Thiol-Substituted Poly(2-oxazoline)s with Photolabile Protecting Groups—Tandem Network Formation by Light


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- a.
- Targeted functionalization of poly(2-oxazoline)s may be conveniently performed utilizing thiol groups;
- b.
- The biological activity of free thiol groups (e.g., after conversion to asymmetric disulfides) as biologically cleavable linker, which has been proposed in advanced transfection methodologies [25];
- c.
- d.
- a.
- 2-Oxazoline monomers bearing free thiol groups are not applicable in cationic ring opening polymerization (CROP), since the thiol is a nucleophile that acts as termination agent for the cationic active centers [30];
- b.
- Further, unprotected thiol groups are susceptible to oxidation to disulfides (e.g., with oxygen from air), which renders them inactive in thiol-ene click reactions and consequently may lead in poly(2-oxazoline)s to uncontrolled crosslinking, making storage and handling under ambient conditions difficult;
- c.
- Free thiol-groups in poly(2-oxazoline)s could also interfere in biological applications with sulfur-containing biomolecules (in particular in living organisms) by nucleophilic or disulfide exchange reactions [10].
2. Materials and Methods
3. Results and Discussion
3.1. Monomer Synthesis
3.2. Polymer Synthesis
- a.
- Low molar mass polymers (fraction 1) with high NbMEtOxa content that can be removed by deprotection (see discussion on photodeprodection further below);
- b.
- Polymer chains (fraction 2) consisting of high EtOxa and low NbMEtOxa content;
- c.
- High molar mass species (fraction 3) with UV-active groups.
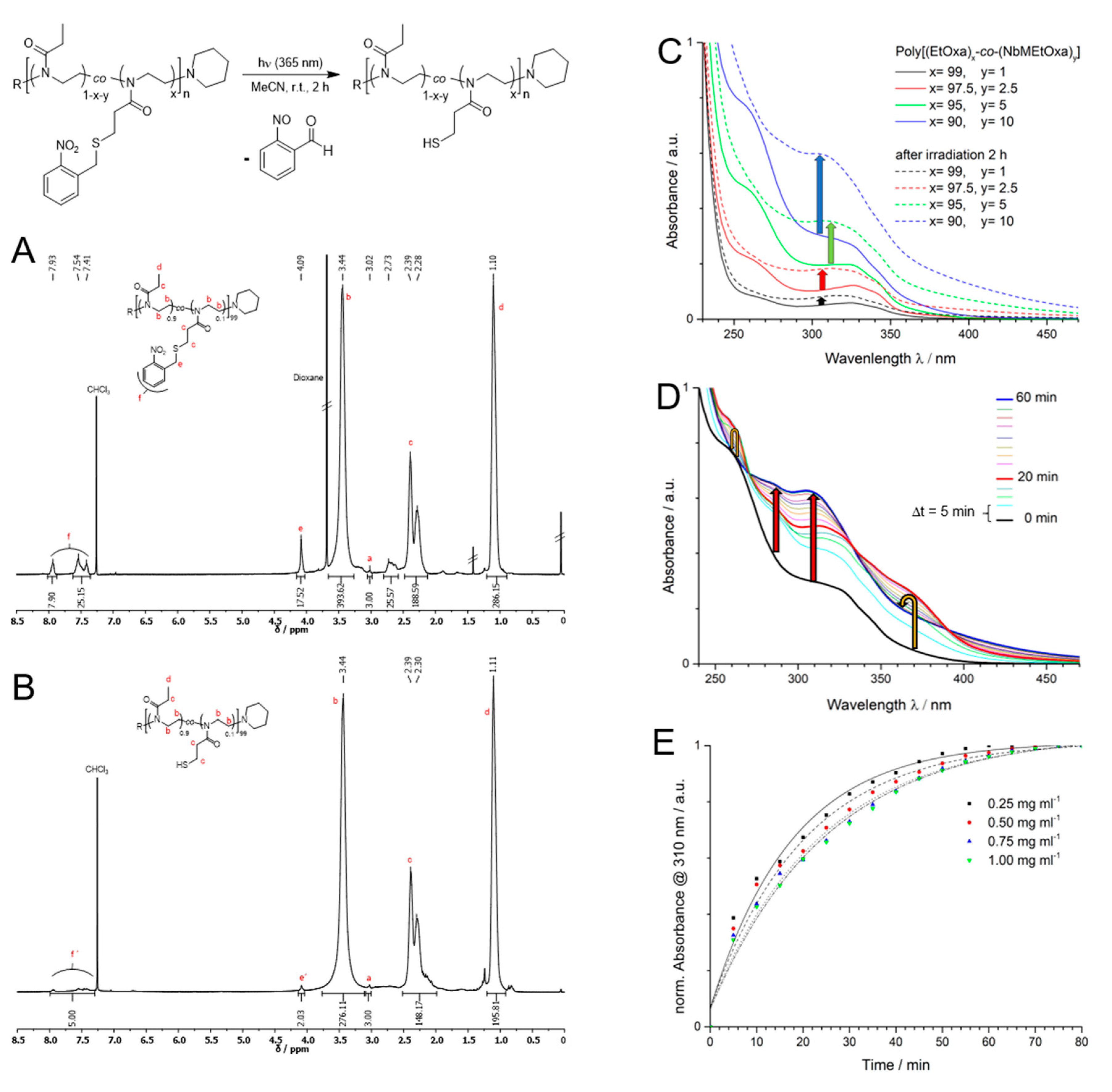
3.3. Photodeprotection
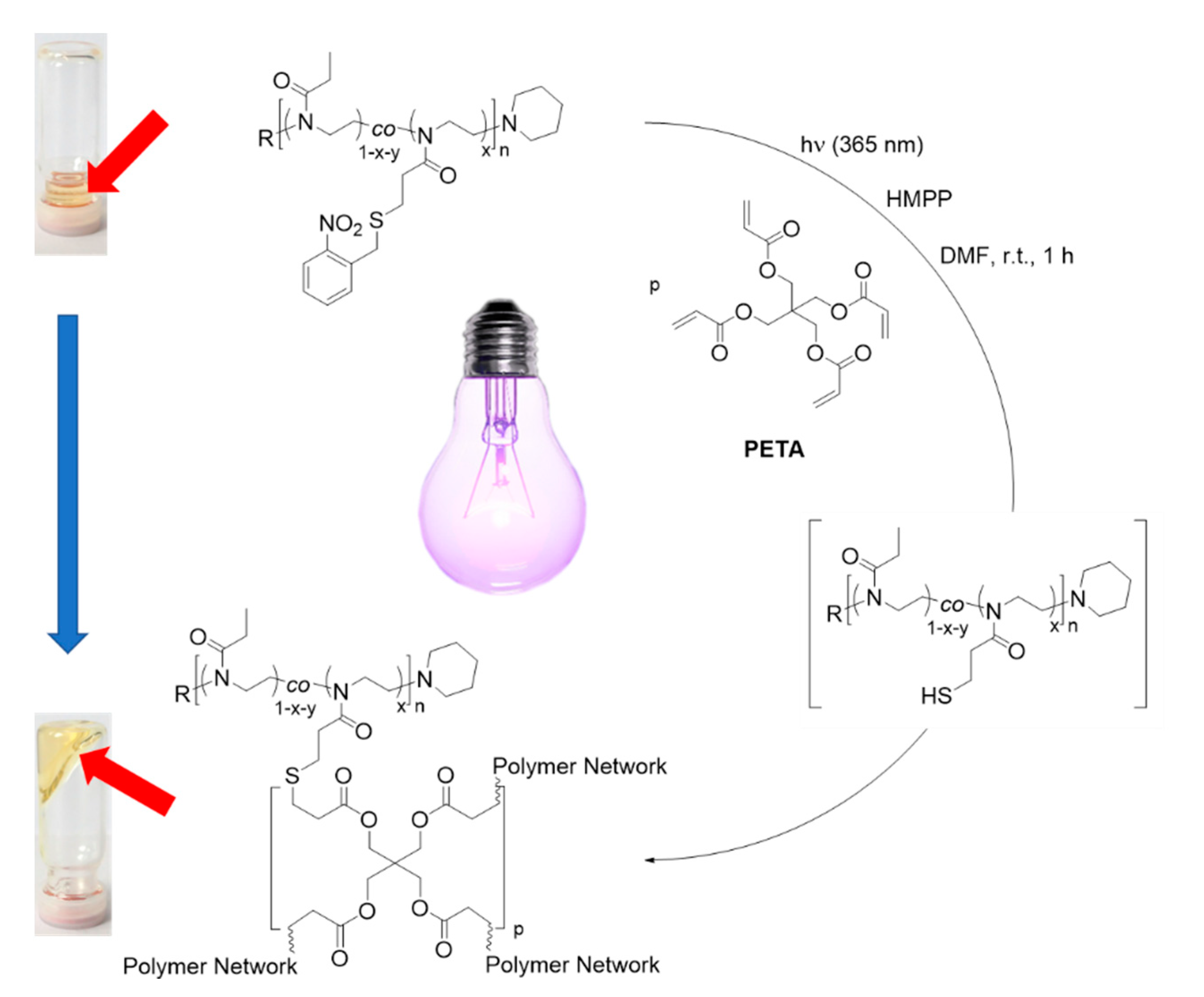
3.4. Tandem Network Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luxenhofer, R.; Han, Y.; Schulz, A.; Tong, J.; He, Z.; Kabanov, A.V.; Jordan, R. Poly(2-oxazoline)s as Polymer Therapeutics. Macromol. Rapid Commun. 2012, 33, 1613–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem. Intern. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Adams, N.; Schubert, U.S. Poly(2-oxazolines) in biological and biomedical application contexts. Adv. Drug Deliv. Rev. 2007, 59, 1504–1520. [Google Scholar] [CrossRef] [PubMed]
- Viegas, T.X.; Bentley, M.D.; Harris, J.M.; Fang, Z.; Yoon, K.; Dizman, B.; Weimer, R.; Mero, A.; Pasut, G.; Veronese, F.M. Polyoxazoline: Chemistry, Properties, and Applications in Drug Delivery. Bioconj. Chem. 2011, 22, 976–986. [Google Scholar] [CrossRef]
- Tauhardt, L.; Kempe, K.; Gottschaldt, M.; Schubert, U.S. Poly(2-oxazoline) functionalized surfaces: From modification to application. Chem. Soc. Rev. 2013, 42, 7998–8011. [Google Scholar] [CrossRef]
- Lorson, T.; Lübtow, M.M.; Wegener, E.; Haider, M.S.; Borova, S.; Nahm, D.; Jordan, R.; Sokolski-Papkov, M.; Kabanov, A.V.; Luxenhofer, R. Poly(2-oxazoline)s based biomaterials: A comprehensive and critical update. Biomaterials 2018, 178, 204–280. [Google Scholar] [CrossRef]
- Hoogenboom, R. Poly(2-oxazoline)s: Alive and Kicking. Macromol. Chem. Phys. 2007, 208, 18–25. [Google Scholar] [CrossRef]
- Hoogenboom, R. Poly(2-oxazoline)s: A Polymer Class with Numerous Potential Applications. Angew. Chem. 2009, 48, 7978–7994. [Google Scholar] [CrossRef]
- Pizzi, D.; Humphries, J.; Morrow, J.P.; Fletcher, N.L.; Bell, C.A.; Thurecht, K.J.; Kempe, K. Poly(2-oxazoline) macromonomers as building blocks for functional and biocompatible polymer architectures. Eur. Polym. J. 2019, 121, 109258. [Google Scholar] [CrossRef]
- Ahmad, N.; Colak, B.; Gibbs, M.J.; Zhang, D.-W.; Gautrot, J.E.; Watkinson, M.; Becer, C.R.; Krause, S. Peptide Cross-Linked Poly(2-oxazoline) as a Sensor Material for the Detection of Proteases with a Quartz Crystal Microbalance. Biomacromolecules 2019, 20, 2506–2514. [Google Scholar] [CrossRef]
- Chujo, Y.; Yoshifuji, Y.; Sada, K.; Saegusa, T. A Novel Nonionic Hydrogel from 2-Methyl-2-oxazoline. Macromolecules 1989, 22, 1074–1077. [Google Scholar] [CrossRef]
- Chujo, Y.; Sada, K.; Saegusa, T. Polyoxazoline Having a Coumarin Moiety as a Pendant Group. Synthesis and Photogelation. Macromolecules 1990, 23, 2693–2697. [Google Scholar] [CrossRef]
- Chujo, Y.; Sada, K.; Saegusa, T. Reversible Gelation of Polyoxazoline by Means of Diels-Alder Reaction. Macromolecules 1990, 23, 2636–2641. [Google Scholar] [CrossRef]
- Chujo, Y.; Sada, K.; Naka, A.; Nomura, R.; Saegusa, T. Synthesis and Redox Gelation of Disulfide-Modified Polyoxazoline. Macromolecules 1993, 26, 883–887. [Google Scholar] [CrossRef]
- Kelly, A.M.; Hecke, A.; Wirnsberger, B.; Wiesbrock, F. Synthesis of Poly(2-oxazoline)-Based Hydrogels with Tailor-Made Swelling Degrees Capable of Stimuli-Triggered Compound Release. Macromol. Rapid Commun. 2011, 32, 1815–1819. [Google Scholar] [CrossRef]
- Hartlieb, M.; Pretzel, D.; Kempe, K.; Fritzsche, C.; Paulus, R.M.; Gottschaldt, M.; Schubert, U.S. Cationic poly(2-oxazoline) hydrogels for reversible DNA binding. Soft Matter 2013, 9, 4693–4704. [Google Scholar] [CrossRef]
- Gress, A.; Völkel, A.; Schlaad, H. Thio-Click Modification of Poly[2-(3-butenyl)-2-oxazoline]. Macromolecules 2007, 40, 7928–7933. [Google Scholar] [CrossRef]
- Kempe, K.; Lobert, M.; Hoogenboom, R.; Schubert, U.S. Synthesis and characterization of a series of diverse poly(2-oxazoline)s. J. Polym. Sci. A Polym. Chem. 2009, 47, 3829–3838. [Google Scholar] [CrossRef]
- Kempe, K.; Vollrath, A.; Schaefer, H.W.; Poehlmann, T.G.; Biskup, C.; Hoogenboom, R.; Hornig, S.; Schubert, U.S. Multifunctional Poly(2-oxazoline) Nanoparticles for Biological Applications. Macromol. Rapid Commun. 2010, 31, 1869–1873. [Google Scholar] [CrossRef]
- Kempe, K.; Hoogenboom, R.; Jaeger, M.; Schubert, U.S. Three-Fold Metal-Free Efficient (“Click”) Reactions onto a Multifunctional Poly(2-oxazoline) Designer Scaffold. Macromolecules 2011, 44, 6424–6432. [Google Scholar] [CrossRef]
- Kempe, K.; Weber, C.; Babiuch, K.; Gottschaldt, M.; Hoogenboom, R.; Schubert, U.S. Responsive Glyco-poly(2-oxazoline)s: Synthesis, Cloud Point Tuning, and Lectin Binding. Biomacromolecules 2011, 12, 2591–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dargaville, T.R.; Forster, R.; Farrugia, B.L.; Kempe, K.; Voorhaar, L.; Schubert, U.S.; Hoogenboom, R. Poly(2-oxazoline) Hydrogel Monoliths via Thiol-ene Coupling. Macromol. Rapid Commun. 2012, 33, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Blöhbaum, J.; Paulus, I.; Pöppler, A.-C.; Tessmar, J.; Groll, J. Influence of charged groups on the cross-linking efficiency and release of guest molecules from thiol–ene cross-linked poly(2-oxazoline) hydrogels. J. Mater. Chem. B 2019, 7, 1782–1794. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, J.; Teßmar, J.; Groll, J. In Situ Polymer Analogue Generation of Azlactone Functions at Poly(oxazoline)s for Peptide Conjugation. Macromol. Chem. Phys. 2020, 221, 1900500. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zheng, M.; Meng, F.; Zhang, J.; Peng, R.; Zhong, Z. Branched Polyethylenimine Derivatives with Reductively Cleavable Periphery for Safe and Efficient In Vitro Gene Transfer. Biomacromolecules 2011, 12, 1032–1040. [Google Scholar] [CrossRef]
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology. Chem. Rev. 2005, 105, 1103–1170. [Google Scholar] [CrossRef]
- Roth, P.J.; Boyer, C.; Lowe, A.B.; Davis, T.P. RAFT Polymerization and Thiol Chemistry: A Complementary Pairing for Implementing Modern Macromolecular Design. Macromol. Rapid Commun. 2011, 32, 1123–1143. [Google Scholar] [CrossRef]
- Yoon, J.A.; Kamada, J.; Koynov, K.; Mohin, J.; Nicolaÿ, R.; Zhang, Y.; Balazs, A.C.; Kowalewski, T.; Matyjaszewski, K. Self-Healing Polymer Films Based on Thiol–Disulfide Exchange Reactions and Self-Healing Kinetics Measured Using Atomic Force Microscopy. Macromolecules 2012, 45, 142–149. [Google Scholar] [CrossRef]
- Pepels, M.; Filot, I.; Klumperman, B.; Goossens, H. Self-healing systems based on disulfide–thiol exchange reactions. Polym. Chem. 2013, 4, 4955–4965. [Google Scholar] [CrossRef]
- Glassner, M.; Vergaelen, M.; Hoogenboom, R. Poly(2-oxazoline)s: A comprehensive overview of polymer structures and their physical properties. Polym. Int. 2018, 67, 32–45. [Google Scholar] [CrossRef]
- Cartwright, I.L.; Hutchinson, D.W.; Armstrong, V.W. The reaction between thiols and 8-azidoadenosine derivatives. Nucleic Acids Res. 1976, 3, 2331–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubmann, C.; Luxenhofer, R.; Cesana, S.; Jordan, R. First Aldehyde-Functionalized Poly(2-oxazoline)s for Chemoselective Ligation. Macromol. Biosci. 2005, 5, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Cesana, S.; Auernheimer, J.; Jordan, R.; Kessler, H.; Nuyken, O. First Poly(2-oxazoline)s with Pendant Amino Groups. Macromol. Chem. Phys. 2006, 207, 183–192. [Google Scholar] [CrossRef]
- Zarka, M.T.; Nuyken, O.; Weberskirch, R. Amphiphilic Polymer Supports for the Asymmetric Hydrogenation of Amino Acid Precursors in Water. Chem. A Eur. J. 2003, 9, 3228–3234. [Google Scholar] [CrossRef]
- Cesana, S.; Kurek, A.; Baur, M.A.; Auernheimer, J.; Nuyken, O. Polymer-Bound Thiol Groups on Poly(2-oxazoline)s. Macromol. Rapid Commun. 2007, 28, 608–615. [Google Scholar] [CrossRef]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef]
- Bochet, C.G. Wavelength-selective cleavage of photolabile protecting groups. Tetrahedron Lett. 2000, 41, 6341–6346. [Google Scholar] [CrossRef]
- Bochet, C.G. Orthogonal Photolysis of Protecting Groups. Angew. Chem. Int. 2001, 40, 2071–2073. [Google Scholar] [CrossRef]
- Bochet, C.G. Photolabile protecting groups and linkers. J. Chem. Soc. Perkin Trans. 2002, 1, 125–142. [Google Scholar]
- Wang, P. Photolabile Protecting Groups: Structure and Reactivity. As. J. Org. Chem. 2013, 2, 452–464. [Google Scholar] [CrossRef]
- Pillai, V.N.R. Photoremoveable Protecting Groups in Organic Synthesis. Synthesis 1980, 1980, 1–26. [Google Scholar] [CrossRef]
- Del Campo, A.; Boos, D.; Spiess, H.W.; Jonas, U. Surface Modification with Orthogonal Photosensitive Silanes for Sequential Chemical Lithography and Site-Selective Particle Deposition. Angew. Chem. Int. 2005, 44, 4707–4712. [Google Scholar] [CrossRef] [PubMed]
- Vorobev, A.Y.; Moskalensky, A.E. Long-wavelength photoremovable protecting groups: On the way to in vivo application. Comput. Struct. Biotechnol. J. 2020, 18, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Hennig, A.-L.K.; Deodato, D.; Asad, N.; Herbivo, C.; Dore, T.M. Two-Photon Excitable Photoremovable Protecting Groups Based on the Quinoline Scaffold for Use in Biology. J. Org. Chem. 2020, 85, 726–744. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Litt, M. Polymerization of cyclic iminoethers. V. 1,3-oxazolines with hydroxy-, acetoxy-, and carboxymethyl-alkyl groups in the 2 position and their polymers. J. Polym. Sci. A Polym. Chem. 1968, 6, 1883–1894. [Google Scholar] [CrossRef]
- Hensarling, R.M.; Hoff, E.A.; LeBlanc, A.P.; Guo, W.; Rahane, S.B.; Patton, D.L. Photocaged pendent thiol polymer brush surfaces for postpolymerization modifications via thiol-click chemistry. J. Polym. Sci. Part. A Polym. Chem. 2013, 51, 1079–1090. [Google Scholar] [CrossRef]
- Pauloehrl, T.; Delaittre, G.; Bastmeyer, M.; Barner-Kowollik, C. Ambient temperature polymer modification by in situ phototriggered deprotection and thiol–ene chemistry. Polym. Chem. 2012, 3, 1740–1749. [Google Scholar] [CrossRef]
- Kobayashi, S.; Iijima, S.; Igarashi, T.; Saegusa, T. Synthesis of a Nonionic Polymer Surfactant from Cyclic Imino Ethers by the Initiator Method. Macromolecules 1987, 20, 1729–1734. [Google Scholar] [CrossRef]
- Kilz, P. Copolymer Analysis by LC Methods, including 2D Chromatography. In Encyclopedia of Chromatography; Cazes, J., Ed.; Marcel Dekker: New York, NY, USA, 2001; pp. 195–200. [Google Scholar]
- Delaittre, G.; Pauloehrl, T.; Bastmeyer, M.; Barner-Kowollik, C. Acrylamide-Based Copolymers Bearing Photoreleasable Thiols for Subsequent Thiol–Ene Functionalization. Macromolecules 2012, 45, 1792–1802. [Google Scholar] [CrossRef]
- Il’ichev, Y.V.; Schwörer, M.A.; Wirz, J. Photochemical Reaction Mechanisms of 2-Nitrobenzyl Compounds: Methyl Ethers and Caged ATP. J. Am. Chem. Soc. 2004, 126, 4581–4595. [Google Scholar] [CrossRef]
- Patchornik, A.; Amit, B.; Woodward, R.B. Photosensitive Protecting Groups. J. Am. Chem. Soc. 1970, 92, 6333–6335. [Google Scholar] [CrossRef]
- Barltrop, J.A.; Plant, P.J.; Schofield, P. Photosensitive protective groups. Chem. Commun. 1966, 822, 822–823. [Google Scholar] [CrossRef]
- Ooi, H.W.; Jack, K.S.; Whittaker, A.K.; Peng, H. Photo-initiated Thiol–Ene “Click” Hydrogels from RAFT-Synthesized Poly(N-isopropylacrylamide). J. Poly. Sci. Part A Poly. Chem. 2013, 51, 4626–4636. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, N.; Diehl, F.; Jonas, U. Thiol-Substituted Poly(2-oxazoline)s with Photolabile Protecting Groups—Tandem Network Formation by Light. Polymers 2020, 12, 1767. https://doi.org/10.3390/polym12081767
Jung N, Diehl F, Jonas U. Thiol-Substituted Poly(2-oxazoline)s with Photolabile Protecting Groups—Tandem Network Formation by Light. Polymers. 2020; 12(8):1767. https://doi.org/10.3390/polym12081767
Chicago/Turabian StyleJung, Niklas, Fiona Diehl, and Ulrich Jonas. 2020. "Thiol-Substituted Poly(2-oxazoline)s with Photolabile Protecting Groups—Tandem Network Formation by Light" Polymers 12, no. 8: 1767. https://doi.org/10.3390/polym12081767