
The Effect of Heat Treatment toward Glycerol-Based, Photocurable Polymeric Scaffold: Mechanical, Degradation and Biocompatibility


Abstract
:1. Introduction
2. Materials and Methods
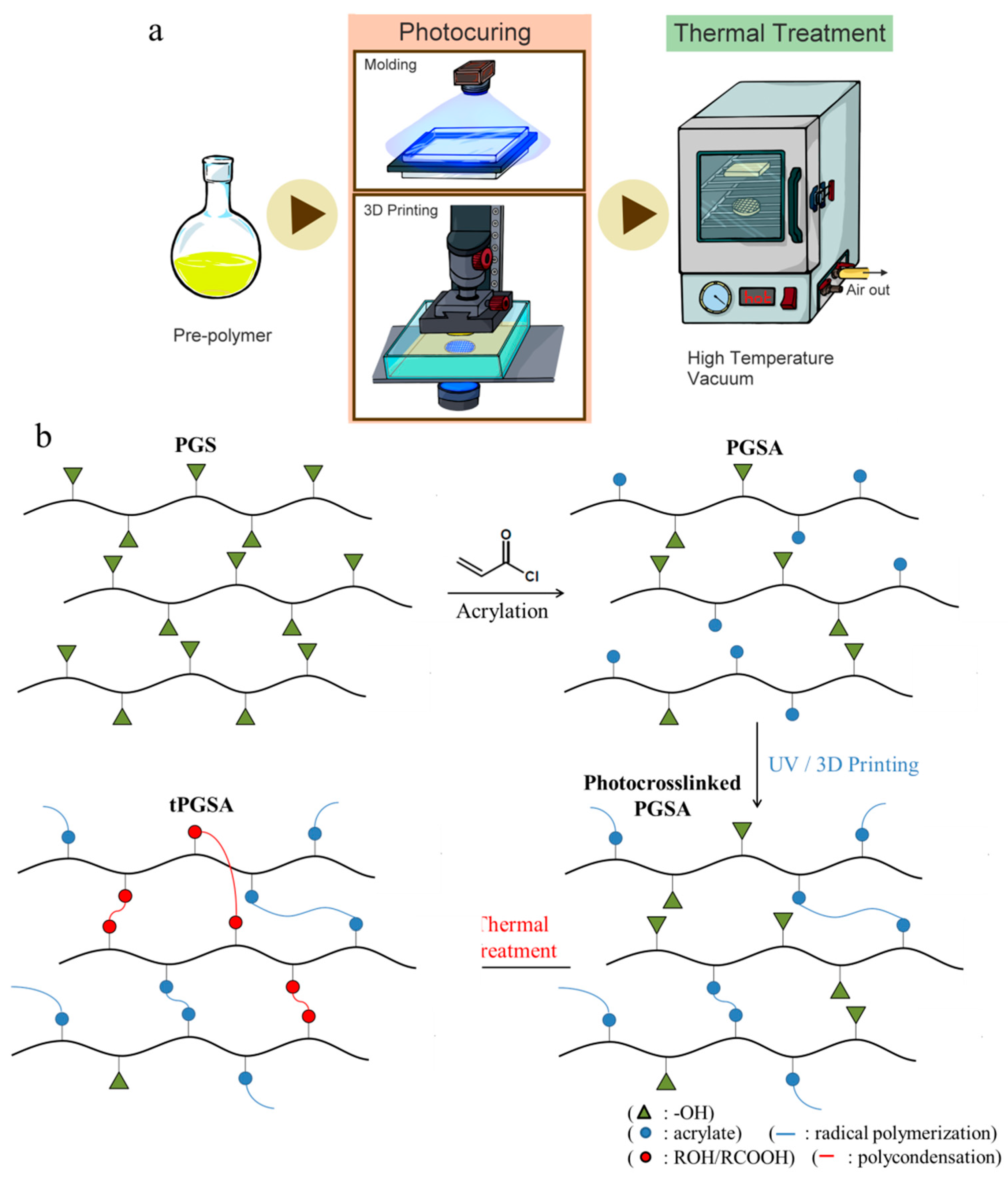
2.1. Synthesis and Characterization of PGSA
2.2. Network Formation and 3D Printing
2.3. Mechanical Measurement of PGSA via Tensile Test
2.4. Thermal Analysis
2.5. In Vitro Degradation Test
2.6. In Vitro Biocompatibility
2.7. Metabolic Activity
2.8. In Vivo Biocompatibility
2.9. Histology and Immunofluorescence
2.10. Statistical Analysis
3. Results and Discussion
3.1. Mechanical Properties of PGSA after Thermal Treatment
3.2. In Vitro Degradation
3.3. In Vitro Biocompatibility
3.4. Metabolic Activity
3.5. Histology
3.6. Immunofluorescence
3.7. Mechanical Properties of Different Treatment Processes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phillips, R. Photopolymerization. J. Photochem. 1984, 25, 79–82. [Google Scholar] [CrossRef]
- Tehfe, M.; Louradour, F.; Lalevée, J.; Fouassier, J.-P. Photopolymerization Reactions: On the Way to a Green and Sustainable Chemistry. Appl. Sci. 2013, 3, 490. [Google Scholar] [CrossRef]
- Bongiovanni, R.; Malucelli, G.; Sangermano, M.; Priola, A. Preparation of coatings via cationic photopolymerisation: Influence of alcoholic additives. Macromol. Symp. 2002, 187. [Google Scholar] [CrossRef]
- Chia, H.N.; Wu, B.M. Recent advances in 3D printing of biomaterials. J. Biol. Eng. 2015, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Ferdous, S.; Mustafa, A.I. Improvement of Physico-mechanical Properties of Chitosan Films by Photocuring with Acrylic Monomers. J. Polym. Environ. 2005, 13, 193–201. [Google Scholar] [CrossRef]
- Cruise, G.M.; Scharp, D.S.; Hubbell, J.A. Characterization of permeability and network structure of interfacially photopolymerized poly (ethylene glycol) diacrylate hydrogels. Biomaterials 1998, 19, 1287–1294. [Google Scholar] [CrossRef]
- Kweon, H.; Yoo, M.K.; Park, I.K.; Kim, T.H.; Lee, H.C.; Lee, H.-S.; Oh, J.-S.; Akaike, T.; Cho, C.-S. A novel degradable polycaprolactone networks for tissue engineering. Biomaterials 2003, 24, 801–808. [Google Scholar] [CrossRef]
- Cai, L.; Wang, S. Poly (ɛ-caprolactone) acrylates synthesized using a facile method for fabricating networks to achieve controllable physicochemical properties and tunable cell responses. Polymer 2010, 51, 164–177. [Google Scholar] [CrossRef]
- Vincent, S.D.V.; Tobias, S.; Geraldine, H.M.S.; Peter, D.; Martin, T.; Jin, X.; Albert, J.J.W.; Katja, L.; Jan, J.; Rudy, F. Biobased Acrylate Photocurable Resin Formulation for Stereolithography 3D Printing. ACS Omega 2018, 3, 1403–1408. [Google Scholar]
- Bonafè, F.; Govoni, M.; Giordano, E.; Caldarera, C.M.; Guarnieri, C.; Muscari, C. Hyaluronan and cardiac regeneration. J. Biomed. Sci. 2014, 21, 100. [Google Scholar] [CrossRef] [Green Version]
- Pateman, C.J.; Harding, A.J.; Glen, A.; Taylor, C.S.; Christmas, C.R.; Robinson, P.P.; Rimmer, S.; Boissonade, F.M.; Claeyssens, F.; Haycock, J.W. Nerve guides manufactured from photocurable polymers to aid peripheral nerve repair. Biomaterials 2015, 49, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, F. Biomaterials and scaffolds for tissue engineering. Mater. Today 2011, 14, 88–95. [Google Scholar] [CrossRef]
- Chan, B.P.; Leong, K.W. Scaffolding in tissue engineering: General approaches and tissue-specific considerations. Eur. Spine J. 2008, 17, 467–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snedeker, J.G.; Niederer, P.; Schmidlin, F.R.; Farshad, M.; Demetropoulos, C.K.; Lee, J.B.; Yang, K.H. Strain-rate dependent material properties of the porcine and human kidney capsule. J. Biomech. 2005, 38, 1011–1021. [Google Scholar] [CrossRef]
- Hollenstein, M.; Nava, A.; Valtorta, D.; Snedeker, J.G.; Mazza, E. Mechanical Characterization of the Liver Capsule and Parenchyma; Springer: Berlin/Heidelberg, Germany, 2006; pp. 150–158. [Google Scholar]
- McKee, C.T.; Last, J.A.; Russell, P.; Murphy, C.J. Indentation versus tensile measurements of Young’s modulus for soft biological tissues. Tissue Eng. Part B Rev. 2011, 17, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Junid, R.; Razak, J.A.; Mahat, M.M. The Effects of Photoinitiator Addition to the Mechanical and Physical Properties of the Epoxy and Vinyl Ester Fiber Glass Laminated Composites. Int. J. Appl. Sci. Technol. 2011, 1, 74–79. [Google Scholar]
- Bonada, J.; Muguruza, A.; Fernández-Francos, X.; Ramis, X. Influence of exposure time on mechanical properties and photocuring conversion ratios for photosensitive materials used in Additive Manufacturing. Proc. Manuf. 2017, 13, 762–769. [Google Scholar] [CrossRef]
- Chang, C.W.; van Spreeuwel, A.; Zhang, C.; Varghese, S. PEG/clay nanocomposite hydrogel: A mechanically robust tissue engineering scaffold. Soft Matter 2010, 6, 5157. [Google Scholar] [CrossRef]
- Morris, V.B.; Nimbalkar, S.; Younesi, M.; McClellan, P.; Akkus, O. Mechanical Properties, Cytocompatibility and Manufacturability of Chitosan:PEGDA Hybrid-Gel Scaffolds by Stereolithography. Ann. Biomed. Eng. 2017, 45, 286–296. [Google Scholar] [CrossRef]
- Suekama, T.C.; Khanlari, A.; Gehrke, S.H. Tuning Mechanical Properties of Chondroitin Sulfate-Based Hydrogels Using the Double-Network Strategy. MRS Proc. 2014, 1622, 79–84. [Google Scholar] [CrossRef]
- Tan, G.X.; Ning, C.Y.; Wang, Y.J.; Guang, Y.X. Synthesis and Mechanical Properties of PEGDA-Based Hydrogels Scaffold. Polym. Mater. Sci. Eng. 2009, 25, 81–83. [Google Scholar]
- Song, S.Y.; Park, M.S.; Lee, J.W.; Yun, J.S. A Study on the Rheological and Mechanical Properties of Photo-Curable Ceramic/Polymer Composites with Different Silane Coupling Agents for SLA 3D Printing Technology. Nanomaterials 2018, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Sean, V.M.; Anthony, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773. [Google Scholar]
- Zhou, D.; Ito, Y. Visible light-curable polymers for biomedical applications. Sci. China Chem. 2014, 57, 510–521. [Google Scholar] [CrossRef]
- Manas, D.; Manas, M.; Stanek, M.; Danek, M. Improvement of plastic properties. Arch. Mater. Sci. Eng. 2008, 32, 69–76. [Google Scholar]
- Almeida-Chetti, V.A.; Macchi, R.L.; Iglesias, M.E. Effect of post-curing treatment on mechanical properties of composite resins. Acta Odontol. Latinoam. 2014, 27, 72–76. [Google Scholar]
- Uzay, C.; Boztepe, M.H.; Bayramoğlu, M.; Geren, N. Effect of post-curing heat treatment on mechanical properties of fiber reinforced polymer (FRP) composites. Mater. Test. 2017, 59, 366–372. [Google Scholar] [CrossRef]
- Tse-Hao, K.; Tsu-Sheng, M. Effect of post-curing on the mechanical properties of carbonized phenolic resins. Polym. Compos. 1998, 19, 456–462. [Google Scholar]
- Grigaleviciute, G.; Baltriukiene, D.; Bukelskiene, V.; Malinauskas, M. Biocompatibility Evaluation and Enhancement of Elastomeric Coatings Made Using Table-Top Optical 3D Printer. Coatings 2020, 10, 254. [Google Scholar] [CrossRef] [Green Version]
- Moritz, T.; Maleksaeedi, S. 4—Additive manufacturing of ceramic components. In Additive Manufacturing; Zhang, J., Jung, Y.-G., Eds.; Butterworth-Heinemann: Oxford, UK, 2018; pp. 105–161. [Google Scholar] [CrossRef]
- Nguyen, Q.B.; Nguyen, N.H.; Rios de Anda, A.; Nguyen, V.H.; Versace, D.-L.; Langlois, V.; Naili, S.; Renard, E. Photocurable bulk epoxy resins based on resorcinol derivative through cationic polymerization. J. Appl. Polym. Sci. 2020, 137, 49051. [Google Scholar] [CrossRef]
- Adeodu, A.; Christopher, A.; Oluwole, L.; Alo, O. Effect of Microwave and Conventional Autoclave Post-Curing on the Mechanical and Micro-structural Properties of Particulate Reinforced Polymer Matrix Composites. Adv. Mater. 2015, 4, 85. [Google Scholar]
- Morancho, J.M.; Fernández-Francos, X.; Ramis, X.; Salla, J.M.; Serra, À. Photocuring and thermal post-curing of a cycloaliphatic epoxide resin with a trithiol and a vinyl epoxy compound. J. Therm. Anal. Calorim. 2015, 121, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Esteves, R.A.; Boaro, L.C.; Gonçalves, F.; Campos, L.M.; Silva, C.M.; Rodrigues-Filho, L.E. Chemical and Mechanical Properties of Experimental Dental Composites as a Function of Formulation and Postcuring Thermal Treatment. BioMed Res. Int. 2018, 2018, 6. [Google Scholar] [CrossRef]
- Nijst, C.L.; Bruggeman, J.P.; Karp, J.M.; Ferreira, L.; Zumbuehl, A.; Bettinger, C.J.; Langer, R. Synthesis and characterization of photocurable elastomers from poly (glycerol-co-sebacate). Biomacromolecules 2007, 8, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Highley, C.B.; Ouyang, L.; Burdick, J.A. 3d printing of photocurable poly (glycerol sebacate) elastomers. Biofabrication 2016, 8, 045004. [Google Scholar] [CrossRef]
- Cheng, Y.; Hsu, Y.; Wang, J. Study on biodegradable photocurable PCL-diacrylate/PGSA for 3D printing. In Proceedings of the 2017 IEEE/SICE International Symposium on System Integration (SII), Taipei, Taiwan, 11–14 December 2017; pp. 102–107. [Google Scholar]
- Yu, C.; Schimelman, J.; Wang, P.; Miller, K.L.; Ma, X.; You, S.; Guan, J.; Sun, B.; Zhu, W.; Chen, S. Photopolymerizable Biomaterials and Light-Based 3D Printing Strategies for Biomedical Applications. Chem. Rev. 2020, 120, 10695–10743. [Google Scholar] [CrossRef] [PubMed]
- Alekya, B.; Rao, S.; Pandya, H.J. Engineering approaches for characterizing soft tissue mechanical properties: A review. Clin. Biomech. 2019, 69, 127–140. [Google Scholar]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Kocatepe, K.; Cerah, M.; Erdogan, M. The tensile fracture behaviour of intercritically annealed and quenched+tempered ferritic ductile iron with dual matrix structure. Mater. Des. 2007, 28, 172–181. [Google Scholar] [CrossRef]
- Cheng, L.; Guo, T.F. Void interaction and coalescence in polymeric materials. Int. J. Solids Struct. 2007, 44, 1787–1808. [Google Scholar] [CrossRef] [Green Version]
- Raj, A.; Samuel, C.; Prashantha, K. Role of Compatibilizer in Improving the Properties of PLA/PA12 Blends. Front. Mater. 2020, 7, 193. [Google Scholar] [CrossRef]
- Wells, R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 2008, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.L.; Chen, J.Y.; Chang, T.L.; Hsiao, S.K.; Hsieh, Y.K.; Villalobos Gorday, K.; Cheng, Y.L.; Wang, J. Design of photocurable, biodegradable scaffolds for liver lobule regeneration via digital light process-additive manufacturing. Biofabrication 2020, 12, 035024. [Google Scholar] [CrossRef]
- Bokhari, M.; Carnachan, R.J.; Cameron, N.R.; Przyborski, S.A. Culture of HepG2 liver cells on three dimensional polystyrene scaffolds enhances cell structure and function during toxicological challenge. J. Anat. 2007, 211, 567–576. [Google Scholar] [CrossRef]
- Ratner, B.D. Reducing capsular thickness and enhancing angiogenesis around implant drug release systems. J. Control. Release 2002, 78, 211–218. [Google Scholar] [CrossRef]
- Ratner, B.D.; Bryant, S.J. Biomaterials: Where we have been and where we are going. Ann. Rev. Biomed. Eng. 2004, 6, 41–75. [Google Scholar] [CrossRef]
- Sundback, C.A.; Shyu, J.Y.; Wang, Y.; Faquin, W.C.; Langer, R.S.; Vacanti, J.P.; Hadlock, T.A. Biocompatibility analysis of poly (glycerol sebacate) as a nerve guide material. Biomaterials 2005, 26, 5454–5464. [Google Scholar] [CrossRef]
- De Fougerolles, A.R.; Koteliansky, V.E. Regulation of monocyte gene expression by the extracellular matrix and its functional implications. Immunol. Rev. 2002, 186, 208–220. [Google Scholar] [CrossRef]
- Hynes, R.O. The Extracellular Matrix: Not Just Pretty Fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [Green Version]
- Korpos, E.; Wu, C.; Song, J.; Hallmann, R.; Sorokin, L. Role of the extracellular matrix in lymphocyte migration. Cell Tissue Res. 2010, 339, 47–57. [Google Scholar] [CrossRef]
- Münker, T.J.A.G.; van de Vijfeijken, S.E.C.M.; Mulder, C.S.; Vespasiano, V.; Becking, A.G.; Kleverlaan, C.J.; Becking, A.G.; Dubois, L.; Karssemakers, L.H.E.; Milstein, D.M.J.; et al. Effects of sterilization on the mechanical properties of poly (methyl methacrylate) based personalized medical devices. J. Mech. Behav. Biomed. Mater. 2018, 81, 168–172. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Young’s Modulus (MPa) | Ultimate Tensile Strength (MPa) | Elongation (%) |
|---|---|---|---|
| PGSA | 4.52 ± 0.37 | 0.82 ± 0.07 | 21.85 ± 1.84 |
| PGSA + UV (10 min) | 5.22 ± 0.30 | 0.84 ± 0.08 | 19.14 ± 1.25 |
| PGSA + Autoclave | 3.89 ± 0.15 | 0.81 ± 0.04 | 25.69 ± 0.78 |
| PGSA + Thermal | 9.36 ± 0.27 | 1.66 ± 0.09 | 23.15 ± 1.51 |
| PGSA + Thermal + Autoclave | 8.95 ± 0.40 | 1.47 ± 0.09 | 20.18 ± 1.38 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ao-Ieong, W.-S.; Chien, S.-T.; Jiang, W.-C.; Yet, S.-F.; Wang, J. The Effect of Heat Treatment toward Glycerol-Based, Photocurable Polymeric Scaffold: Mechanical, Degradation and Biocompatibility. Polymers 2021, 13, 1960. https://doi.org/10.3390/polym13121960
Ao-Ieong W-S, Chien S-T, Jiang W-C, Yet S-F, Wang J. The Effect of Heat Treatment toward Glycerol-Based, Photocurable Polymeric Scaffold: Mechanical, Degradation and Biocompatibility. Polymers. 2021; 13(12):1960. https://doi.org/10.3390/polym13121960
Chicago/Turabian StyleAo-Ieong, Wai-Sam, Shin-Tian Chien, Wei-Cheng Jiang, Shaw-Fang Yet, and Jane Wang. 2021. "The Effect of Heat Treatment toward Glycerol-Based, Photocurable Polymeric Scaffold: Mechanical, Degradation and Biocompatibility" Polymers 13, no. 12: 1960. https://doi.org/10.3390/polym13121960
APA StyleAo-Ieong, W. -S., Chien, S. -T., Jiang, W. -C., Yet, S. -F., & Wang, J. (2021). The Effect of Heat Treatment toward Glycerol-Based, Photocurable Polymeric Scaffold: Mechanical, Degradation and Biocompatibility. Polymers, 13(12), 1960. https://doi.org/10.3390/polym13121960