2016, 10, 516–534. [Google Scholar] [CrossRef]
- Reith, D.; Milchev, A.; Virnau, P.; Binder, K. Computer Simulation Studies of Chain Dynamics in Polymer Brushes. Macromolecules 2012, 45, 4381–4393. [Google Scholar] [CrossRef]
- Gao, X.; Feng, W.; Zhu, S.P.; Sheardown, H.; Brash, J.L. Kinetic Modeling of Surface-Initiated Atom Transfer Radical Polymerization. Macromol. React. Eng. 2010, 4, 235–250. [Google Scholar] [CrossRef]
- Zhou, D.P.; Mastan, E.; Zhu, S.P. Termination of Surface Radicals and Kinetic Analysis of Surface-Initiated RAFT Polymerization on Flat Surfaces. Macromol. Theory Simul. 2012, 21, 602–614. [Google Scholar] [CrossRef]
- Genzer, J. In silico polymerization: Computer simulation of controlled radical polymerization in bulk and on flat surfaces. Macromolecules 2006, 39, 7157–7169. [Google Scholar] [CrossRef]
- Turgman-Cohen, S.; Genzer, J. Computer Simulation of Controlled Radical Polymerization: Effect of Chain Confinement Due to Initiator Grafting Density and Solvent Quality in “Grafting From” Method. Macromolecules 2010, 43, 9567–9577. [Google Scholar] [CrossRef]
- Turgman-Cohen, S.; Genzer, J. Computer Simulation of Surface-Initiated Controlled Radical Polymerization: Effect of Free-Monomer Model on Brush Properties. Macromol. Theory Simul. 2019, 28, 1900033. [Google Scholar] [CrossRef]
- Mastan, E.; Xi, L.; Zhu, S. Factors Affecting Grafting Density in Surface-Initiated ATRP: A Simulation Study. Macromol. Theory Simul. 2016, 25, 220–228. [Google Scholar] [CrossRef]
- Polanowski, P.; Hałagan, K.; Pietrasik, J.; Jeszka, J.K.; Matyjaszewski, K. Growth of polymer brushes by “grafting from” via ATRP—Monte Carlo simulations. Polymers 2017, 130, 267–279. [Google Scholar] [CrossRef]
- Polanowski, P.; Jeszka, J.K.; Matyjaszewski, K. Polymer brush relaxation during and after polymerization—Monte Carlo simulation study. Polymers 2019, 173, 190–196. [Google Scholar] [CrossRef]
- Chen, C.M.; Fwu, Y.A. Monte Carlo simulations of polymer brushes. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2001, 63, 011506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, K.; Lai, P.Y.; Wittmer, J. Monte-Carlo Simulations of Chain Dynamics in Polymer Brushes. Faraday Discuss. 1994, 98, 97–109. [Google Scholar] [CrossRef]
- Shaffer, J.S. Effects of Chain Topology on Polymer Dynamics—Bulk Melts. J. Chem. Phys. 1994, 101, 4205–4213. [Google Scholar] [CrossRef]
- Zhong, M.; Matyjaszewski, K. How Fast Can a CRP Be Conducted with Preserved Chain End Functionality? Macromolecules 2011, 44, 2668–2677. [Google Scholar] [CrossRef]
- Johnston-Hall, G.; Monteiro, M.J. Bimolecular radical termination: New perspectives and insights. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3155–3173. [Google Scholar] [CrossRef]
- Zhou, D.P.; Gao, X.; Wang, W.J.; Zhu, S.P. Termination of Surface Radicals and Kinetic Modeling of ATRP Grafting from Flat Surfaces by Addition of Deactivator. Macromolecules 2012, 45, 1198–1207. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Van Steenberge, P.H.M.; Derboven, P.; Reyniers, M.F.; Marin, G.B. Model-based design of the polymer microstructure: Bridging the gap between polymer chemistry and engineering. Polym. Chem. 2015, 6, 7081–7096. [Google Scholar] [CrossRef]
- Devlaminck, D.J.G.; Van Steenberge, P.H.M.; De Keer, L.; Reyniers, M.F.; D’hooge, D.R. A detailed mechanistic study of bulk MADIX of styrene and its chain extension. Polym. Chem. 2017, 8, 6948–6963. [Google Scholar] [CrossRef] [Green Version]
- D’hooge, D.R.; Van Steenberge, P.H.M.; Reyniers, M.F.; Marin, G.B. Fed-Batch Control and Visualization of Monomer Sequences of Individual ICAR ATRP Gradient Copolymer Chains. Polymers 2014, 6, 1074–1095. [Google Scholar] [CrossRef] [Green Version]
- Van Steenberge, P.H.M.; D’hooge, D.R.; Reyniers, M.F.; Marin, G.B. Improved kinetic Monte Carlo simulation of chemical composition-chain length distributions in polymerization processes. Chem. Eng. Sci. 2014, 110, 185–199. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.M.; D’hooge, D.R.; Wang, Y.; Zhong, M.; Reyniers, M.-F.; Konkolewicz, D.; Matyjaszewski, K.; Marin, G.B. Linear Gradient Quality of ATRP Copolymers. Macromolecules 2012, 45, 8519–8531. [Google Scholar] [CrossRef]
- Arraez, F.J.; Xu, X.W.; Van Steenberge, P.H.M.; Jerca, V.V.; Hoogenboom, R.; D’hooge, D.R. Macropropagation Rate Coefficients and Branching Levels in Cationic Ring-Opening Polymerization of 2-Ethyl-2-oxazoline through Prediction of Size Exclusion Chromatography Data. Macromolecules 2019, 52, 4067–4078. [Google Scholar] [CrossRef]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Krys, P.; Matyjaszewski, K. Kinetics of Atom Transfer Radical Polymerization. Eur. Polym. J. 2017, 89, 482–523. [Google Scholar] [CrossRef] [Green Version]
- Derboven, P.; D’hooge, D.R.; Reyniers, M.-F.; Marin, G.B.; Barner-Kowollik, C. The Long and the Short of Radical Polymerization. Macromolecules 2015, 48, 492–501. [Google Scholar] [CrossRef]
- Feng, W.; Brash, J.L.; Zhu, S. Non-biofouling materials prepared by atom transfer radical polymerization grafting of 2-methacryloloxyethyl phosphorylcholine: Separate effects of graft density and chain length on protein repulsion. Biomaterials 2006, 27, 847–855. [Google Scholar] [CrossRef]
- Yu, W.H.; Kang, E.T.; Neoh, K.G.; Zhu, S.P. Controlled grafting of well-defined polymers on hydrogen-terminated silicon substrates by surface-initiated atom transfer radical polymerization. J. Phys. Chem. B 2003, 107, 10198–10205. [Google Scholar] [CrossRef]
- Huang, J.Y.; Murata, H.; Koepsel, R.R.; Russell, A.J.; Matyjaszewski, K. Antibacterial polypropylene via surface-initiated atom transfer radical polymerization. Biomacromolecules 2007, 8, 1396–1399. [Google Scholar] [CrossRef]
- Griffiths, M.C.; Strauch, J.; Monteiro, M.J.; Gilbert, R.G. Measurement of Diffusion Coefficients of Oligomeric Penetrants in Rubbery Polymer Matrixes. Macromolecules 1998, 31, 7835–7844. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Russell, G.T. Chain-length-dependent termination in radical polymerization: Subtle revolution in tackling a long-standing challenge. Progress Polym. Sci. 2009, 34, 1211–1259. [Google Scholar] [CrossRef]
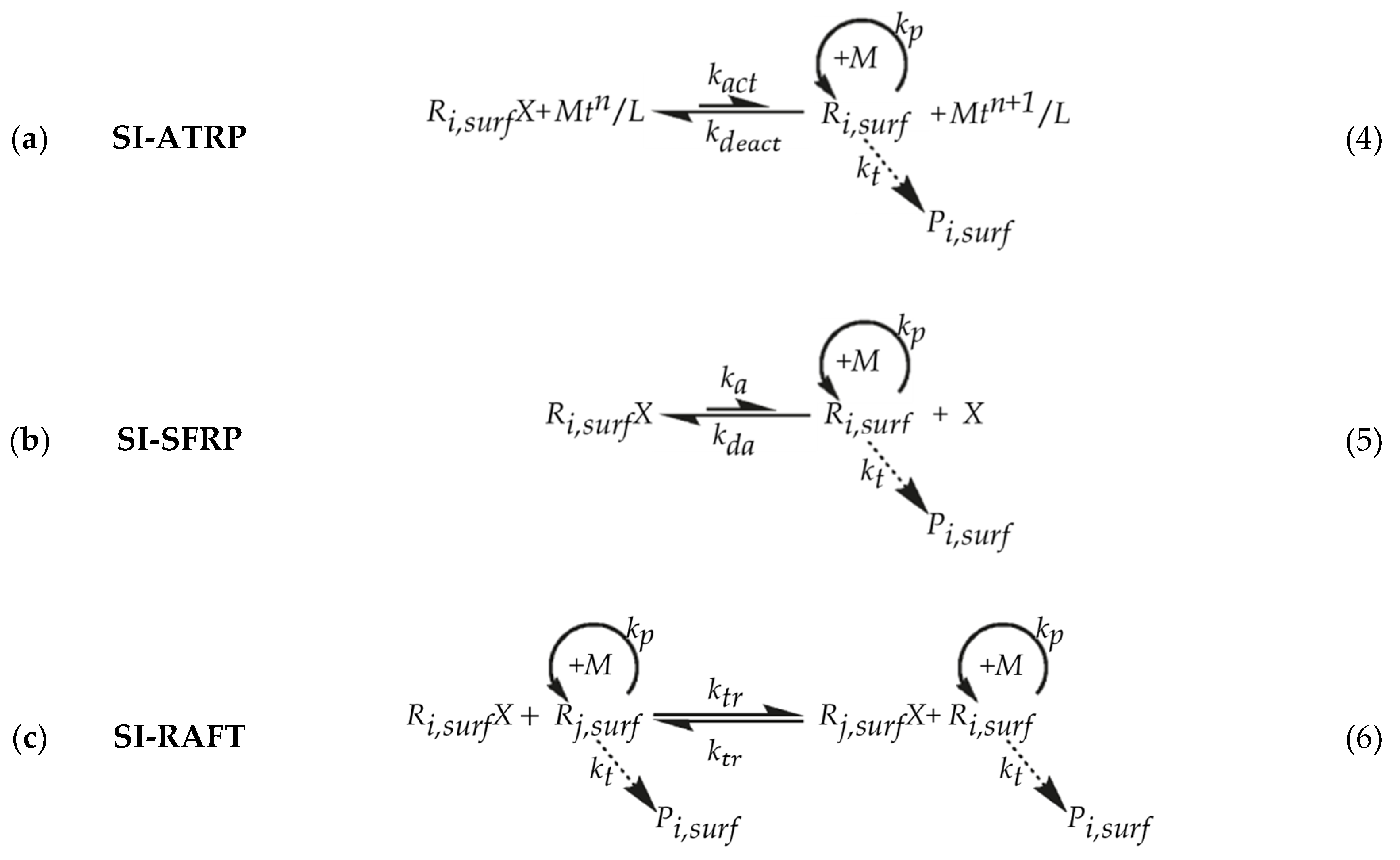
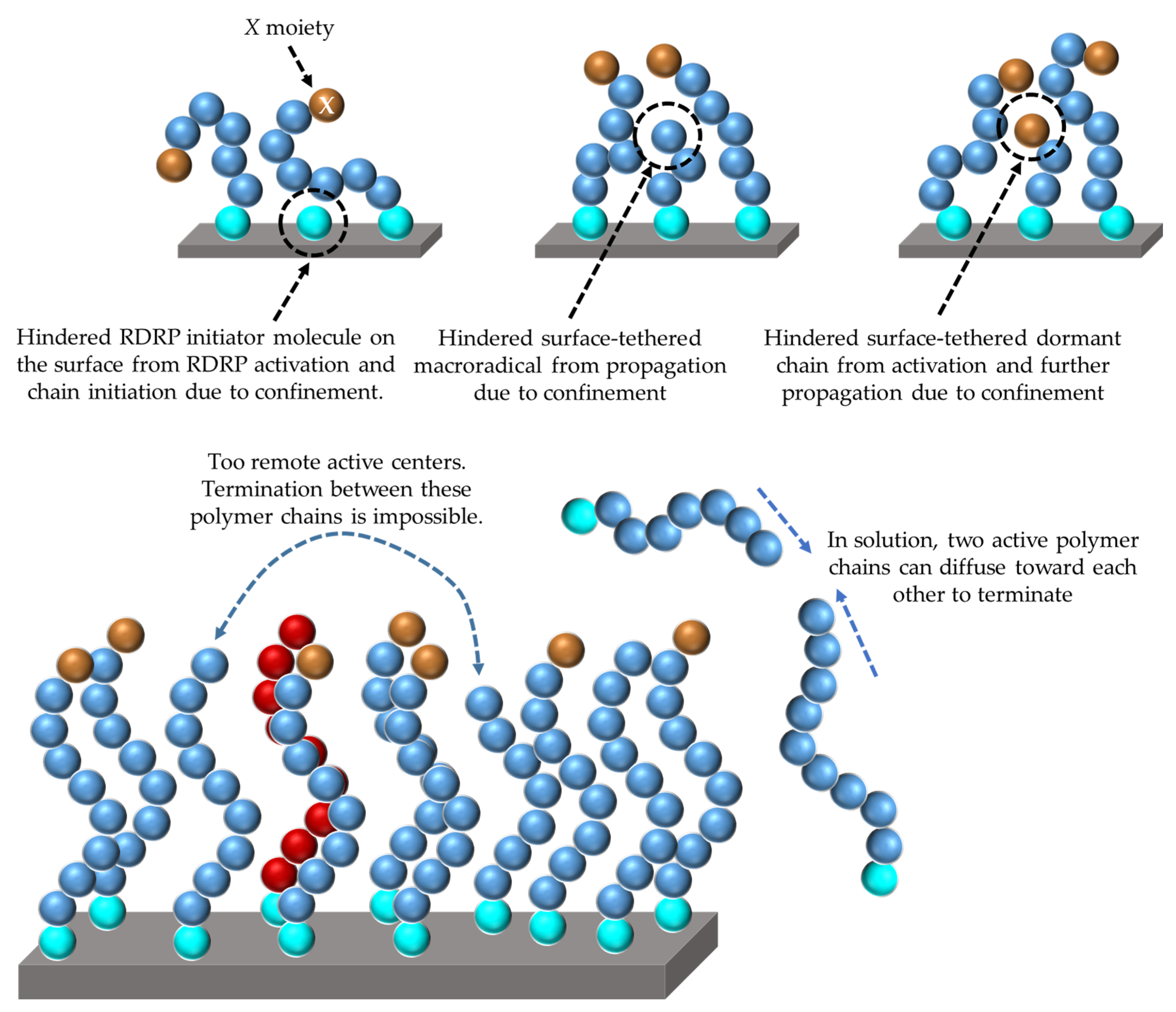














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lumped Reaction | Reaction Equation | n a | Ref | ||
|---|---|---|---|---|---|
| Chain initiation | c | 3 | [58] | ||
| 3 | [58] | ||||
| Propagation | c | 3 | [58] | ||
| 3 | [58] | ||||
| Termination by recombination | 2 | This work | |||
| c | 3 | [59] | |||
| b | 3 | [59] | |||
| Termination by disproportionation | 2 | This work | |||
| c | 3 | [59] | |||
| b | 3 | [59] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arraez, F.J.; Van Steenberge, P.H.M.; D’hooge, D.R. The Competition of Termination and Shielding to Evaluate the Success of Surface-Initiated Reversible Deactivation Radical Polymerization. Polymers 2020, 12, 1409. https://doi.org/10.3390/polym12061409
Arraez FJ, Van Steenberge PHM, D’hooge DR. The Competition of Termination and Shielding to Evaluate the Success of Surface-Initiated Reversible Deactivation Radical Polymerization. Polymers. 2020; 12(6):1409. https://doi.org/10.3390/polym12061409
Chicago/Turabian StyleArraez, Francisco J., Paul H. M. Van Steenberge, and Dagmar R. D’hooge. 2020. "The Competition of Termination and Shielding to Evaluate the Success of Surface-Initiated Reversible Deactivation Radical Polymerization" Polymers 12, no. 6: 1409. https://doi.org/10.3390/polym12061409