
Effect of Solution Viscosity on the Precipitation of PSaMA in Aqueous Phase Separation-Based Membrane Formation



Abstract
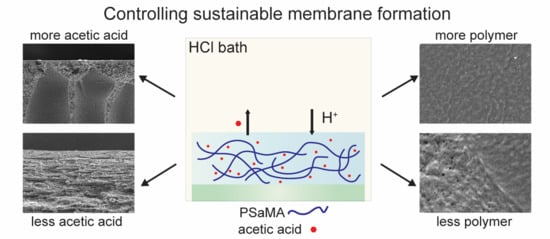
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Membrane Preparation
2.3. Membrane Performance Tests
2.4. Scanning Electron Microscopy (SEM)
2.5. Dynamic Viscosity Measurements
3. Results and Discussion
3.1. Reduced Acid Concentration
3.2. Polymer Concentration
3.3. Acetic Acid Concentration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loeb, S.; Sourirajan, S. Sea water demineralization by means of an osmotic membrane. Adv. Chem. 1962, 38, 117–132. [Google Scholar]
- Strathmann, H.; Kock, K. Formation Mechanism of Phase Inversion Membranes. Desalination 1977, 21, 241–255. [Google Scholar] [CrossRef]
- Strathmann, H.; Giorno, L.; Drioli, E. Introduction to Membrane Science and Technology; Wiley: Weinheim, Germany, 2011. [Google Scholar]
- Guillen, G.R.; Pan, Y.J.; Li, M.H.; Hoek, E.M.V. Preparation and Characterization of Membranes Formed by Nonsolvent Induced Phase Separation: A Review. Ind. Eng. Chem. Res. 2011, 50, 3798–3817. [Google Scholar] [CrossRef]
- Baker, R.W. Membrane Technology and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Mulder, J. Basic Principles of Membrane Technology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Strathmann, H.; Kock, K.; Amar, P.; Baker, R.W. Formation Mechanism of Asymmetric Membranes. Desalination 1975, 16, 179–203. [Google Scholar] [CrossRef]
- Mosqueda-Jimenez, D.B.; Narbaitz, R.M.; Matsuura, T.; Chowdhury, G.; Pleizier, G.; Santerre, J.P. Influence of processing conditions on the properties of ultrafiltration membranes. J. Membr. Sci. 2004, 231, 209–224. [Google Scholar] [CrossRef]
- Boom, R.; Wienk, I.; Van den Boomgaard, T.; Smolders, C. Microstructures in phase inversion membranes. Part 2. The role of a polymeric additive. J. Membr. Sci. 1992, 73, 277–292. [Google Scholar] [CrossRef]
- Ma, Y.; Shi, F.; Ma, J.; Wu, M.; Zhang, J.; Gao, C. Effect of PEG additive on the morphology and performance of polysulfone ultrafiltration membranes. Desalination 2011, 272, 51–58. [Google Scholar] [CrossRef]
- Yoo, S.H.; Kim, J.H.; Jho, J.Y.; Won, J.; Kang, Y.S. Influence of the addition of PVP on the morphology of asymmetric polyimide phase inversion membranes: Effect of PVP molecular weight. J. Membr. Sci. 2004, 236, 203–207. [Google Scholar] [CrossRef]
- Sukitpaneenit, P.; Chung, T.-S. Molecular elucidation of morphology and mechanical properties of PVDF hollow fiber membranes from aspects of phase inversion, crystallization and rheology. J. Membr. Sci. 2009, 340, 192–205. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Z. Preparation and characterization of polyacrylonitrile ultrafiltration membranes. J. Membr. Sci. 2003, 222, 87–98. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Jeong, B.-H.; Huang, X.; Hoek, E.M.V. Impacts of reaction and curing conditions on polyamide composite reverse osmosis membrane properties. J. Membr. Sci. 2008, 311, 34–45. [Google Scholar] [CrossRef]
- Zheng, Q.-Z.; Wang, P.; Yang, Y.-N. Rheological and thermodynamic variation in polysulfone solution by PEG introduction and its effect on kinetics of membrane formation via phase-inversion process. J. Membr. Sci. 2006, 279, 230–237. [Google Scholar] [CrossRef]
- Tsai, H.A.; Li, L.D.; Lee, K.R.; Wang, Y.C.; Li, C.L.; Huang, J.; Lai, J.Y. Effect of surfactant addition on the morphology and pervaporation performance of asymmetric polysulfone membranes. J. Membr. Sci. 2000, 176, 97–103. [Google Scholar] [CrossRef]
- Nunes, S.P.; Culfaz-Emecen, P.Z.; Ramon, G.Z.; Visser, T.; Koops, G.H.; Jin, W.; Ulbricht, M. Thinking the future of membranes: Perspectives for advanced and new membrane materials and manufacturing processes. J. Membr. Sci. 2019, 117761. [Google Scholar] [CrossRef]
- Figoli, A.; Marino, T.; Simone, S.; Di Nicolo, E.; Li, X.M.; He, T.; Tornaghi, S.; Drioli, E. Towards non-toxic solvents for membrane preparation: A review. Green Chem. 2014, 16, 4034–4059. [Google Scholar] [CrossRef]
- Razali, M.; Kim, J.F.; Attfield, M.; Budd, P.M.; Drioli, E.; Lee, Y.M.; Szekely, G. Sustainable wastewater treatment and recycling in membrane manufacturing. Green Chem. 2015, 17, 5196–5205. [Google Scholar] [CrossRef]
- Alexowsky, C.; Bojarska, M.; Ulbricht, M. Porous poly(vinylidene fluoride) membranes with tailored properties by fast and scalable non-solvent vapor induced phase separation. J. Membr. Sci. 2019, 577, 69–78. [Google Scholar] [CrossRef]
- Huang, Q.; Seibig, B.; Paul, D. Polycarbonate hollow fiber membranes by melt extrusion. J. Membr. Sci. 1999, 161, 287–291. [Google Scholar] [CrossRef]
- Kim, D.; Salazar, O.R.; Nunes, S.P. Membrane manufacture for peptide separation. Green Chem. 2016, 18, 5151–5159. [Google Scholar] [CrossRef]
- Marino, T.; Blasi, E.; Tornaghi, S.; Di Nicolò, E.; Figoli, A. Polyethersulfone membranes prepared with Rhodiasolv®Polarclean as water soluble green solvent. J. Membr. Sci. 2018, 549, 192–204. [Google Scholar] [CrossRef]
- Marino, T.; Galiano, F.; Molino, A.; Figoli, A. New frontiers in sustainable membrane preparation: Cyrene™ as green bioderived solvent. J. Membr. Sci. 2019, 580, 224–234. [Google Scholar] [CrossRef]
- Sadman, K.; Delgado, D.E.; Won, Y.; Wang, Q.; Gray, K.A.; Shull, K.R. Versatile and High-throughput Polyelectrolyte Complex Membranes via Phase Inversion. ACS Appl. Mater. Interfaces 2019, 11, 16018–16026. [Google Scholar] [CrossRef]
- Nielen, W.M.; Willott, J.D.; de Vos, W.M. Aqueous Phase Separation of Responsive Copolymers for Sustainable and Mechanically Stable Membranes. ACS Appl. Polym. Mater. 2020, 2, 1702–1710. [Google Scholar] [CrossRef]
- Durmaz, E.N.; Baig, M.I.; Willott, J.D.; de Vos, W.M. Polyelectrolyte Complex Membranes via Salinity Change Induced Aqueous Phase Separation. ACS Appl. Polym. Mater. 2020, 2, 2612–2621. [Google Scholar] [CrossRef]
- Willott, J.D.; Nielen, W.M.; de Vos, W.M. Stimuli-Responsive Membranes through Sustainable Aqueous Phase Separation. ACS Appl. Polym. Mater. 2020, 2, 659–667. [Google Scholar] [CrossRef]
- Baig, M.I.; Durmaz, E.N.; Willott, J.D.; de Vos, W.M. Sustainable Membrane Production through Polyelectrolyte Complexation Induced Aqueous Phase Separation. Adv. Funct. Mater. 2020, 30, 1907344. [Google Scholar] [CrossRef]
- Kamp, J.; Emonds, S.; Borowec, J.; Restrepo Toro, M.A.; Wessling, M. On the organic solvent free preparation of ultrafiltration and nanofiltration membranes using polyelectrolyte complexation in an all aqueous phase inversion process. J. Membr. Sci. 2021, 618, 118632. [Google Scholar] [CrossRef]
- Chun, K.-Y.; Jang, S.-H.; Kim, H.-S.; Kim, Y.-W.; Han, H.-S.; Joe, Y.-I. Effects of solvent on the pore formation in asymmetric 6FDA–4,4′ODA polyimide membrane: Terms of thermodynamics, precipitation kinetics, and physical factors. J. Membr. Sci. 2000, 169, 197–214. [Google Scholar] [CrossRef]
- Susanto, H.; Stahra, N.; Ulbricht, M. High performance polyethersulfone microfiltration membranes having high flux and stable hydrophilic property. J. Membr. Sci. 2009, 342, 153–164. [Google Scholar] [CrossRef]
- Baig, M.I.; Willott, J.D.; de Vos, W.M. Tuning the structure and performance of polyelectrolyte complexation based aqueous phase separation membranes. J. Membr. Sci. 2020, 615, 118502. [Google Scholar] [CrossRef]
- Moghaddam, S.Z.; Thormann, E. The Hofmeister series: Specific ion effects in aqueous polymer solutions. J. Colloid Interface Sci. 2019, 555, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Nielen, W.M.; Willott, J.D.; Esguerra, Z.M.; de Vos, W.M. Ion specific effects on aqueous phase separation of responsive copolymers for sustainable membranes. J. Colloid Interface Sci. 2020, 576, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, E.N.; Willott, J.D.; Fatima, A.; de Vos, W.M. Weak polyanion and strong polycation complex based membranes: Linking aqueous phase separation to traditional membrane fabrication. Eur. Polym. J. 2020, 139, 110015. [Google Scholar] [CrossRef]
- Pal, P.; Nayak, J. Acetic acid production and purification: Critical review towards process intensification. Sep. Purif. Rev. 2017, 46, 44–61. [Google Scholar] [CrossRef]
- Fujioka, T.; Khan, S.J.; McDonald, J.A.; Nghiem, L.D. Nanofiltration of trace organic chemicals: A comparison between ceramic and polymeric membranes. Sep. Purif. Technol. 2014, 136, 258–264. [Google Scholar] [CrossRef]
- Xu, P.; Drewes, J.E.; Bellona, C.; Amy, G.; Kim, T.-U.; Adam, M.; Heberer, T. Rejection of Emerging Organic Micropollutants in Nanofiltration–Reverse Osmosis Membrane Applications. Water Environ. Res. 2005, 77, 40–48. [Google Scholar] [CrossRef]
- Kimura, K.; Amy, G.; Drewes, J.E.; Heberer, T.; Kim, T.-U.; Watanabe, Y. Rejection of organic micropollutants (disinfection by-products, endocrine disrupting compounds, and pharmaceutically active compounds) by NF/RO membranes. J. Membr. Sci. 2003, 227, 113–121. [Google Scholar] [CrossRef]
- Sehgal, D.; Vijay, I.K. A method for the high efficiency of water-soluble carbodiimide-mediated amidation. Anal. Biochem. 1994, 218, 87–91. [Google Scholar] [CrossRef]
- Ilyas, S.; Abtahi, S.M.; Akkilic, N.; Roesink, H.D.W.; de Vos, W.M. Weak polyelectrolyte multilayers as tunable separation layers for micro-pollutant removal by hollow fiber nanofiltration membranes. J. Membr. Sci. 2017, 537, 220–228. [Google Scholar] [CrossRef]
- Ball, P. Patterns in Nature: Why the Natural World Looks the Way It Does; University of Chicago Press: Chicago, IL, USA, 2016. [Google Scholar]
- Goehring, L.; Conroy, R.; Akhter, A.; Clegg, W.J.; Routh, A.F. Evolution of mud-crack patterns during repeated drying cycles. Soft Matter 2010, 6, 3562–3567. [Google Scholar] [CrossRef]
- Guvendiren, M.; Lu, H.D.; Burdick, J.A. Shear-thinning hydrogels for biomedical applications. Soft Matter 2012, 8, 260–272. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Solution Composition | Coagulation Bath Conditions [HCl] | |
|---|---|---|
| PSaMA% w/v | Acetic Acid% v/v | |
| 20 | 40 | 0.05, 0.1, 0.2, 0.3, 0.4, 0.5 |
| 20 | 30 | 0.1, 0.2, 0.1 |
| 20 | 25 | 0.1, 0.2, 0.1 |
| 22.5 | 40 | 0.1, 0.2, 0.1 |
| 24 | 40 | 0.1, 0.2, 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nielen, W.M.; Willott, J.D.; Galicia, J.A.R.; de Vos, W.M. Effect of Solution Viscosity on the Precipitation of PSaMA in Aqueous Phase Separation-Based Membrane Formation. Polymers 2021, 13, 1775. https://doi.org/10.3390/polym13111775
Nielen WM, Willott JD, Galicia JAR, de Vos WM. Effect of Solution Viscosity on the Precipitation of PSaMA in Aqueous Phase Separation-Based Membrane Formation. Polymers. 2021; 13(11):1775. https://doi.org/10.3390/polym13111775
Chicago/Turabian StyleNielen, Wouter M., Joshua D. Willott, Julia A. R. Galicia, and Wiebe M. de Vos. 2021. "Effect of Solution Viscosity on the Precipitation of PSaMA in Aqueous Phase Separation-Based Membrane Formation" Polymers 13, no. 11: 1775. https://doi.org/10.3390/polym13111775
APA StyleNielen, W. M., Willott, J. D., Galicia, J. A. R., & de Vos, W. M. (2021). Effect of Solution Viscosity on the Precipitation of PSaMA in Aqueous Phase Separation-Based Membrane Formation. Polymers, 13(11), 1775. https://doi.org/10.3390/polym13111775