Block Copolymers of Poly(ω-Pentadecalactone) in Segmented Polyurethanes: Novel Biodegradable Shape Memory Polyurethanes




Abstract
:
1. Introduction
2. Materials and Characterization
2.1. Materials
2.2. Characterization
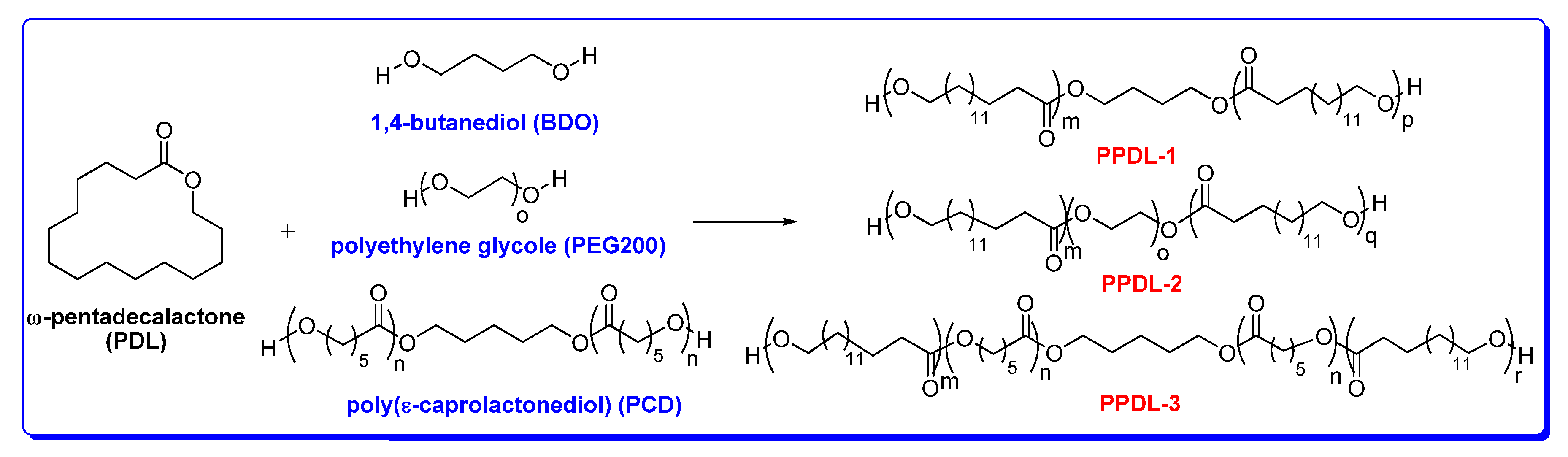
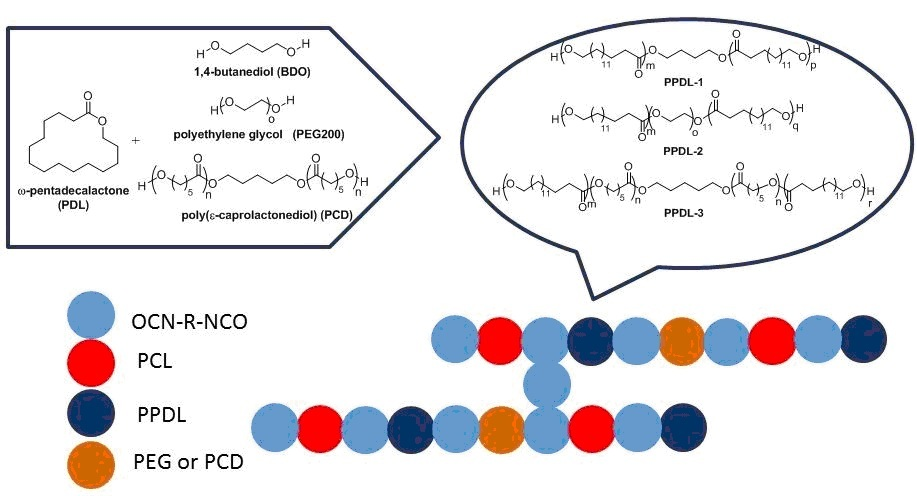
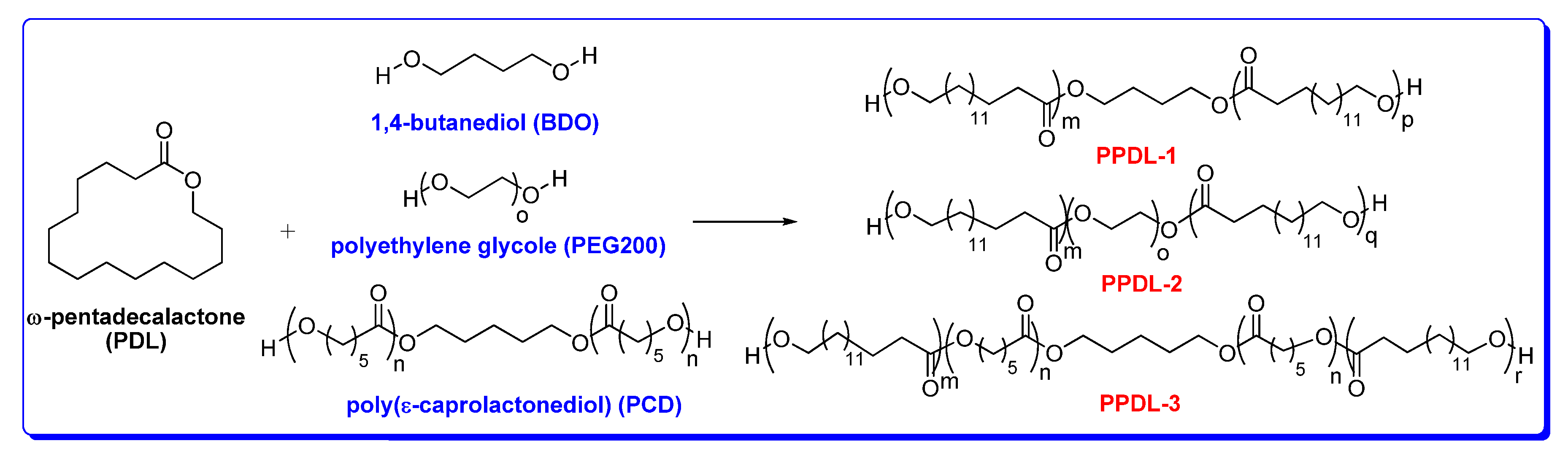
3. Polymerization of ω-Pentadecalactone (PDL) by ROP
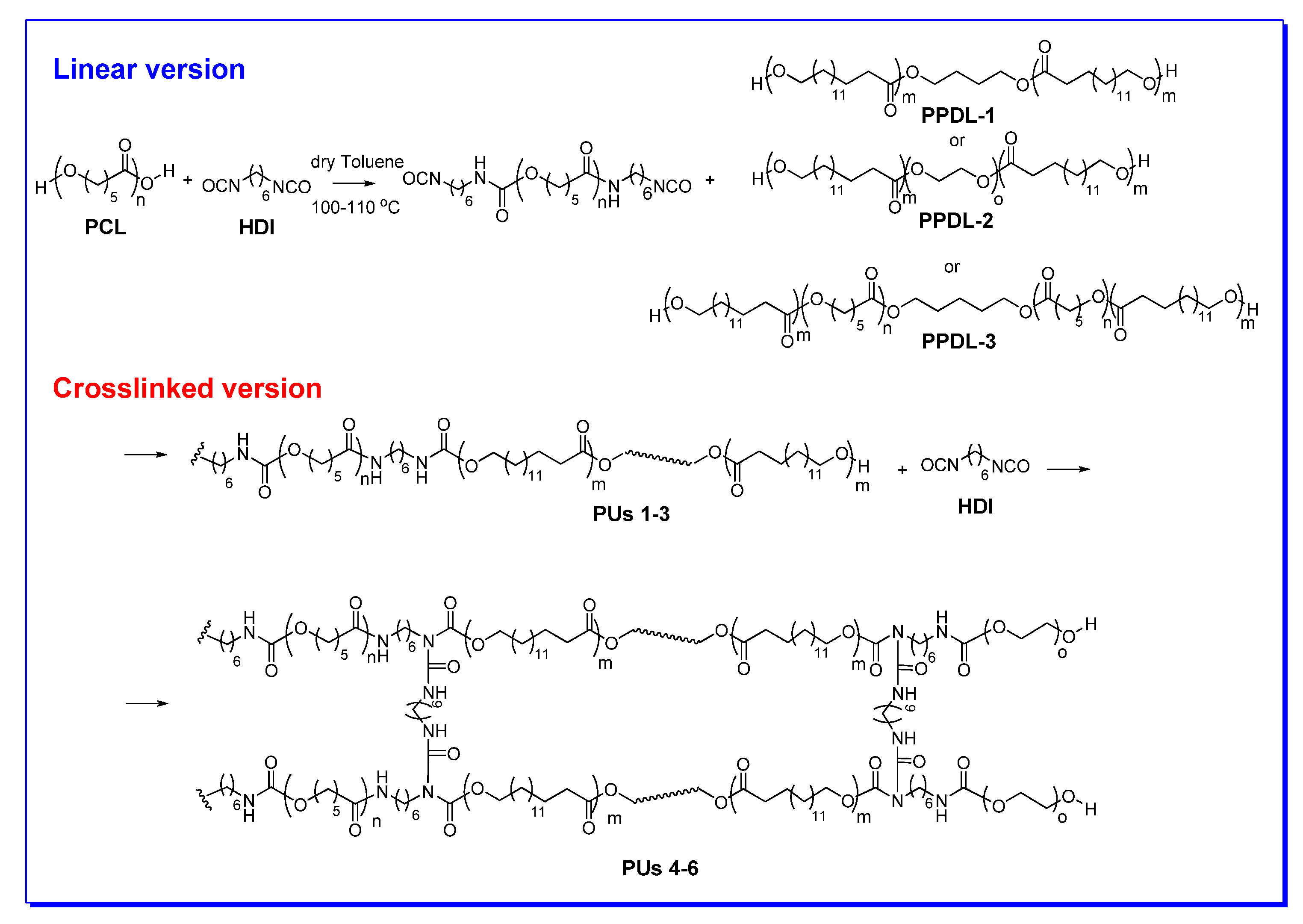
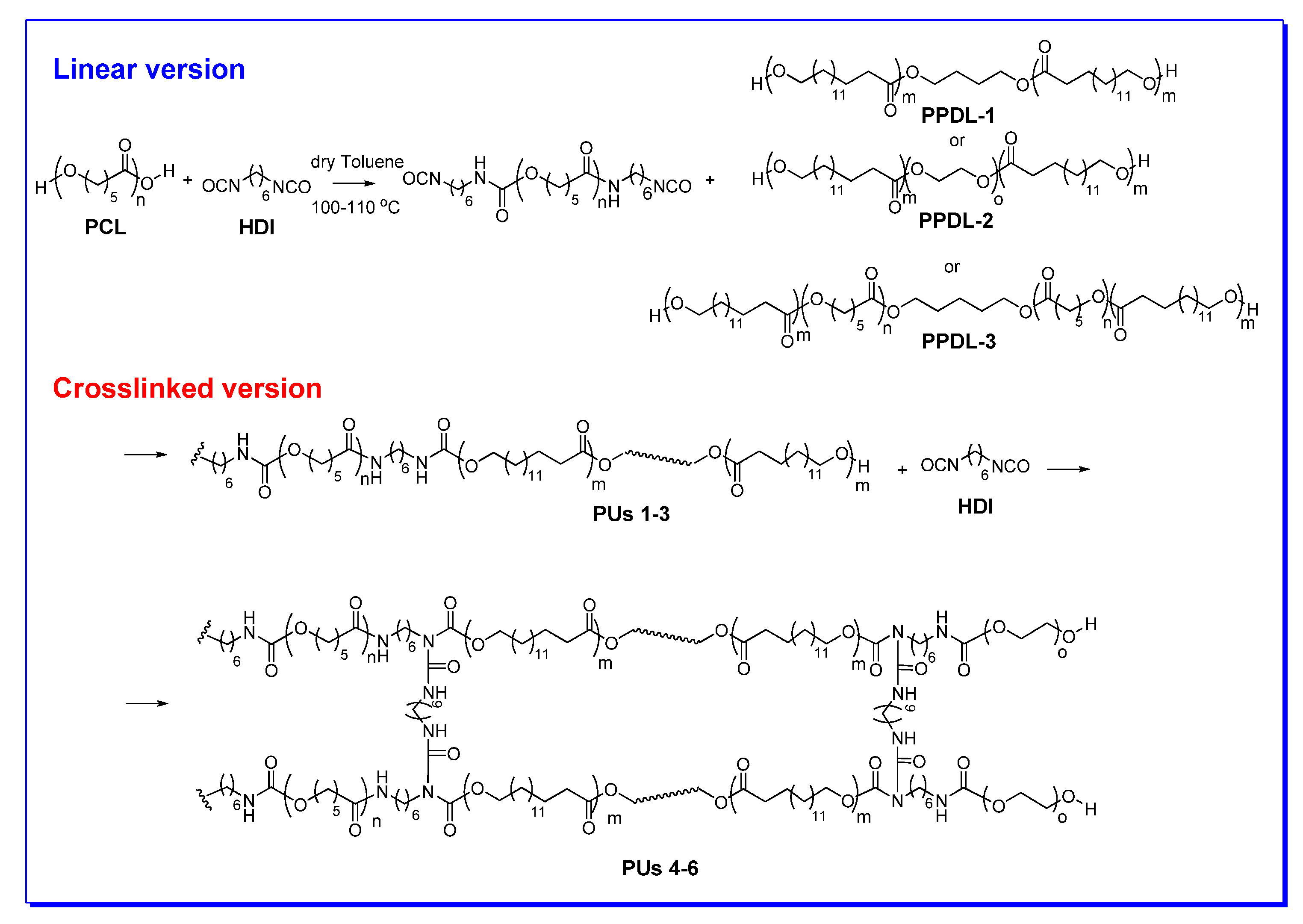
4. Synthesis of PUs
5. Results and Discussion
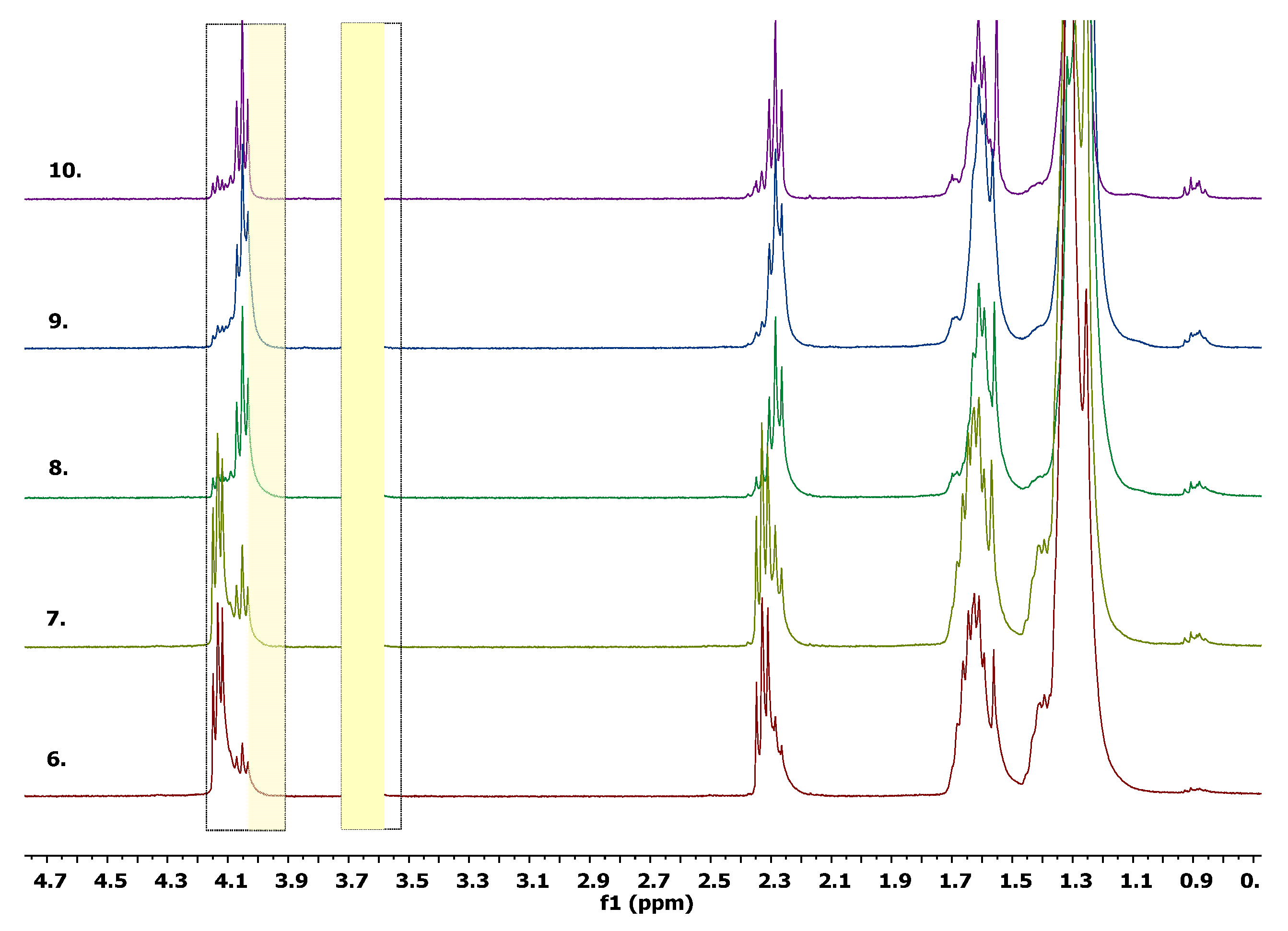
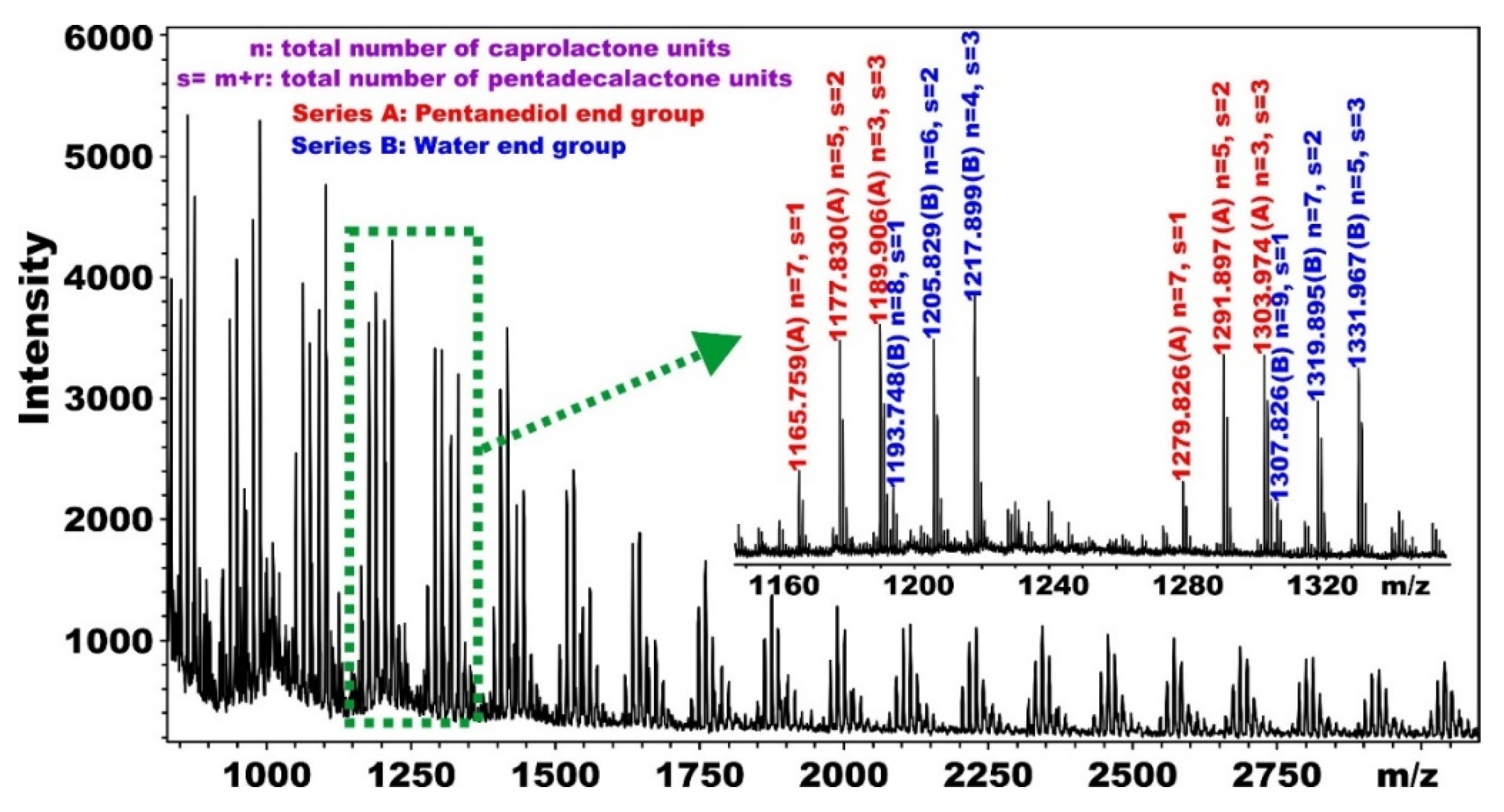
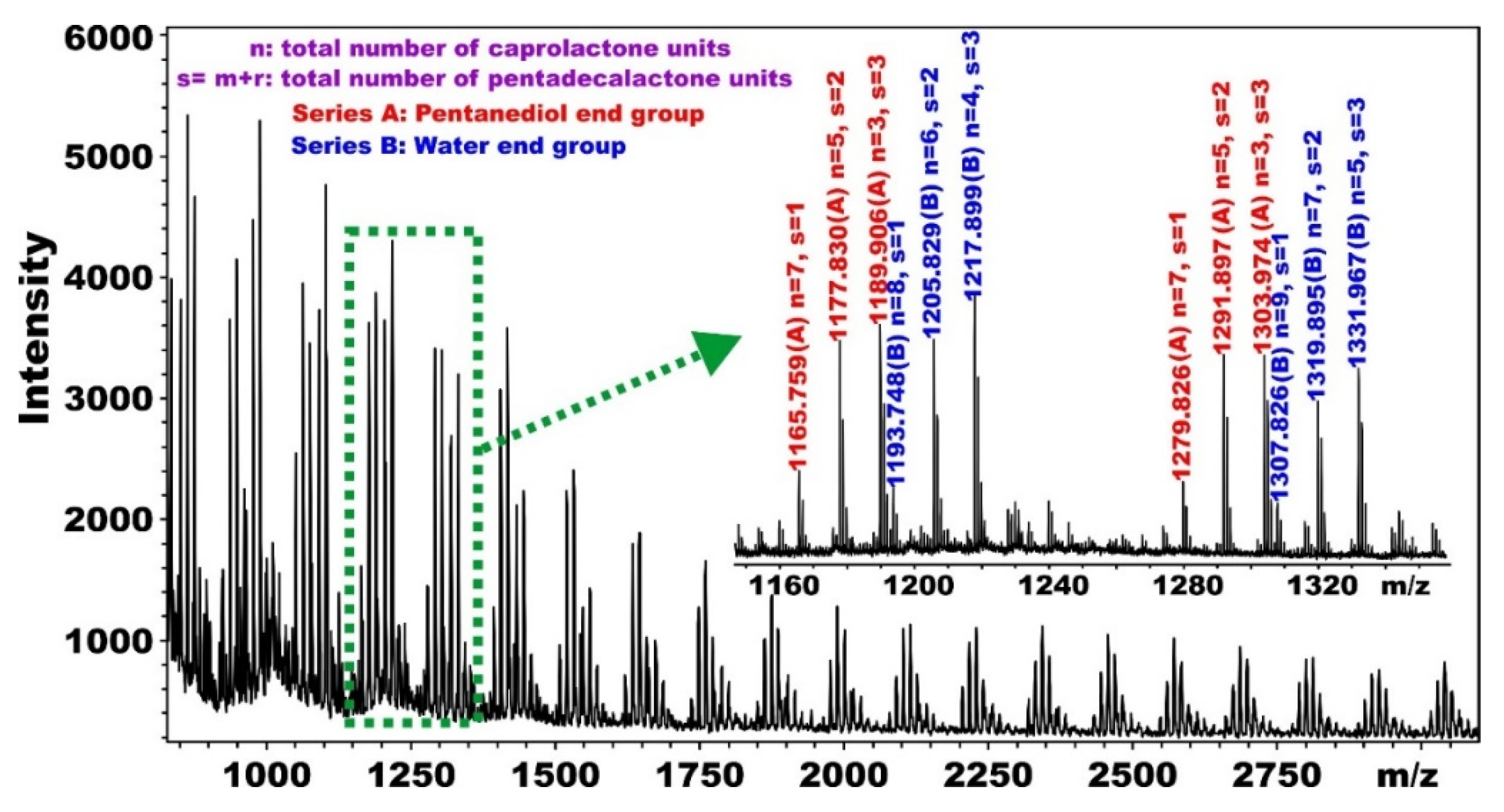
5.1. Synthesis and Characterization of α, ω-PPDL Diols by ROP
5.2. Synthesis and Characterization of PPDL Containing Urethanes
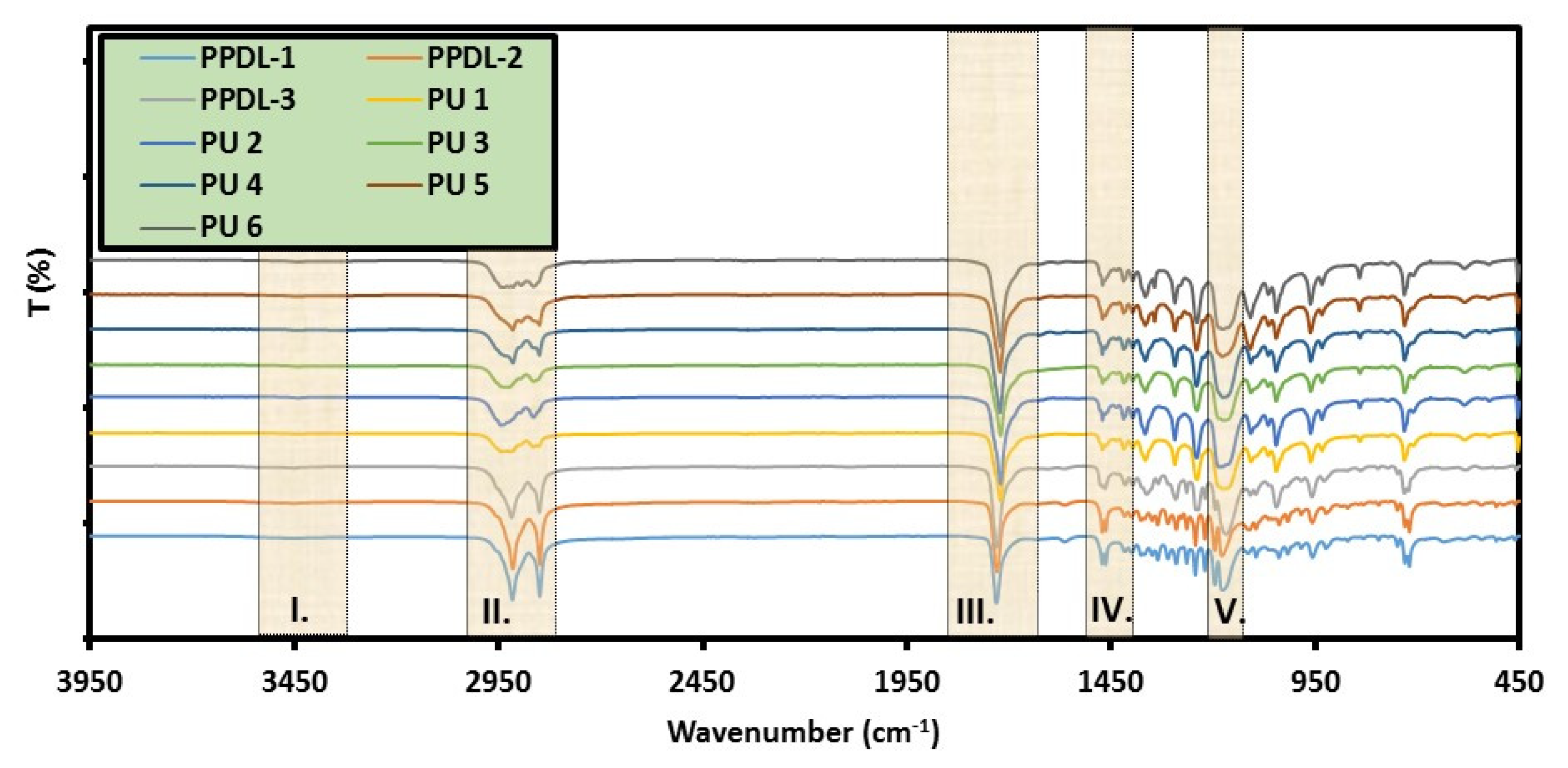
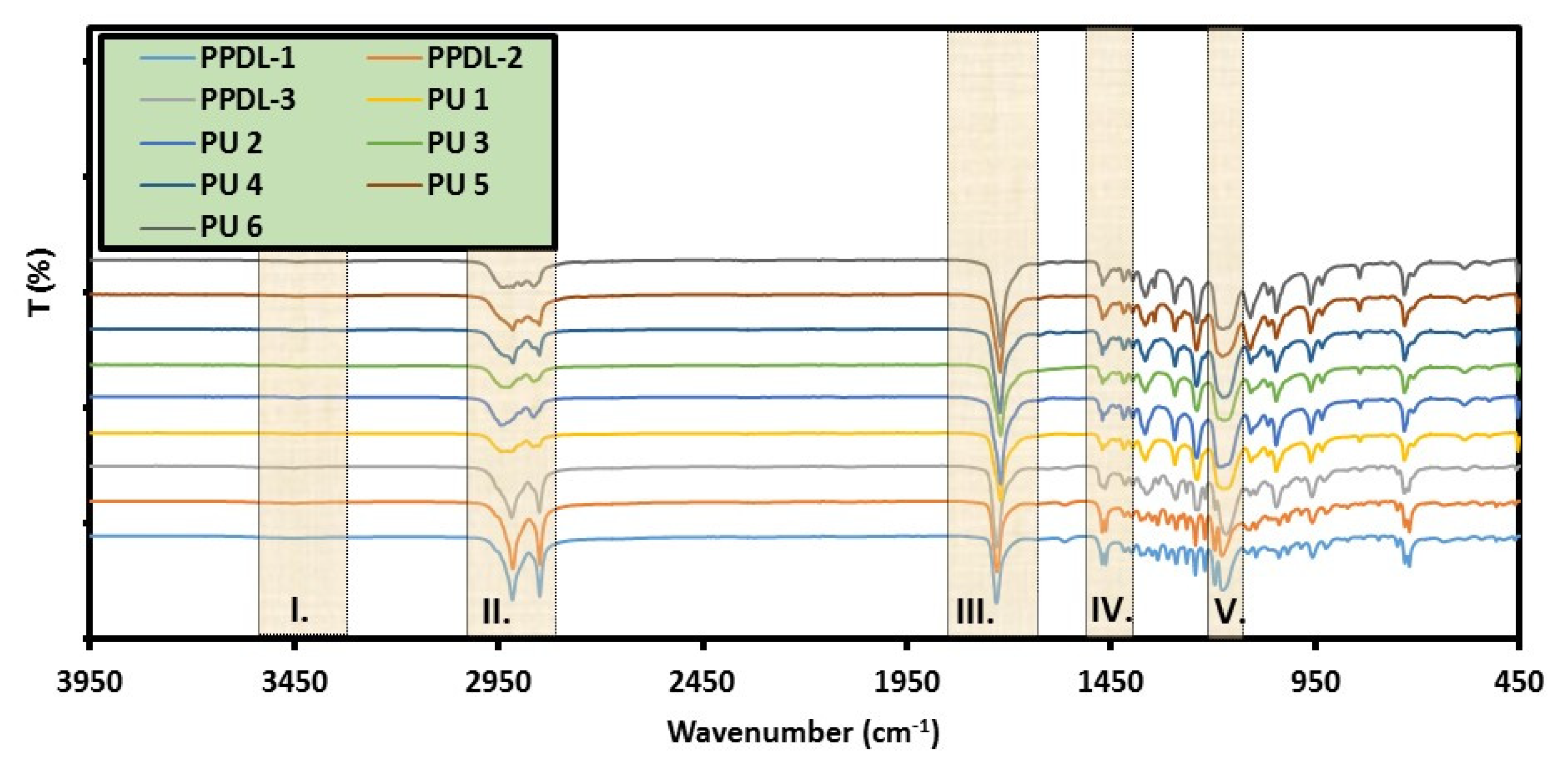
5.3. Infrared Spectroscopy (FT-IR)
5.4. Morphology of PUs by SEM
5.5. Mechanical Properties of PUs
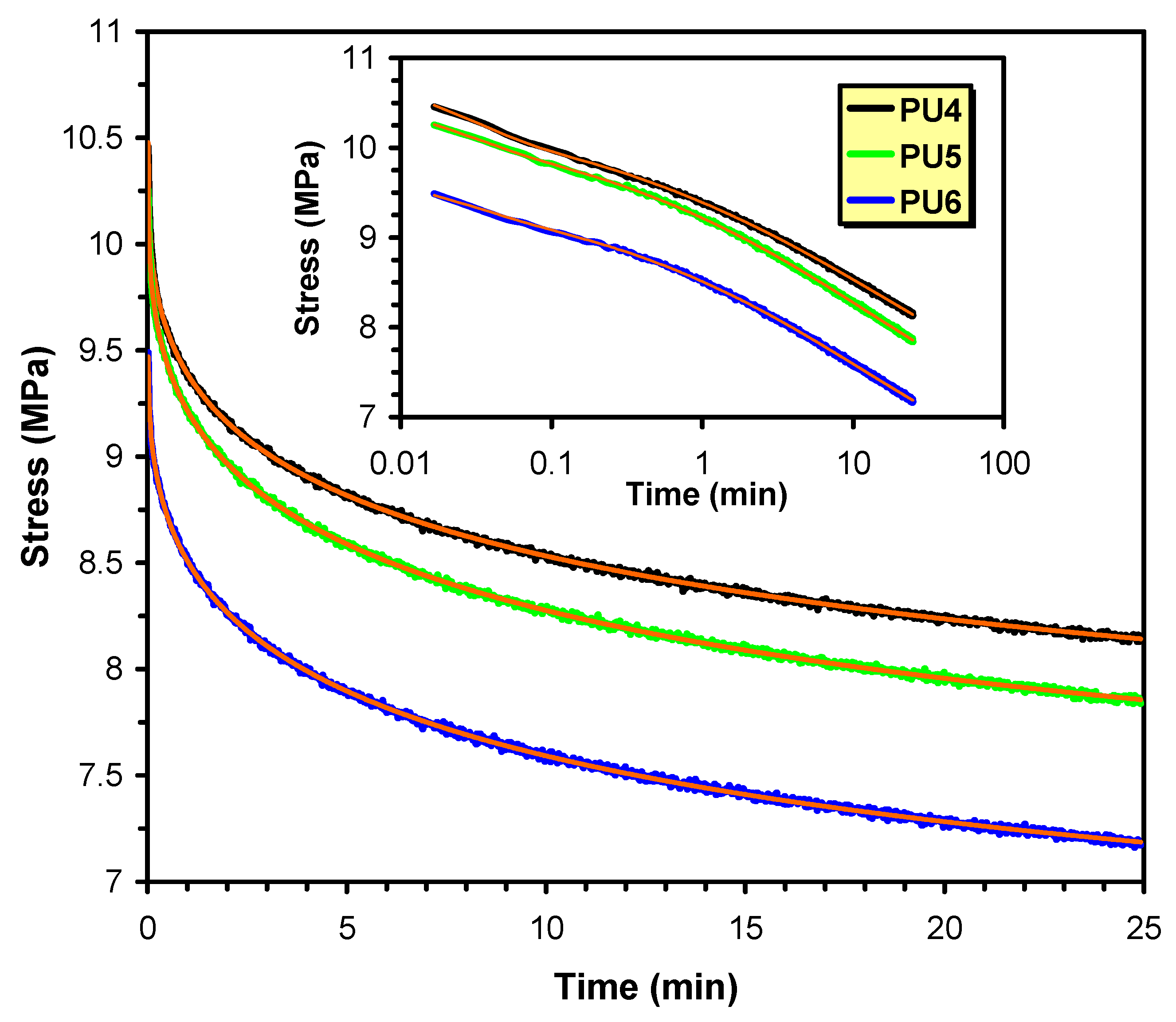
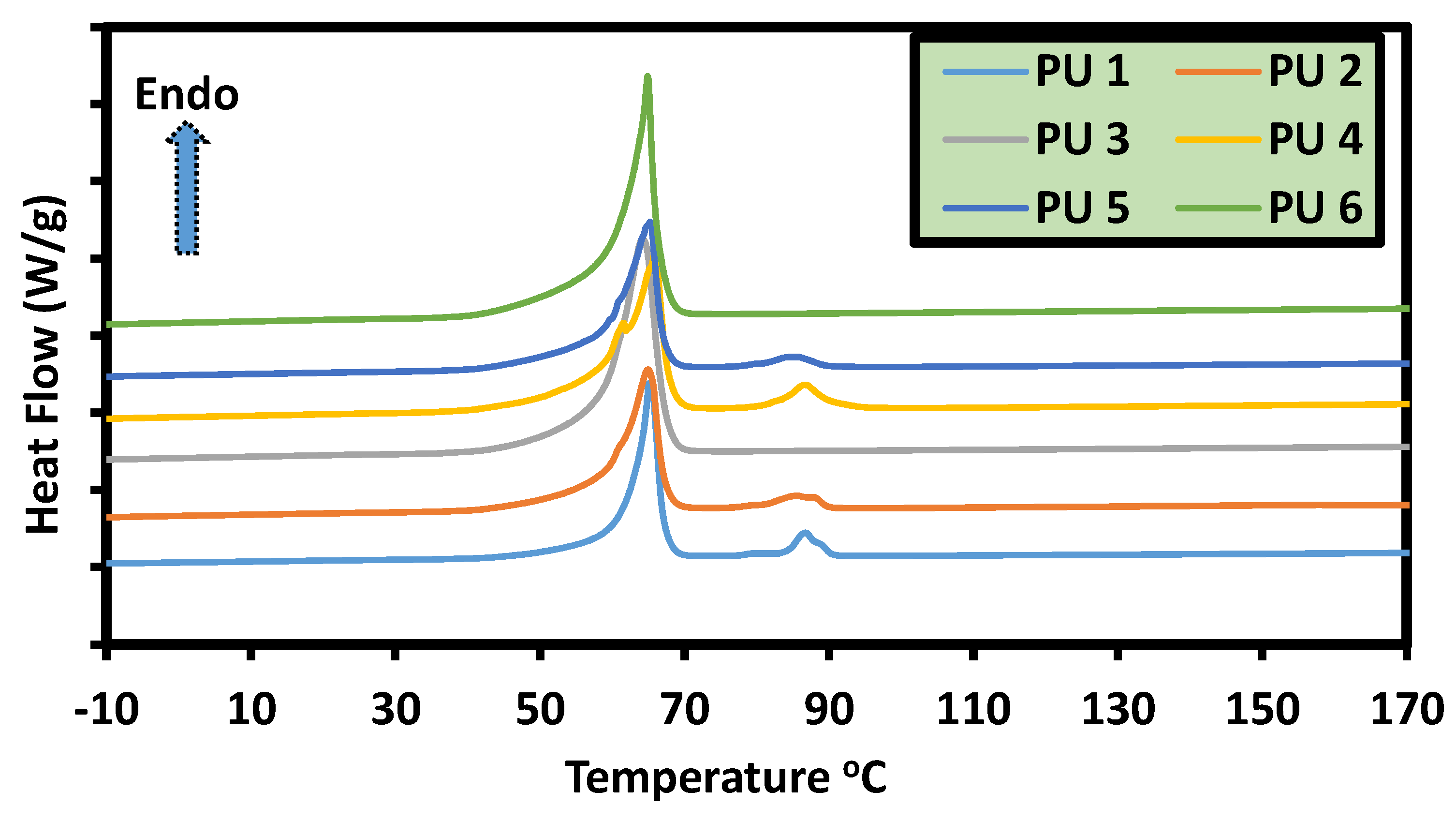
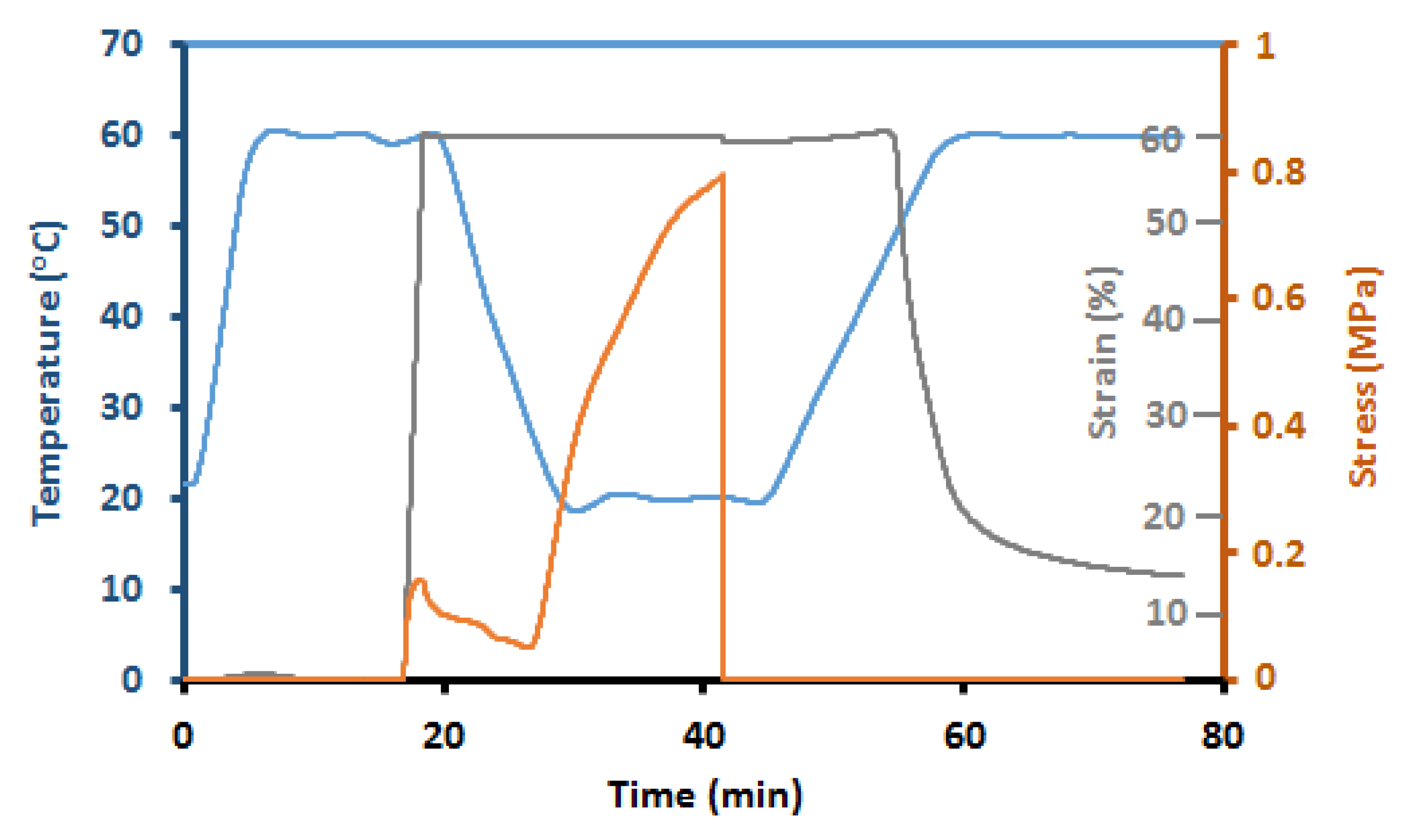
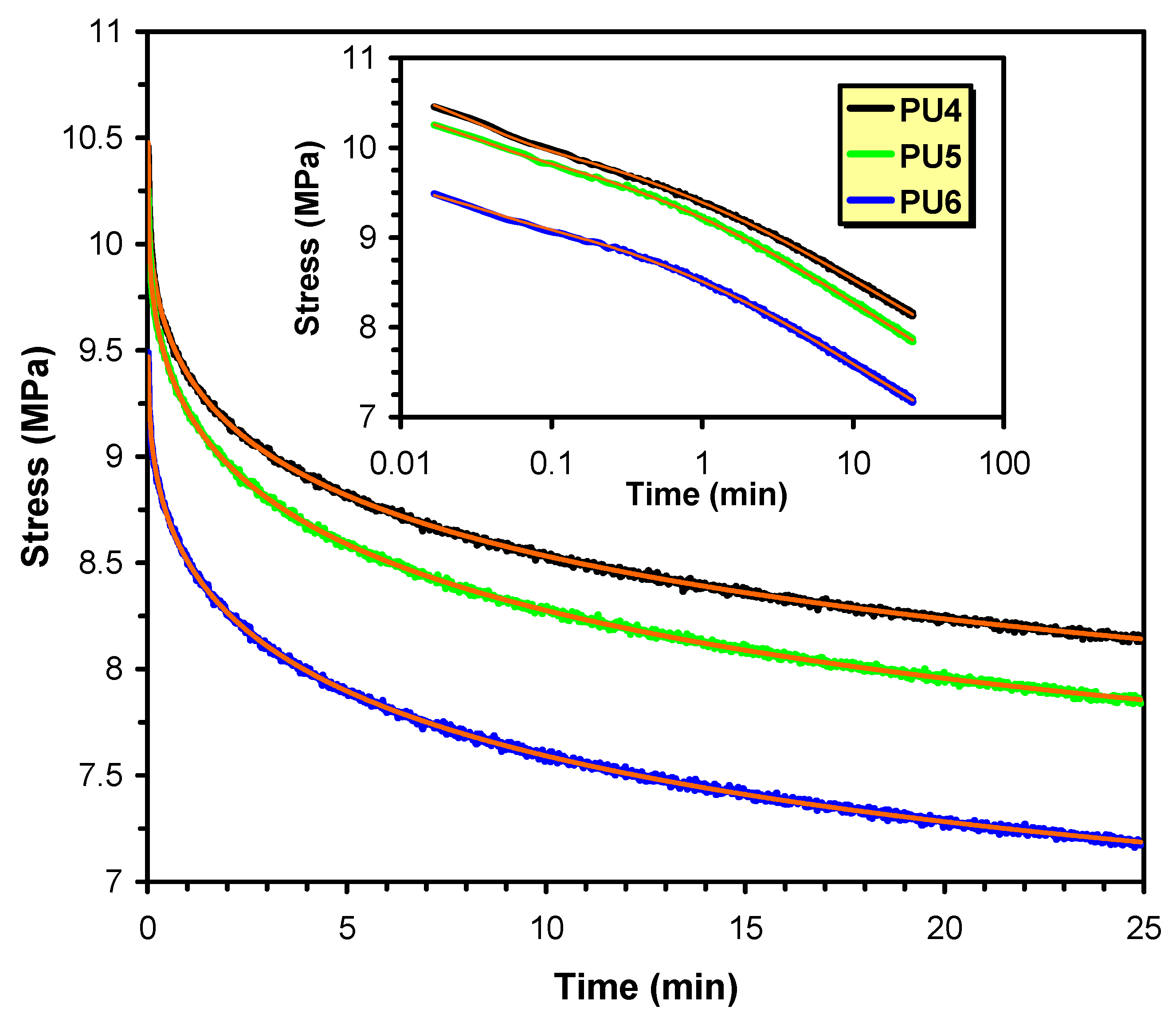
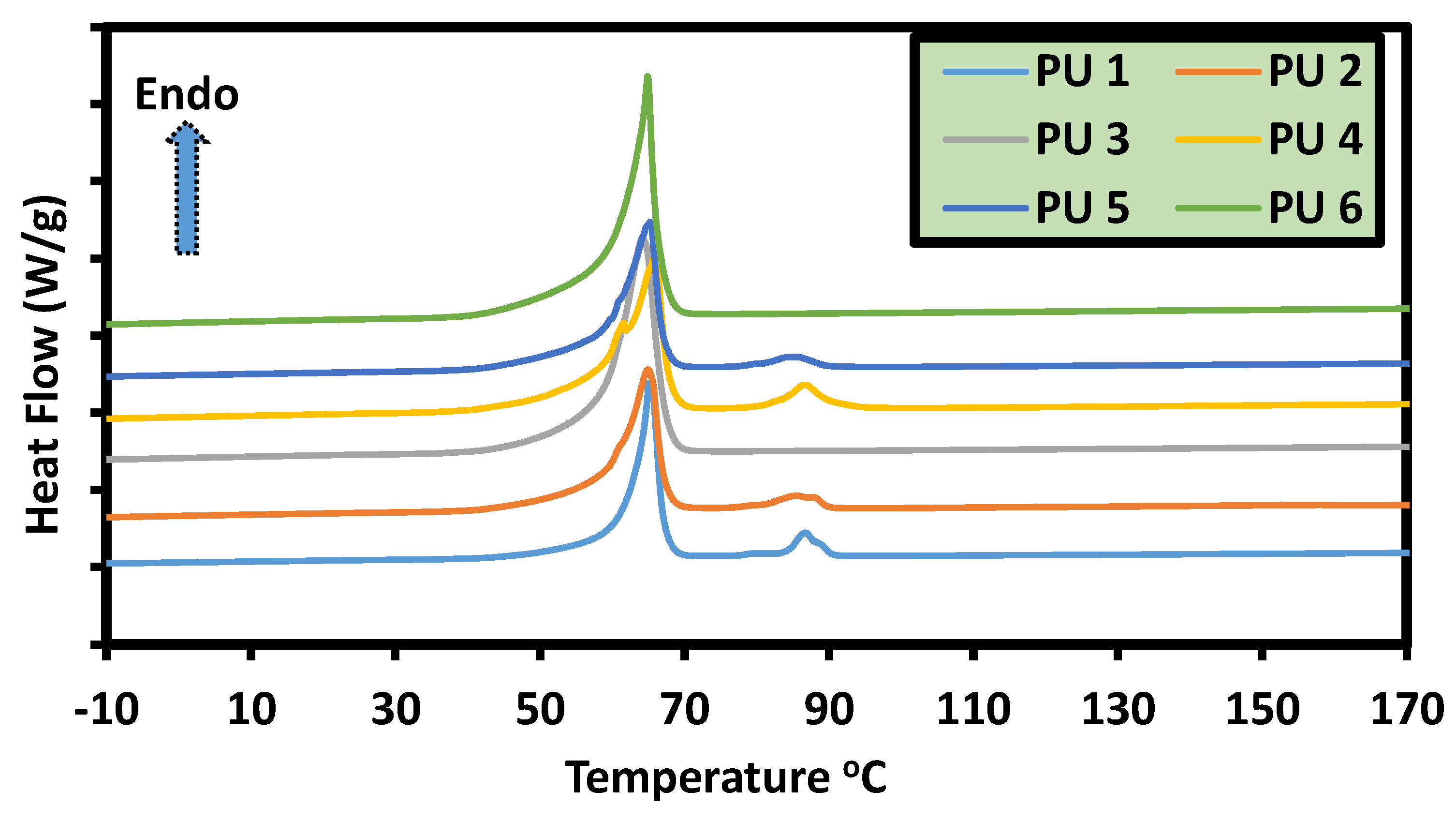
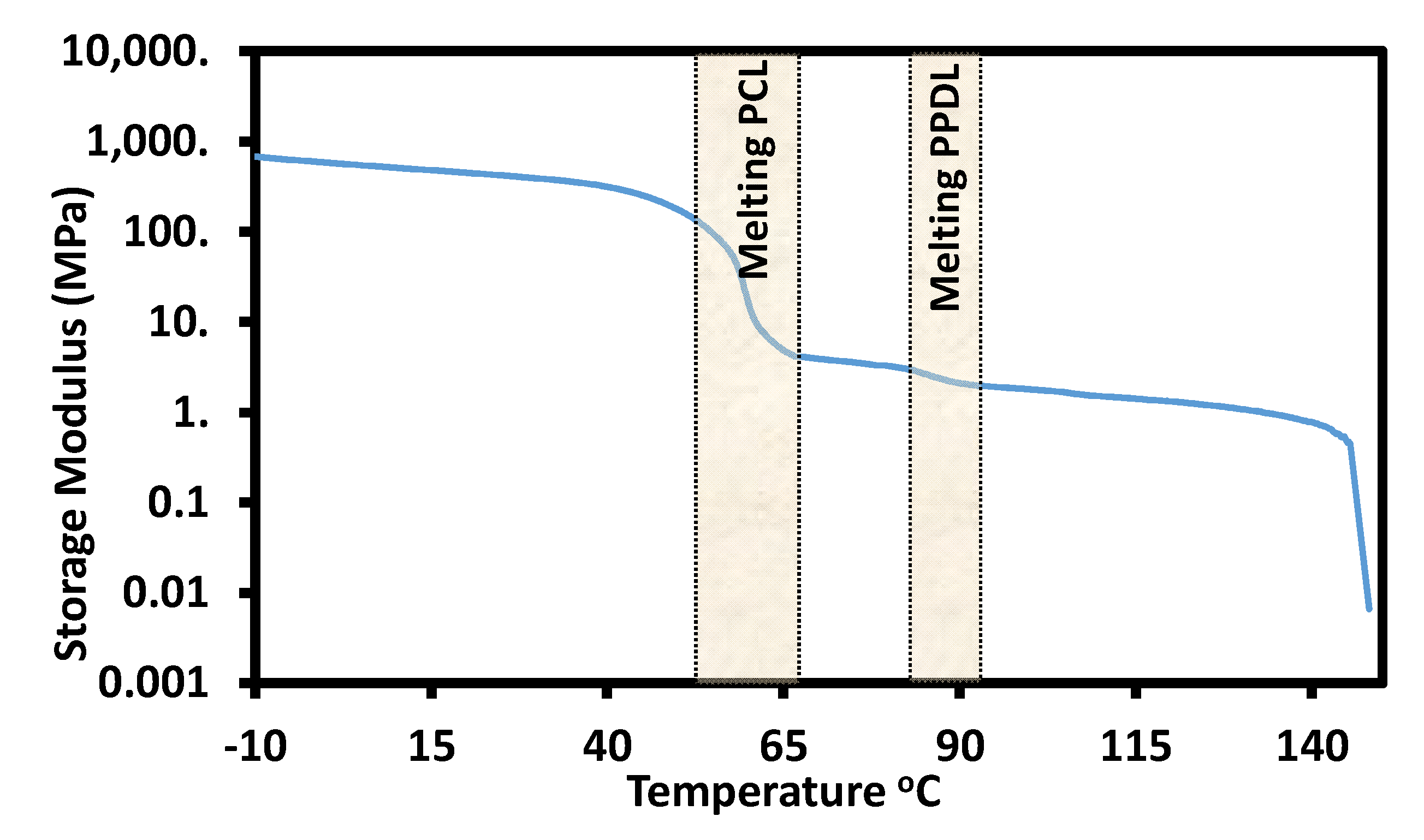
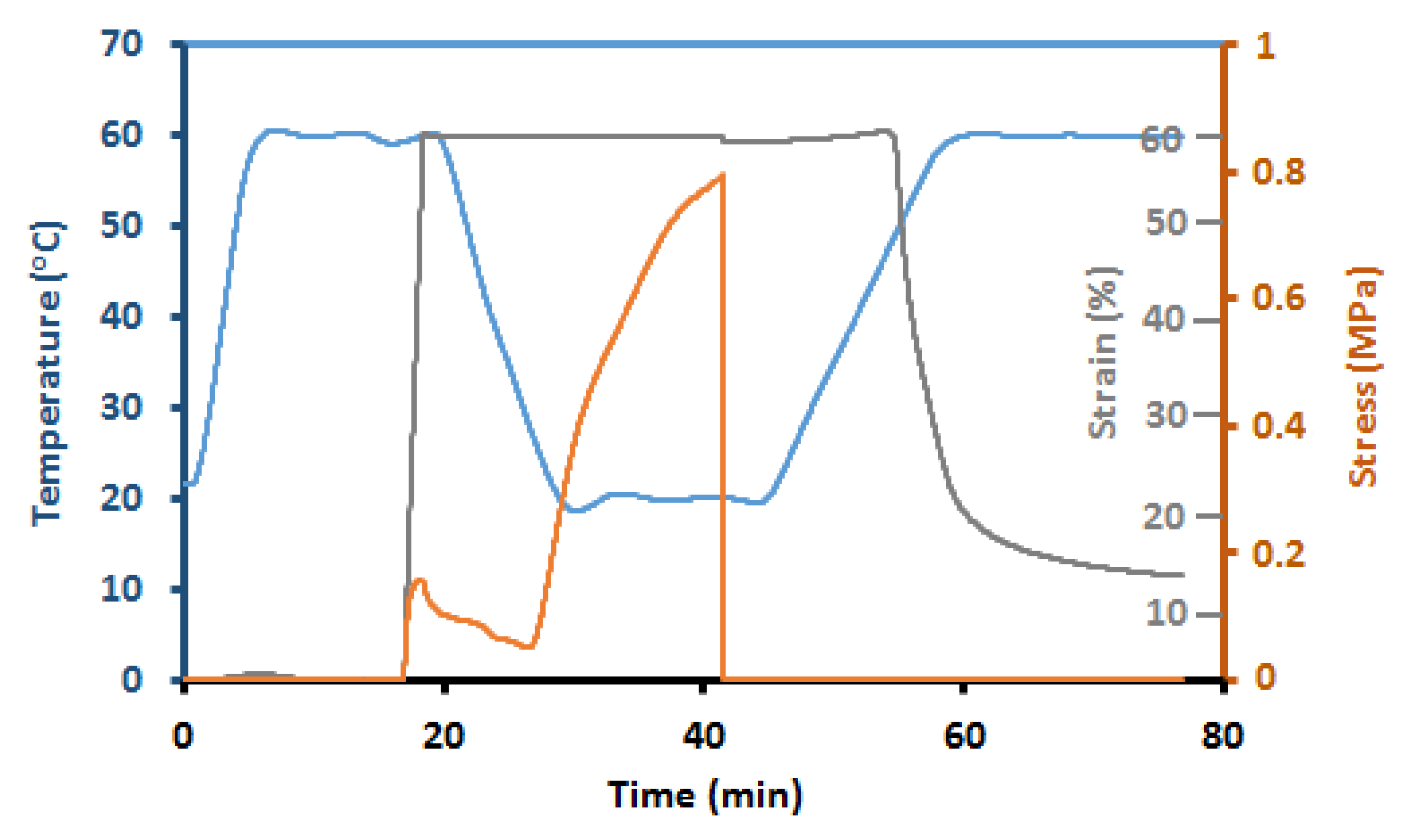
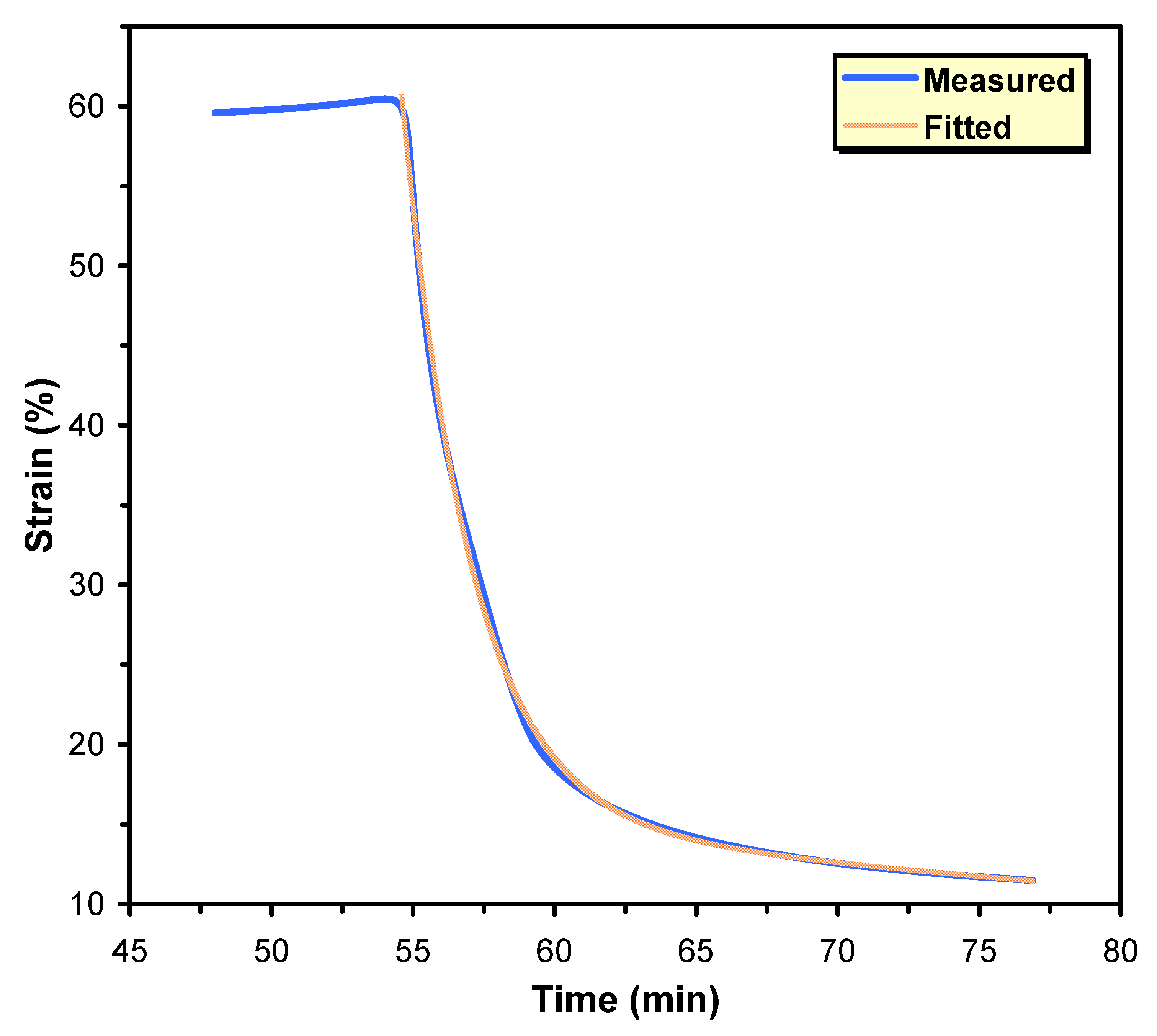
5.6. Thermal, Thermomechanical, and Shape Memory Behavior
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McGinty, D.; Letizia, C.S.; Api, A.M. Fragrance material review on ω-pentadecalactone. Food Chem. Toxicol. 2011, 49, S193–S201. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Etxeberria, A.; Varga, A.L.; Sarasua, J.-R. Synthesis and characterization of ω-pentadecalactone-co-ε-decalactone copolymers: Evaluation of thermal, mechanical and biodegradation properties. Polymer 2015, 81, 12–22. [Google Scholar] [CrossRef]
- De Geus, M.; van der Meulen, I.; Goderis, B.; van Hecke, K.; Dorschu, M.; van der Werff, H.; Koninga, C.E.; Heise, A. Performance polymers from renewable monomers: High molecular weight poly(pentadecalactone) for fiber applications. Polym. Chem. 2010, 1, 525–533. [Google Scholar] [CrossRef]
- Cai, J.; Hsiao, B.S.; Gross, R.A. Polypentadecalactone prepared by lipase catalysis: Crystallization kinetics and morphology. Polym. Int. 2009, 58, 944–953. [Google Scholar] [CrossRef]
- Van der Meulen, I.; de Geus, M.; Antheunis, H.; Deumens, R.; Joosten, E.A.J.; Koning, C.E.; Heise, A. Polymers from Functional Macrolactones as Potential. Biomaterials: Enzymatic Ring Opening Polymerization, Biodegradation, and Biocompatibility. Biomacromolecules 2008, 9, 3404–3410. [Google Scholar] [CrossRef]
- Pascual, A.; Leiza, J.R.; Mecerreyes, D. Acid catalyzed polymerization of macrolactones in bulk and aqueous miniemulsion: Ring opening vs. Condensation Eur. Polym. J. 2013, 49, 1601–1609. [Google Scholar] [CrossRef]
- Van der Mee, L.; Helmich, F.; de Bruijn, R.; Vekemans, J.; Palmans, A.; Meijer, E.W. Investigation of lipase-catalyzed ring-opening polymerizations of lactones with various ring sizes: Kinetic evaluation. Macromolecules 2006, 39, 5021–5027. [Google Scholar] [CrossRef]
- Nakayama, Y.; Watanabe, N.; Kusaba, K.; Sasaki, K.; Cai, Z.; Shiono, T.; Tsutsumi, C. High activity of rare earth tetrahydroborates for ring-opening polymerization of x-pentadecalactone. J. Appl. Polym. Sci. 2011, 121, 2098–2103. [Google Scholar] [CrossRef]
- Zhong, Z.; Dijkstra, P.J.; Feijen, J. Controlled ring-opening polymerization of x-pentadecalactone with yttrium isopropoxide as an initiator. Macromol. Chem. Phys. 2000, 201, 1329–1333. [Google Scholar] [CrossRef]
- Van der Meulen, I.; Gubbels, E.; Huijser, S.; Sablong, R.; Koning, C.E.; Heise, A.; Duchateau, R. Catalytic ring-opening polymerization of renewable macrolactones to high molecular weight polyethylene-like polymers. Macromolecules 2011, 44, 4301–4305. [Google Scholar] [CrossRef]
- Van der Meulen, I.; Li, Y.; Deumens, R.; Joosten, E.A.; Koning, C.E.; Heise, A. Copolymers from unsaturated macrolactones: Toward the design of cross-linked biodegradable polyesters. Biomacromolecules 2011, 12, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Jérôme, C.; Lecomte, P. Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization. Adv. Drug Deliv. Rev. 2008, 60, 1056–1076. [Google Scholar] [CrossRef] [PubMed]
- Panlawan, P.; Luangthongkam, P.; Wiemann, L.O.; Sieber, V.; Marie, E.; Durand, A.; Inprakhon, P. Lipase-catalyzed interfacial polymerization of ω-pentadecalactone in aqueous biphasic medium: A mechanistic study. J. Mol. Cat. B Enzym. 2013, 88, 69–76. [Google Scholar] [CrossRef]
- Walther, P.; Naumann, S. N-Heterocyclic Olefin-Based (Co)polymerization of a Challenging Monomer: Homopolymerization of ω-Pentadecalactone and Its Copolymers with γ-Butyrolactone, δ-Valerolactone, and ε-Caprolactone. Macromolecules 2017, 50, 8406–8416. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, W.; He, J.; Zhang, Y.; Chen, E.Y.-X. Living Ring-Opening Polymerization of Lactones by N-Heterocyclic Olefin/Al(C6F5)3 Lewis Pairs: Structures of Intermediates, Kinetics, and Mechanism. Macromolecules 2016, 50, 123–136. [Google Scholar] [CrossRef]
- Sonnenschein, M.F.; Lysenko, Z.; Brune, D.A.; Wendt, B.L.; Schrock, A.K. Enhancing polyurethane properties via soft segment crystallization. Polymer 2005, 46, 10158–10166. [Google Scholar] [CrossRef]
- Martin, D.J.; Meijs, G.F.; Renwick, G.M.; McCarthy, S.J.; Gunatillake, P.A. The Effect of Average Soft Segment Length on Morphology and Properties of a Series of Polyurethane Elastomers. 1. Characterization of the Series. J. App. Polym. Sci. 1996, 62, 1377–1386. [Google Scholar] [CrossRef]
- Brunette, C.M.; Hsu, S.L.; Rossman, M.; MacKnight, W.J.; Schneider, N.S. Thermal and Mechanical Properties of linear Segmented Polyurethanes with Butadiene Soft Segments. Polym. Eng. Sci. 1981, 21, 668–674. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, H.; Ishiguro, T.; Konosu, Y.; Minagawa, M.; Tanioka, A.; Richau, K.; Kratz, K.; Lendlein, A. Shape-memory properties of electrospun non-woven fabrics prepared from degradable polyesterurethanes containing poly(ω-pentadecalactone) hard segments. Eur. Polym. J. 2012, 48, 1866–1874. [Google Scholar] [CrossRef] [Green Version]
- Karger-Kocsis, J.; Kéki, S. Biodegradable polyester-based shape memory polymers:Concepts of (supra)molecular architecturing. Exp. Polym. Lett. 2014, 8, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Nguyen, T.D. Thermo-mechanics of Amorphous Shape-Memory Polymers. Proc. IUTAM 2015, 12, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Schöne, A.-C.; Kratz, K.; Schulz, B.; Lendlein, A. The relevance of hydrophobic segments in multiblock copolyesterurethanes for their enzymatic degradation at the air-water interface. Polymer 2016, 102, 92–98. [Google Scholar] [CrossRef]
- Guelcher, S.A. Biodegradable Polyurethanes: Synthesis and Applications in Regenerative Medicine. Tissue Eng. Part B 2008, 14, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Gorna, K.; Polowinski, S.; Gogolewski, S. Synthesis and Characterization of Biodegradable Poly(ε-caprolactone urethane)s. I. Effect of the Polyol Molecular Weight, Catalyst, and Chain Extender on the Molecular and Physical Characteristics. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 156–170. [Google Scholar] [CrossRef]
- Jiang, X.; Li, J.; Ding, M.; Tan, H.; Ling, Q.; Zhong, Y.; Fu, Q. Synthesis and degradation of nontoxic biodegradable waterborne polyurethanes elastomer with poly(ε-caprolactone) and poly(ethylene glycol) as soft segment. Eur. Polym. J. 2007, 43, 1838–1846. [Google Scholar] [CrossRef]
- Jiang, S.; Ji, X.; An, L.; Jiang, B. Crystallization behavior of PCL in hybrid confined environment. Polymer 2001, 42, 3901–3907. [Google Scholar] [CrossRef]
- Crescenzi, V.; Manzini, G.; Calzolari, B.; Borri, C. Thermodynamics of fusion of poly-β-propiolactone and poly-ε-caprolactone. Comparative analysis of the melting of aliphatic polylactone and polyester chains. Eur. Polym. J. 1972, 8, 449–463. [Google Scholar] [CrossRef]
- Focarete, M.L.; Scandola, M.; Kumar, A.; Gross, R.A. Physical Characterization of Poly(ω-pentadecalactone) Synthesized by Lipase-Catalyzed Ring-Opening Polymerization. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 1721–1729. [Google Scholar] [CrossRef]
- Báez, J.E.; Marcos-Fernández, Á.; Martínez-Richa, A.; Galindo-Iranzo, P. Poly(ε-caprolactone) Diols (HOPCLOH) and Their Poly(ester-urethanes) (PEUs): The Effect of Linear Aliphatic Diols [HO–(CH2)m–OH] as Initiators. Polym. Plast. Technol. Eng. 2017, 56, 889–898. [Google Scholar]
- Stjerndahl, A.; Wistrand, A.F.; Albertsson, A.-C. Industrial Utilization of Tin-Initiated Resorbable Polymers: Synthesis on a Large Scale with a Low Amount of Initiator Residue. Biomacromolecules 2007, 8, 937–940. [Google Scholar] [CrossRef]
- Borda, J.; Bodnár, I.; Kéki, S.; Sipos, L.; Zsuga, M. Optimum conditions for the synthesis of linear polylactic acid based urethanes. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2925–2933. [Google Scholar] [CrossRef]
- Kima, K.S.; Ryu, C.M.; Parkc, C.S.; Surd, G.S.; Parka, C.E. Investigation of crystallinity effects on the surface of oxygen plasma. treated low density polyethylene using X-ray photoelectron spectroscopy. Polymer 2003, 44, 6287–6295. [Google Scholar] [CrossRef]
- Paajanen, A.; Vaari, J.; Verho, T. Crystallization of cross-linked polyethylene by molecular dynamics simulation. Polymer 2019, 171, 80–86. [Google Scholar] [CrossRef]
- Lakatos, C.; Czifrák, K.; Karger-Kocsis, J.; Daróczi, L.; Zsuga, M.; Kéki, S. Shape memory crosslinked polyurethanes containing thermoreversible. Diels-Alder couplings. J. Appl. Polym. Sci. 2016, 133, 44145. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Composition in the Feed/Molar Ratio |
|---|---|
| PU 1 | PCL(50)-HDI-(PPDL-1)/1-3-1 |
| PU 2 | PCL(50)-HDI-(PPDL-2)/1-3-1 |
| PU 3 | PCL(50)-HDI-(PPDL-3)/1-3-1 |
| PU 4 | PCL(50)-HDI-(PPDL-1)-HDI/1-3-1-1 |
| PU 5 | PCL(50)-HDI-(PPDL-2)-HDI/1-3-1-1 |
| PU 6 | PCL(50)-HDI-(PPDL-3)-HDI/1-3-1-1 |
| Sample | PDL/Initiator (mol ratio) a | Catalyst (mol%) | Conv. (%) b | DP b,c | Mn (NMR) b,c |
|---|---|---|---|---|---|
| 1 | 10/1 | 0.1 | 9.1 | 4.4 | 1150 |
| 2 | 20/1 | 0.1 | 12.2 | 6.5 | 1650 |
| 3 | 30/1 | 0.1 | 13.5 | 18.2 | 4460 |
| 4 | 40/1 | 0.1 | 12.9 | 12 | 2970 |
| 5 | 50/1 | 0.1 | 10.8 | 17.4 | 4270 |
| 6 | 10/1 | 0.5 | 24 | 10 | 2490 |
| 7 | 10/1 | 1.0 | 31.4 | 12.4 | 3070 |
| 8 | 10/1 | 1.5 | 80.6 | 22 | 5370 |
| 9 | 10/1 | 2.0 | 85 | 22 | 5370 |
| 10 | 10/1 | 2.5 | 80 | 20.2 | 4940 |
| Sample | Initiator a | Conv.(%) (NMR) b | DPn | Mn (NMR) c | Mn (GPC) | Mw/Mn |
|---|---|---|---|---|---|---|
| PPDL-1 | BDO | 92.6 | 20 | 4890 * | - | - |
| PPDL-2 | PEG200 | ~100 | 12 | 2990 * | - | - |
| PPDL-3 | PCD | ~100 | - | - | 5880 ** | 1.5 |
| Code | E (MPa) | εm (%) | σm (MPa) | εR (%) | σR (MPa) |
|---|---|---|---|---|---|
| PU 1 | 427 ± 52 | 8.6 ± 1.8 | 18.5 ± 5.4 | 428 ± 26 | 21.5 ± 3.2 |
| PU 2 | 397 ± 35 | 9.9 ± 0.4 | 18.5 ± 1.9 | 596 ± 125 | 29.3 ± 4.1 |
| PU 3 | 292 ± 4 | 9.4 ± 0.1 | 15.3 ± 0.3 | 749 ± 13 | 30.8 ± 1.1 |
| PU 4 | 235 ± 17 | 11.5 ± 1.3 | 12.4 ± 0.8 | 892 ± 122 | 29.9 ± 3.2 |
| PU 5 | 329 ± 35 | 10.6 ± 0.2 | 15.9 ± 1.3 | 627 ± 127 | 26.2 ± 1.5 |
| PU 6 | 334 ± 2 | 8.9 ± 0.5 | 14.8 ± 1.1 | 756 ± 126 | 27.2 ± 0.5 |
| Code | Tm(°C) | Hm (J/g) | Cr(PCL) (%) | Tm(°C) | Hm (J/g) | Cr (PPDL) (%) |
|---|---|---|---|---|---|---|
| PU 1 | 65.3 | 87.7 | 70.7 | 86.7 | 9.7 | 91.6 |
| PU 2 | 64.9 | 84.4 | 66.7 | 85.5 | 6.8 | 87.8 |
| PU 3 | 64.2 | 96.0 | 76.0 | - | - | - |
| PU 4 | 65.9 | 75.5 | 61.0 | 86.5 | 8.7 | 83.4 |
| PU 5 | 65.2 | 84.8 | 65.6 | 85.4 | 5.0 | 64.5 |
| PU 6 | 64.9 | 104.4 | 82.8 | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czifrák, K.; Lakatos, C.; Árpád Kordován, M.; Nagy, L.; Daróczi, L.; Zsuga, M.; Kéki, S. Block Copolymers of Poly(ω-Pentadecalactone) in Segmented Polyurethanes: Novel Biodegradable Shape Memory Polyurethanes. Polymers 2020, 12, 1928. https://doi.org/10.3390/polym12091928
Czifrák K, Lakatos C, Árpád Kordován M, Nagy L, Daróczi L, Zsuga M, Kéki S. Block Copolymers of Poly(ω-Pentadecalactone) in Segmented Polyurethanes: Novel Biodegradable Shape Memory Polyurethanes. Polymers. 2020; 12(9):1928. https://doi.org/10.3390/polym12091928
Chicago/Turabian StyleCzifrák, Katalin, Csilla Lakatos, Marcell Árpád Kordován, Lajos Nagy, Lajos Daróczi, Miklós Zsuga, and Sándor Kéki. 2020. "Block Copolymers of Poly(ω-Pentadecalactone) in Segmented Polyurethanes: Novel Biodegradable Shape Memory Polyurethanes" Polymers 12, no. 9: 1928. https://doi.org/10.3390/polym12091928
APA StyleCzifrák, K., Lakatos, C., Árpád Kordován, M., Nagy, L., Daróczi, L., Zsuga, M., & Kéki, S. (2020). Block Copolymers of Poly(ω-Pentadecalactone) in Segmented Polyurethanes: Novel Biodegradable Shape Memory Polyurethanes. Polymers, 12(9), 1928. https://doi.org/10.3390/polym12091928