Facile Synthesis of Mixed-Mode Weak Anion-Exchange Microspheres via One-Step Pickering Emulsion Polymerization for Efficient Simultaneous Extraction of Strongly and Weakly Acidic Drugs from Reservoir Water

Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals and Materials
2.2. Preparation of Mixed-Mode WAX Microspheres
2.3. Sample Collection
2.4. Characterization of the Sorbent
2.5. Chromatographic Analysis
3. Results and Discussion
3.1. Preparation of the Poly(DEAEMA-co-DVB) Microspheres
Characterization of the Poly(DEAEMA-co-DVB) Microspheres
3.2. Optimization of SPE Procedures
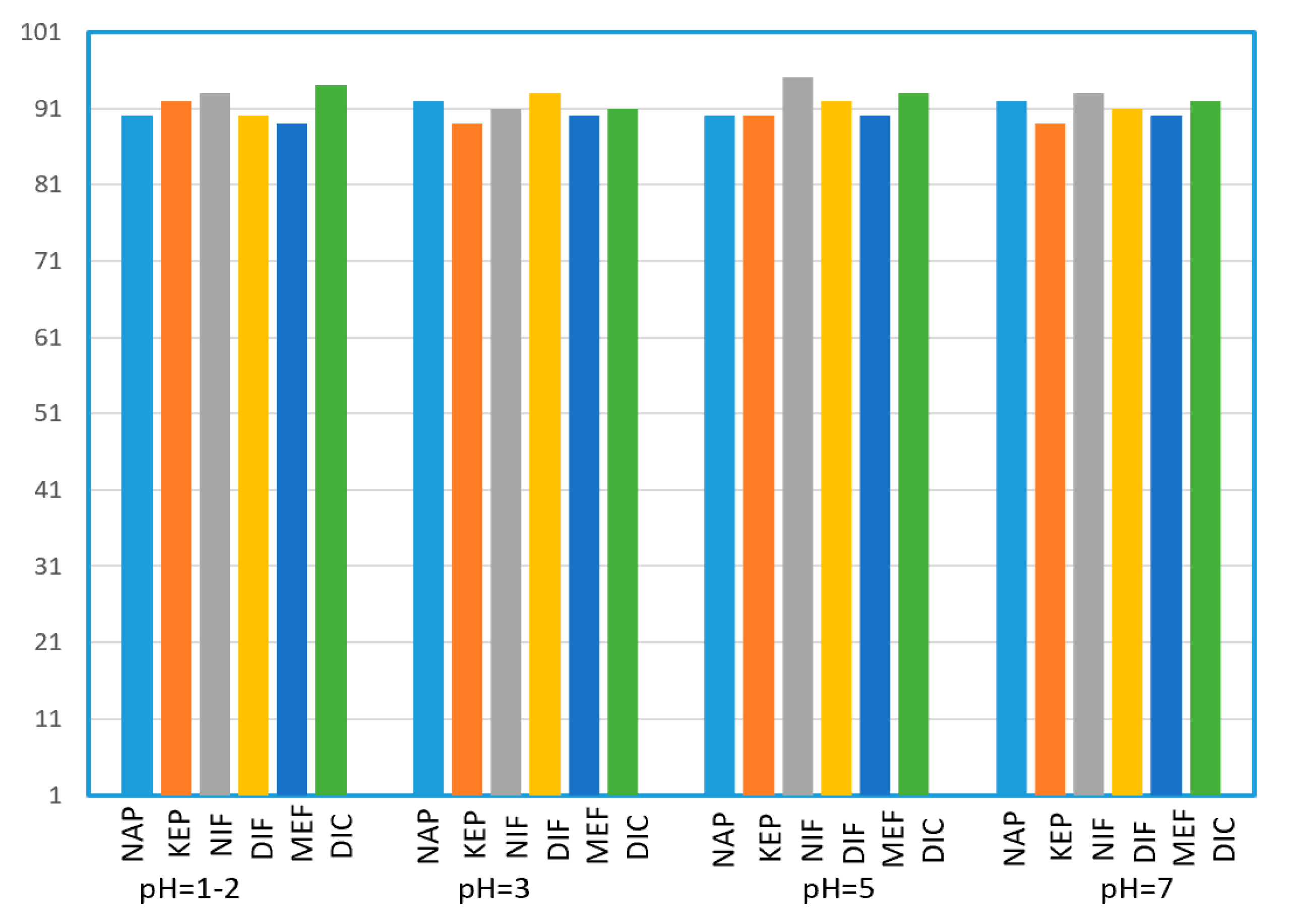
3.2.1. Effect of Sample pH
3.2.2. Effect of Elution Volume
3.2.3. Sample Breakthrough Volume
3.3. Validation of the WAX Mixed-Mode SPE/HPLC-UV Method
3.4. Analysis of Real Samples
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fu, X.; Liao, Y.; Liu, H. Sample preparation for pharmaceutical analysis. Anal. Bioanal. Chem. 2005, 381, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Qiao, Y.; Xing, J. Ternary mixed-mode silica sorbent of solid-phase extraction for determination of basic, neutral and acidic drugs in human serum. Anal. Bioanal. Chem. 2018, 410, 3731–3742. [Google Scholar] [CrossRef]
- Augusto, F.; Hantao, L.W.; Mogollón, N.G.S.; Braga, S.C.G.N. New materials and trends in sorbents for solid-phase extraction. Trac Trends Anal. Chem. 2013, 43, 14–23. [Google Scholar] [CrossRef]
- Plotka-Wasylka, J.; Szczepanska, N.; Miguel, D.L.G.; Namiesnik, J. Modern trends in solid phase extraction: New sorbent media. Trac Trends Anal. Chem. 2016, 77, 23–43. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, Z.; Lv, S.; Du, Z.; Liu, X. Simultaneous determination of 16 polycyclic aromatic hydrocarbons in reclaimed water using solid-phase extraction followed by ultra-performance convergence chromatography with photodiode array detection. J. Sep. Sci. 2016, 39, 993–999. [Google Scholar] [CrossRef]
- Picó, Y.; Fernández, M.; Ruiz, M.J.; Font, G. Current trends in solid-phase-based extraction techniques for the determination of pesticides in food and environment. J. Biochem. Biophys. Methods 2008, 70, 117–131. [Google Scholar] [CrossRef]
- Clarke, S.J.; Rivory, L.P.; Li, K.M. Solid-phase extraction (SPE) techniques for sample preparation in clinical and pharmaceutical analysis: A brief overview. Curr. Pharm. Anal. 2006, 2, 95–102. [Google Scholar]
- Calvo, M.V.; Ramos, L.; Fontecha, J. Determination of cholesterol oxides content in milk products by solid phase extraction and gas chromatography-mass spectrometry. J. Sep. Sci. 2003, 26, 927–931. [Google Scholar] [CrossRef]
- Bing, S.; Dan, D.; Wu, Y.; Hu, J.; Meng, J.; Tu, X. Simultaneous determination of 17 sulfonamide residues in porcine meat, kidney and liver by solid-phase extraction and liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2005, 546, 174–181. [Google Scholar]
- Leitner, A.; Zöllner, P.; Lindner, W. Determination of the metabolites of nitrofuran antibiotics in animal tissue by high-performance liquid chromatography–tandem mass spectrometry. Food Sci. 2008, 939, 49–58. [Google Scholar] [CrossRef]
- Chen, H.C.; Wang, S.P.; Ding, W.H. Determination of fluorescent whitening agents in environmental waters by solid-phase extraction and ion pair liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2006, 1102, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Stubbings, G.; Tarbin, J.; Cooper, A.; Sharman, M.; Bigwood, T.; Robb, P. A multi-residue cation-exchange clean up procedure for basic drugs in produce of animal origin. Anal. Chim. Acta 2005, 547, 262–268. [Google Scholar] [CrossRef]
- Jacobsen, A.M.; Halling-Sorensen, B.; Ingerslev, F.; Steen, H.H. Simultaneous extraction of tetracycline, macrolide and sulfonamide antibiotics from agricultural soils using pressurised liquid extraction, followed by solid-phase extraction and liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2004, 1038, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Nazario, C.E.D.; Lancas, F.M. Analysis of fluoxetine and norfluoxetine in human plasma by HPLC-UV using a high purity C18 silica-based SPE sorbent. Anal. Methods 2014, 6, 4181–4187. [Google Scholar] [CrossRef]
- Núria, F.; Rosa, M.M.; Borrull, F.; Cormack, P.A.G. Mixed-mode ion-exchange polymeric sorbents: Dual-phase materials that improve selectivity and capacity. Trac Trends Anal. Chem. 2010, 29, 765–779. [Google Scholar]
- Cai, X.; Guo, Z.; Xue, X.; Xu, J.; Zhang, X.; Liang, X. Two-dimensional liquid chromatography separation of peptides using reversed-phase/weak cation-exchange mixed-mode column in first dimension. J. Chromatogr. A 2012, 1228, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, Z.; Sheng, Q.; Xue, X.; Liang, X. Sequential elution of multiply and singly phosphorylated peptides with polar-copolymerized mixed-mode RP18/SCX material. Analyst 2012, 137, 2774–2776. [Google Scholar] [CrossRef] [PubMed]
- Núria, F.; Cormack, P.A.G.; Sherrington, D.C.; Rosa, M.M.; Borrull, F. Weak anion-exchange hypercrosslinked sorbent in on-line solid-phase extraction–liquid chromatography coupling to achieve automated determination with an effective clean-up. J. Chromatogr. A 2010, 1217, 2855–2861. [Google Scholar]
- Hu, P.; Zhang, H.Y.; Mao, D.Y.; Ning, F.H.; Wang, R.J.; Wang, Y.R. Determination of homovanilic acid in human urine by weak anion-exchange hypercrosslinked polymer resin-solid phase extraction-high performance liquid chromatography-ultraviolet detection method. Chin. J. Anal. Chem. 2012, 40, 1175–1180. [Google Scholar] [CrossRef]
- Huang, C.; Li, Y.; Yang, J.; Peng, J.; Jin, J.; Dhanjai; Wang, J.; Chen, J. Preparation of a reversed-phase/anion-exchange mixed-mode spherical sorbent by pickering emulsion polymerization for highly selective solid-phase extraction of acidic pharmaceuticals from wastewater. J. Chromatogr. A 2017, 1521, 1–9. [Google Scholar] [CrossRef]
- He, J.; Ma, Z.; Yang, Y.; Hemar, Y.; Zhao, T. Extraction of tetracycline in food samples using biochar microspheres prepared by a pickering emulsion method. Food Chem. 2020, 329, 127162. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Huang, C.; Jiao, Y.; Chen, J. A phenolphthalein-dummy template molecularly imprinted polymer for highly selective extraction and clean-up of bisphenol a in complex biological, environmental and food samples. Polymers 2018, 10, 1150. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SBET (m2 g−1) | Pore Volume (cm3 g−1) | Pore Size (nm) | |
|---|---|---|---|
| Poly(DVB-co-DEAEMA) | 575 | 0.94 | 10–30 |
| Pharmaceutical | CAS Number | Structure | pKa | Log P | Water Solubility (mg L−1) |
|---|---|---|---|---|---|
| niflumic acid | 4394-00-7 |  | 1.68 | 4.43 | 19 |
| naproxen | 22204-53-1 |  | 4.15 | 3.18 | 15.9 |
| diflunisal | 22494-42-4 |  | 3.3 | 4.44 | 14.5 |
| diclofenac | 15307-86-5 |  | 4.15 | 4.51 | 2.37 |
| ketoprofen | 22071-15-4 |  | 4.45 | 3.12 | 51 |
| mefenamic acid | 61-68-7 |  | 4.2 | 5.12 | 20 |
| Analyte | Liner Range (μg L−1) | Correlation of Determination (R2) | LOD a (μg L−1) | LOQ a (μg L−1) | Intra-Day RSD (%, n = 3) | Inter-Day RSD (%, n = 3) |
|---|---|---|---|---|---|---|
| naproxen | 0.01–10.0 | 0.995 | 0.011 | 0.032 | 0.3 | 3.3 |
| ketoprofen | 0.05–10.0 | 0.994 | 0.025 | 0.044 | 0.5 | 2.7 |
| diflunisal | 0.05–10.0 | 0.996 | 0.018 | 0.076 | 0.9 | 3.1 |
| mefenamic acid | 0.05–10.0 | 0.995 | 0.016 | 0.088 | 0.9 | 2.8 |
| diclofenac | 0.05–10.0 | 0.997 | 0.008 | 0.006 | 1.3 | 4.0 |
| niflumic acid | 0.05–10.0 | 0.997 | 0.002 | 0.003 | 2.1 | 3.3 |
| Analyte | Spiked Levels a | |||
|---|---|---|---|---|
| (1) | (2) | |||
| Eluate b | RSD% | Eluate | RSD% | |
| naproxen | 101.3 | 2.7 | 92.0 | 3.6 |
| ketoprofen | 93.3 | 1.8 | 101.7 | 4.4 |
| diflunisal | 89.5 | 7.7 | 98.4 | 5.6 |
| mefenamic acid | 92.4 | 1.4 | 93.6 | 7.2 |
| diclofenac | 96.6 | 2.2 | 88.3 | 2.7 |
| niflumic acid | 90.6 | 4.6 | 90.1 | 3.5 |
| Analyte | Poly(DEAEMA-co-DVB) a | Oasis MAX a | C18 a |
|---|---|---|---|
| naproxen | 90 | 93 | 70 |
| ketoprofen | 98 | 93 | 73 |
| diflunisal | 92 | 90 | 66 |
| mefenamic acid | 89 | 88 | 83 |
| diclofenac | 88 | 90 | 62 |
| niflumic acid | 93 | 7 | 69 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gou, X.; Li, Y.; Huang, C.; Zhu, X.; Chen, J. Facile Synthesis of Mixed-Mode Weak Anion-Exchange Microspheres via One-Step Pickering Emulsion Polymerization for Efficient Simultaneous Extraction of Strongly and Weakly Acidic Drugs from Reservoir Water. Polymers 2020, 12, 2089. https://doi.org/10.3390/polym12092089
Gou X, Li Y, Huang C, Zhu X, Chen J. Facile Synthesis of Mixed-Mode Weak Anion-Exchange Microspheres via One-Step Pickering Emulsion Polymerization for Efficient Simultaneous Extraction of Strongly and Weakly Acidic Drugs from Reservoir Water. Polymers. 2020; 12(9):2089. https://doi.org/10.3390/polym12092089
Chicago/Turabian StyleGou, Xiaoyi, Yun Li, Chaonan Huang, Xiuhua Zhu, and Jiping Chen. 2020. "Facile Synthesis of Mixed-Mode Weak Anion-Exchange Microspheres via One-Step Pickering Emulsion Polymerization for Efficient Simultaneous Extraction of Strongly and Weakly Acidic Drugs from Reservoir Water" Polymers 12, no. 9: 2089. https://doi.org/10.3390/polym12092089
APA StyleGou, X., Li, Y., Huang, C., Zhu, X., & Chen, J. (2020). Facile Synthesis of Mixed-Mode Weak Anion-Exchange Microspheres via One-Step Pickering Emulsion Polymerization for Efficient Simultaneous Extraction of Strongly and Weakly Acidic Drugs from Reservoir Water. Polymers, 12(9), 2089. https://doi.org/10.3390/polym12092089