
Solution Self-Assembly of Coil-Crystalline Diblock Copolypeptoids Bearing Alkyl Side Chains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
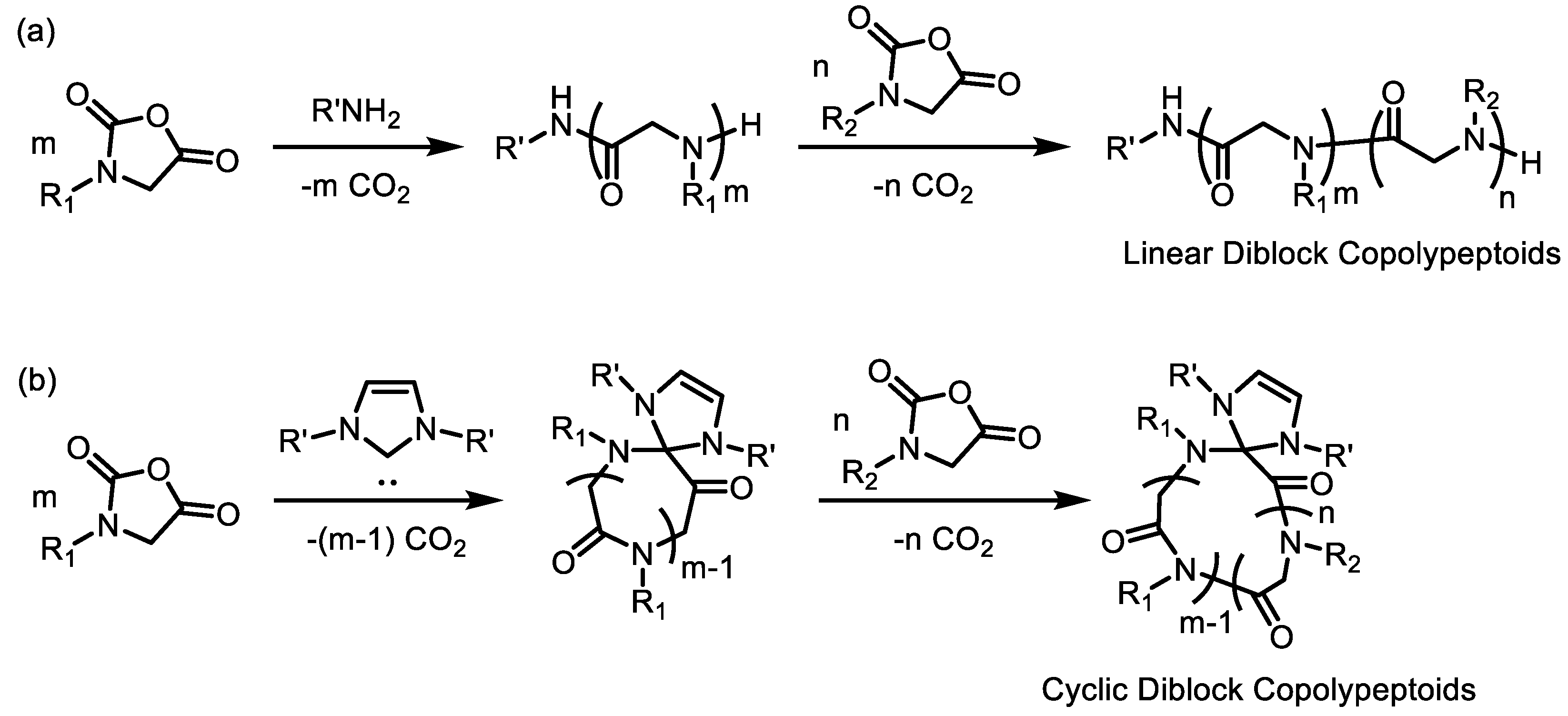
2. Polypeptoids Bearing Alkyl Side Chains: Synthetic Methods and Their Phase Behavior
2.1. Controlled Ring-Opening Polymerization (ROP) for Polypeptoids
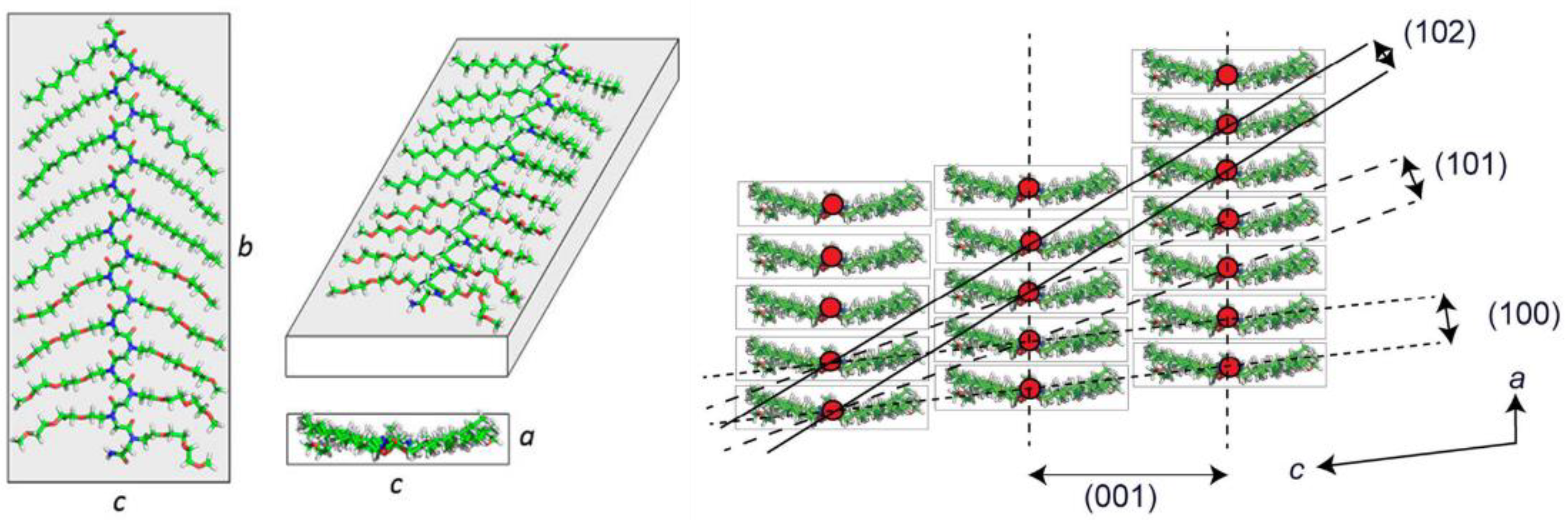
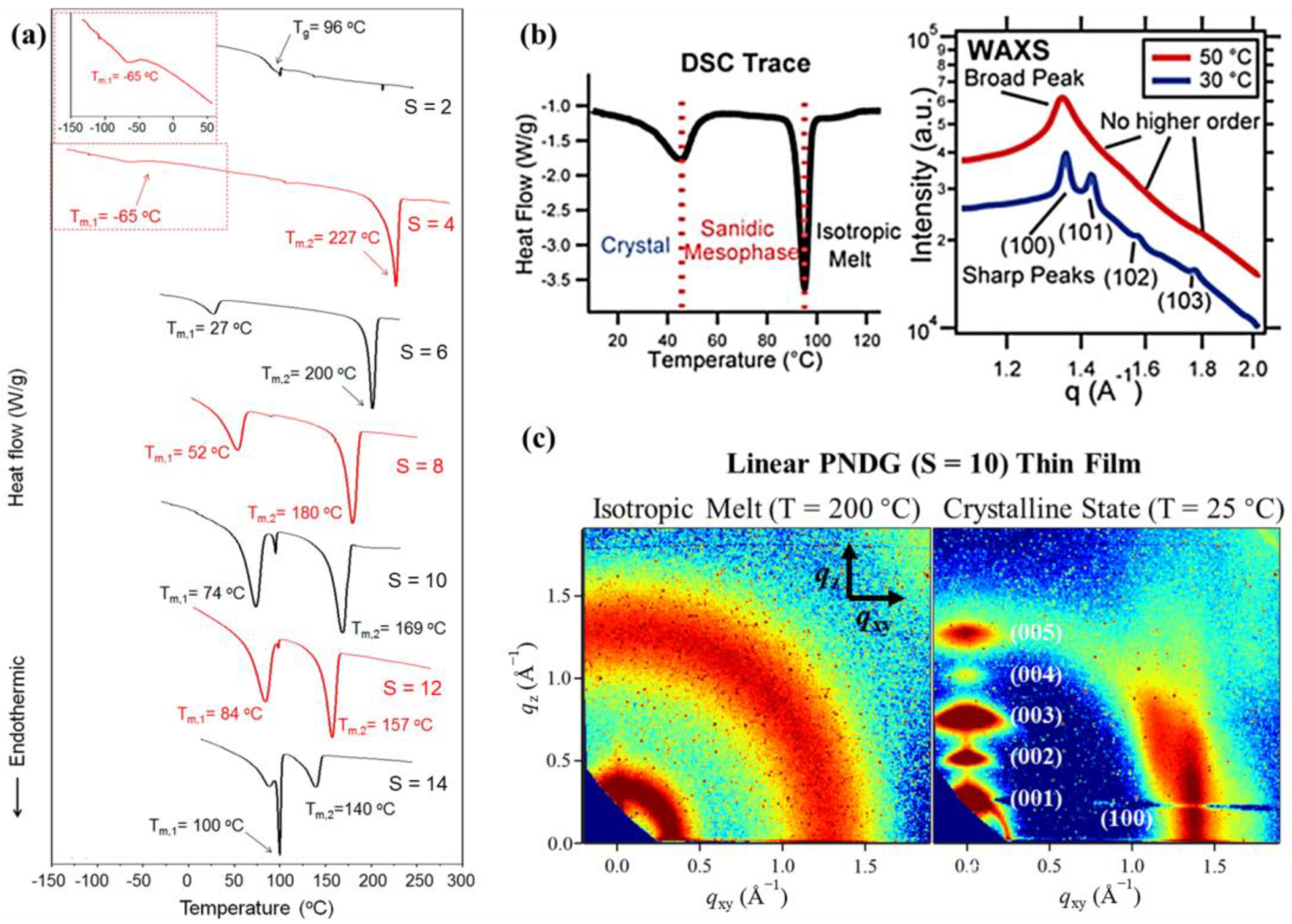
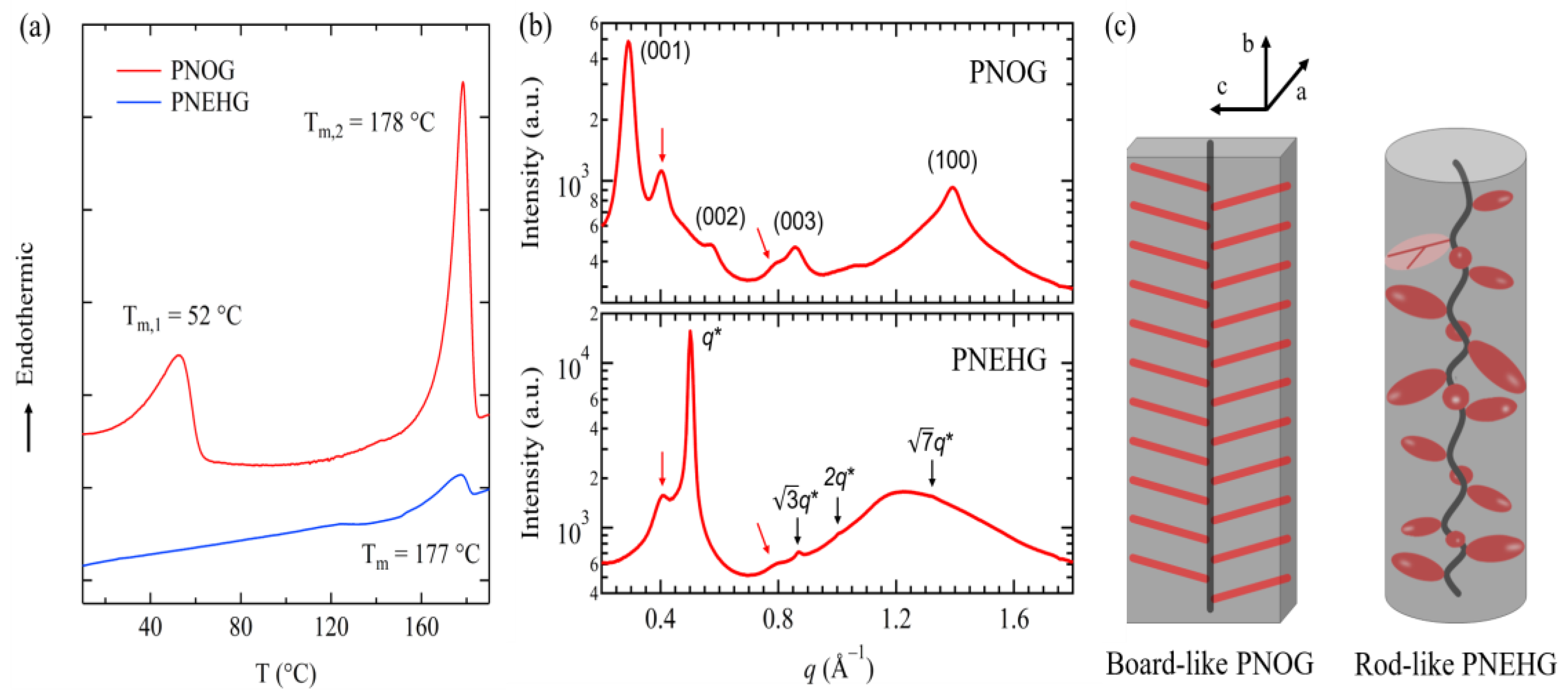
2.2. Molecular Packing and Phase Behavior of Polypeptoid Homopolymers Bearing Alkyl Side Chains
3. Solution Self-Assembly of Coil-Crystalline Diblock Copolypeptoids Bearing Alkyl Side Chains
3.1. Sample Preparation and Characterization of Coil-Crystalline Diblock Copolypeptoid Solutions
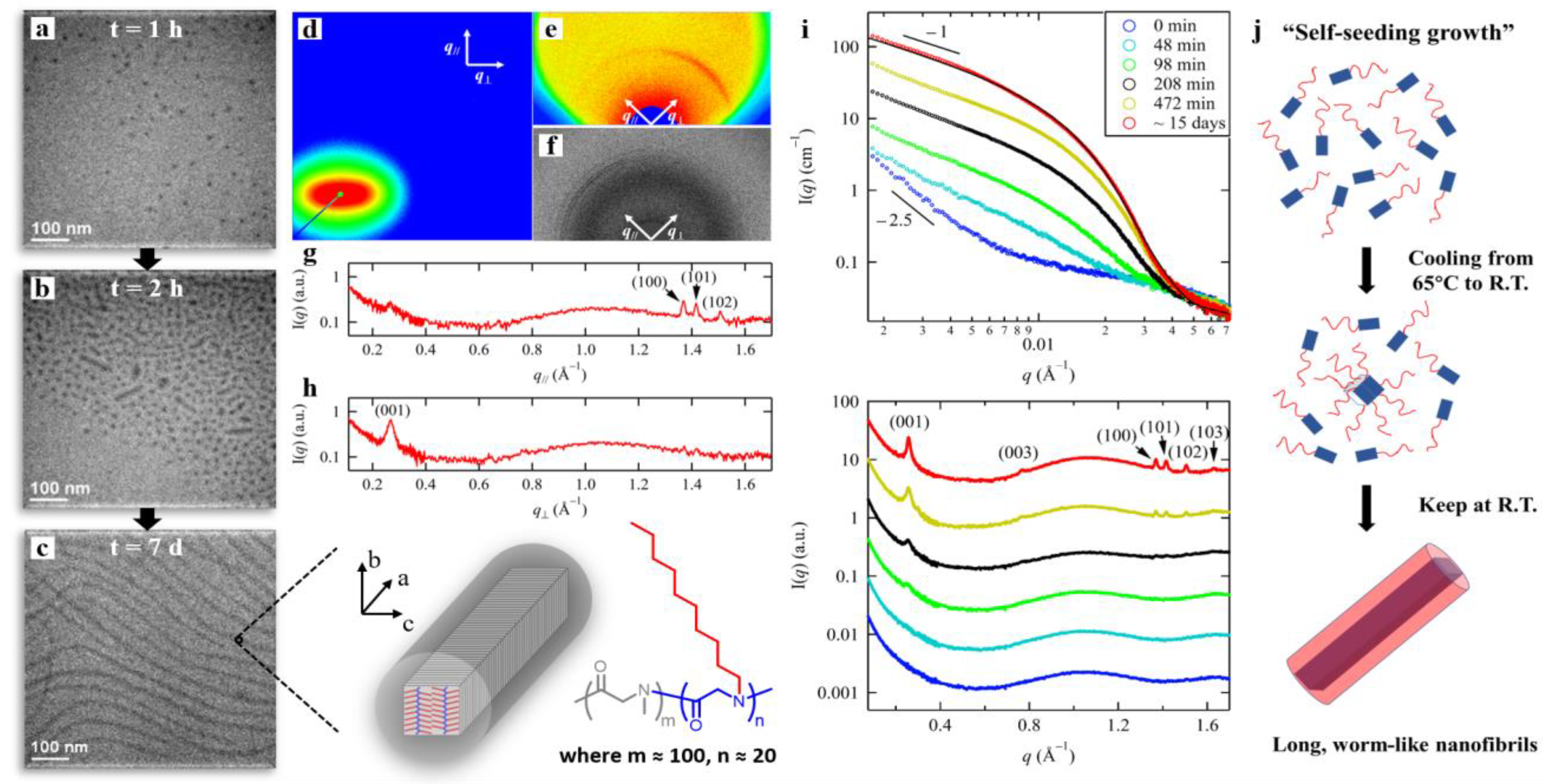
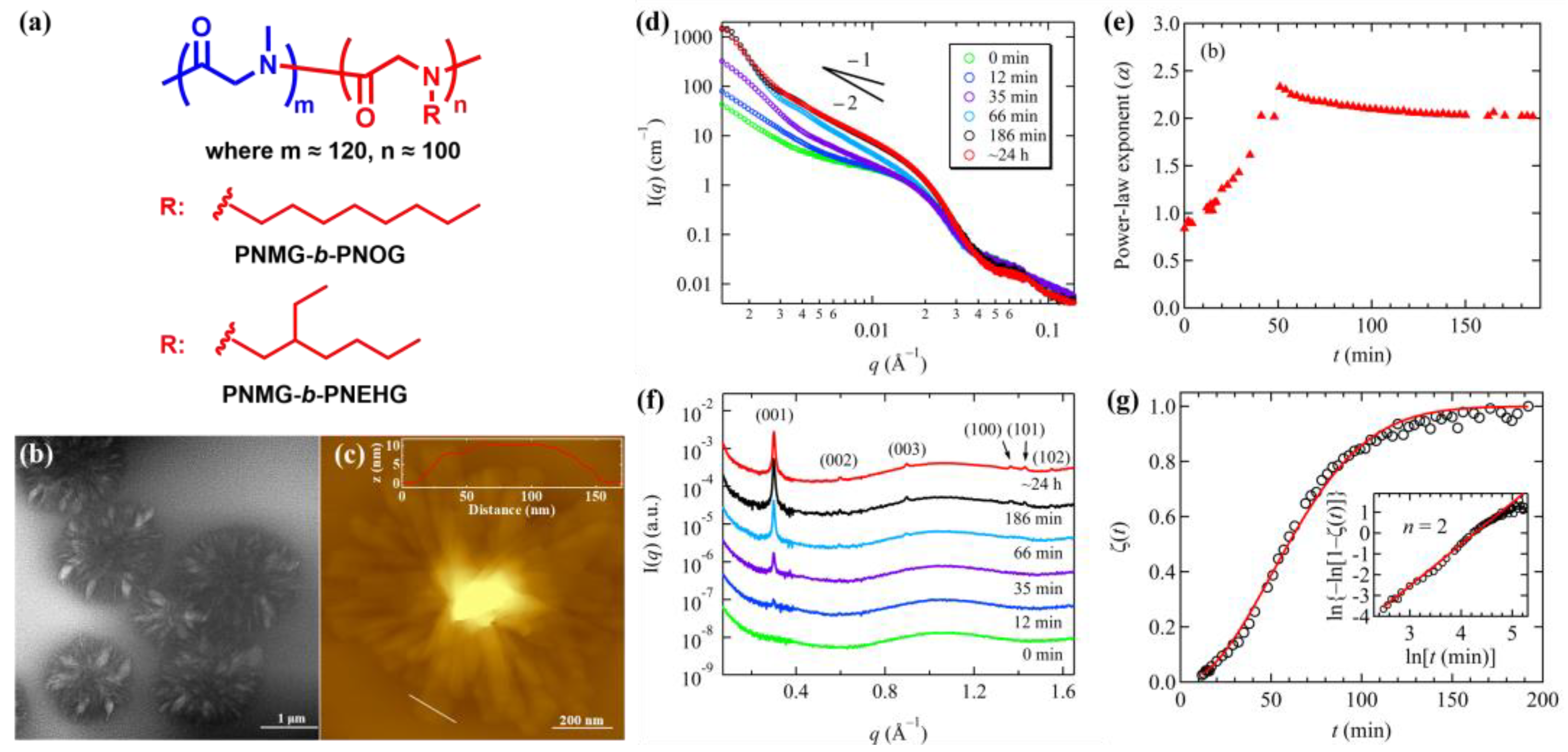
3.2. Crystallization-Driven Self-Assembly of 1D nanofibrils
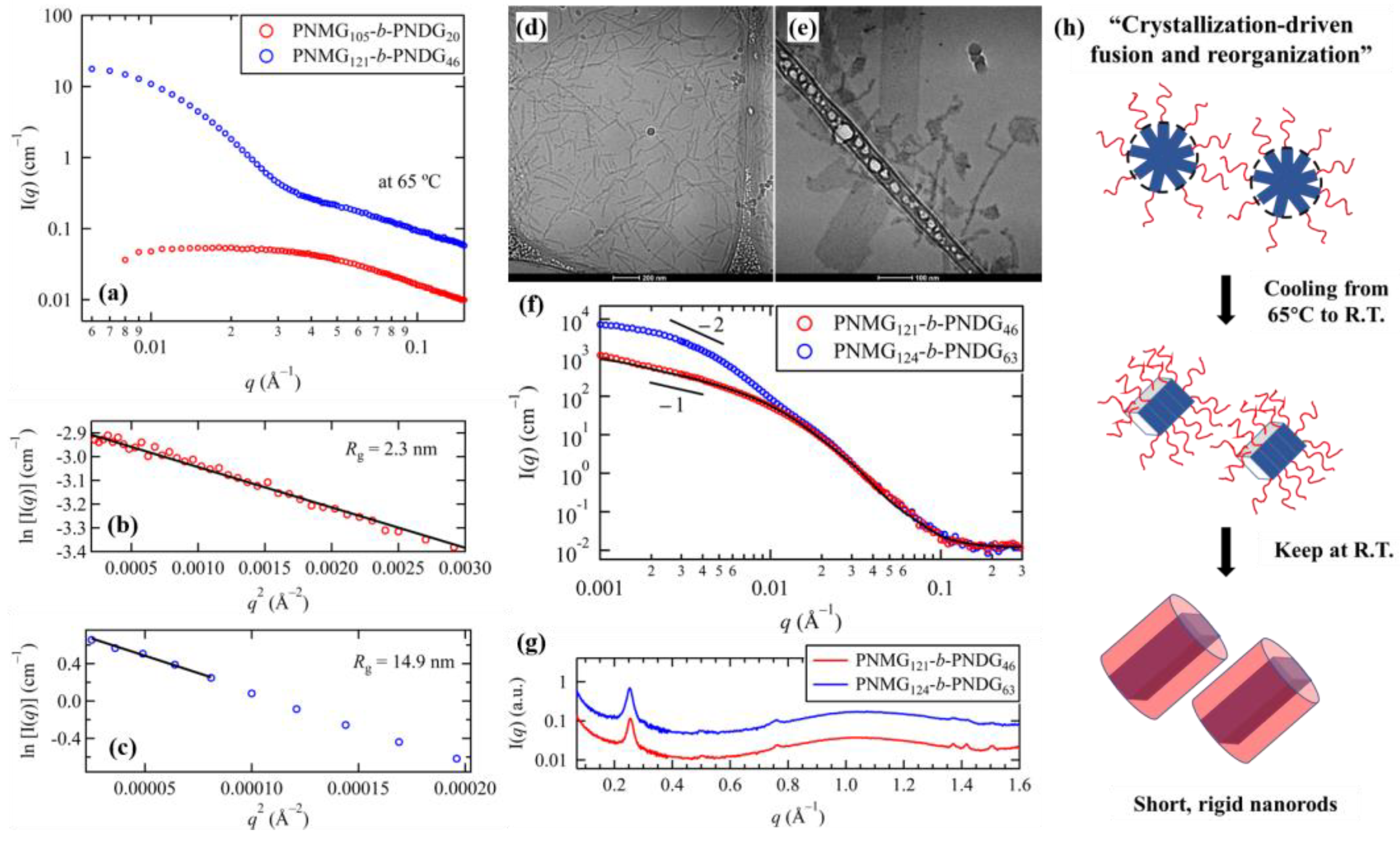
3.3. Effect of Block Composition on the Solution Self-Assemblies
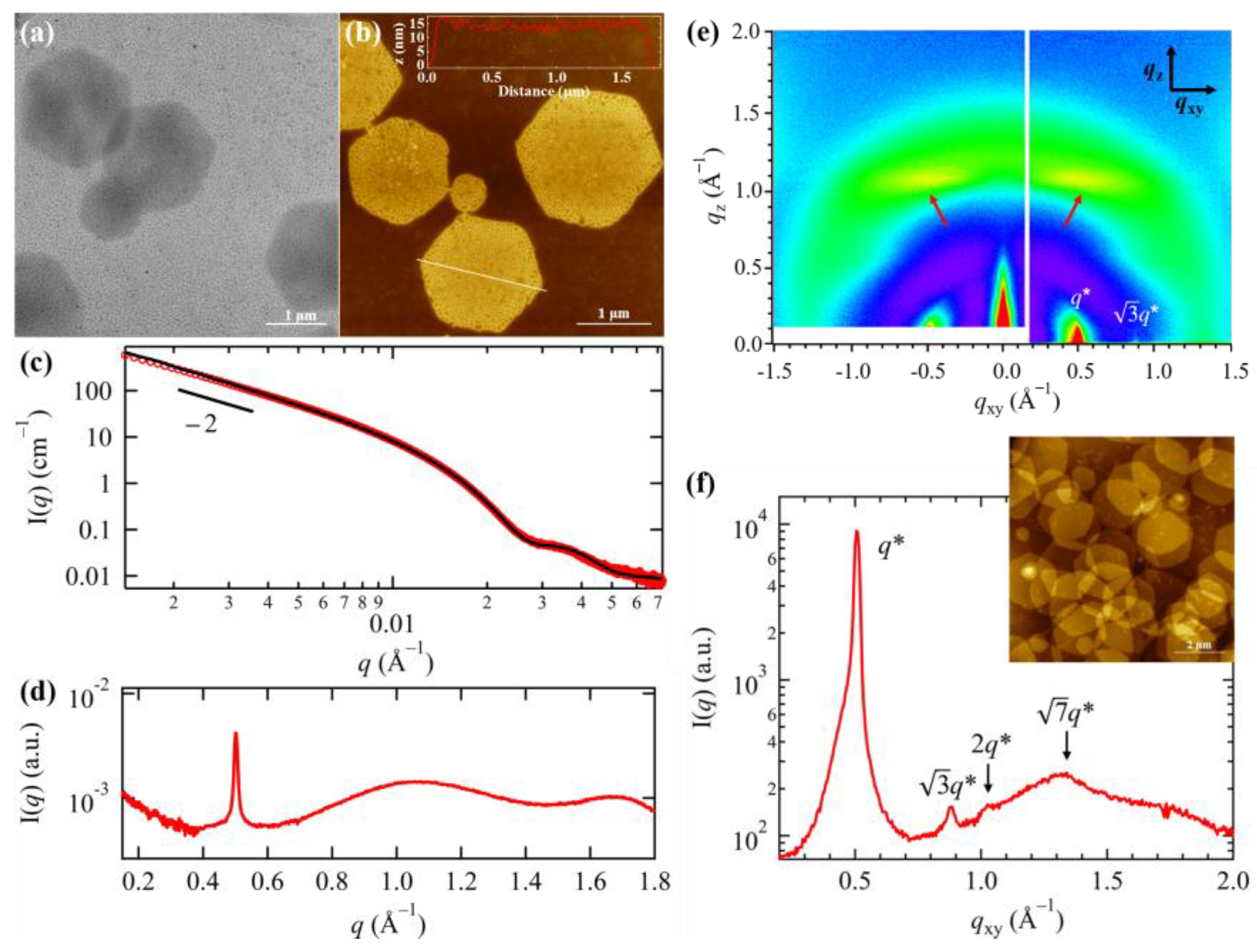
3.4. Effect of Side Chain Branching on the Solution Self-Assemblies
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blanazs, A.; Armes, S.P.; Ryan, A.J. Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and their Biological Applications. Macromol. Rapid Commun. 2009, 30, 267–277. [Google Scholar] [CrossRef]
- Hayward, R.C.; Pochan, D.J. Tailored Assemblies of Block Copolymers in Solution: It Is All about the Process. Macromolecules 2010, 43, 3577–3584. [Google Scholar] [CrossRef]
- Vilgis, T.; Halperin, A. Aggregation of coil-crystalline block copolymers: Equilibrium crystallization. Macromolecules 1991, 24, 2090–2095. [Google Scholar] [CrossRef]
- Massey, J.A.; Temple, K.; Cao, L.; Rharbi, Y.; Raez, J.; Winnik, M.A.; Manners, I. Self-Assembly of Organometallic Block Copolymers: The Role of Crystallinity of the Core-Forming Polyferrocene Block in the Micellar Morphologies Formed by Poly(ferrocenylsilane-b-dimethylsiloxane) in n-Alkane Solvents. J. Am. Chem. Soc. 2000, 122, 11577–11584. [Google Scholar] [CrossRef]
- Gilroy, J.B.; Gädt, T.; Whittell, G.R.; Chabanne, L.; Mitchels, J.M.; Richardson, R.M.; Winnik, M.A.; Manners, I. Monodisperse cylindrical micelles by crystallization-driven living self-assembly. Nat. Chem. 2010, 2, 566–570. [Google Scholar] [CrossRef]
- Wang, X.; Guerin, G.; Wang, H.; Wang, Y.; Manners, I.; Winnik, M.A. Cylindrical Block Copolymer Micelles and Co-Micelles of Controlled Length and Architecture. Science 2007, 317, 644–647. [Google Scholar] [CrossRef] [Green Version]
- He, X.; He, Y.; Hsiao, M.-S.; Harniman, R.L.; Pearce, S.; Winnik, M.A.; Manners, I. Complex and Hierarchical 2D Assemblies via Crystallization-Driven Self-Assembly of Poly(l-lactide) Homopolymers with Charged Termini. J. Am. Chem. Soc. 2017, 139, 9221–9228. [Google Scholar] [CrossRef] [Green Version]
- Inam, M.; Cambridge, G.; Pitto-Barry, A.; Laker, Z.P.L.; Wilson, N.R.; Mathers, R.T.; Dove, A.P.; O’Reilly, R.K. 1D vs. 2D shape selectivity in the crystallization-driven self-assembly of polylactide block copolymers. Chem. Sci. 2017, 8, 4223–4230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arno, M.C.; Inam, M.; Coe, Z.; Cambridge, G.; Macdougall, L.J.; Keogh, R.; Dove, A.P.; O’Reilly, R.K. Precision Epitaxy for Aqueous 1D and 2D Poly(ε-caprolactone) Assemblies. J. Am. Chem. Soc. 2017, 139, 16980–16985. [Google Scholar] [CrossRef]
- Qiu, H.; Hudson, Z.M.; Winnik, M.A.; Manners, I. Multidimensional hierarchical self-assembly of amphiphilic cylindrical block comicelles. Science 2015, 347, 1329–1332. [Google Scholar] [CrossRef] [Green Version]
- Nicolai, T.; Colombani, O.; Chassenieux, C. Dynamic polymeric micelles versus frozen nanoparticles formed by block copolymers. Soft Matter 2010, 6, 3111–3118. [Google Scholar] [CrossRef]
- Jain, S.; Bates, F.S. Consequences of Nonergodicity in Aqueous Binary PEO−PB Micellar Dispersions. Macromolecules 2004, 37, 1511–1523. [Google Scholar] [CrossRef]
- Tritschler, U.; Pearce, S.; Gwyther, J.; Whittell, G.R.; Manners, I. 50th Anniversary Perspective: Functional Nanoparticles from the Solution Self-Assembly of Block Copolymers. Macromolecules 2017, 50, 3439–3463. [Google Scholar] [CrossRef] [Green Version]
- Lunn, D.J.; Finnegan, J.R.; Manners, I. Self-assembly of “patchy” nanoparticles: A versatile approach to functional hierarchical materials. Chem. Sci. 2015, 6, 3663–3673. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Lodge, T.P.; Hillmyer, M.A. A Stepwise “Micellization–Crystallization” Route to Oblate Ellipsoidal, Cylindrical, and Bilayer Micelles with Polyethylene Cores in Water. Macromolecules 2012, 45, 9460–9467. [Google Scholar] [CrossRef]
- Schmelz, J.; Karg, M.; Hellweg, T.; Schmalz, H. General Pathway toward Crystalline-Core Micelles with Tunable Morphology and Corona Segregation. ACS Nano 2011, 5, 9523–9534. [Google Scholar] [CrossRef]
- He, W.-N.; Zhou, B.; Xu, J.-T.; Du, B.-Y.; Fan, Z.-Q. Two Growth Modes of Semicrystalline Cylindrical Poly(ε-caprolactone)-b-poly(ethylene oxide) Micelles. Macromolecules 2012, 45, 9768–9778. [Google Scholar] [CrossRef]
- Pitto-Barry, A.; Kirby, N.; Dove, A.P.; O’Reilly, R.K. Expanding the scope of the crystallization-driven self-assembly of polylactide-containing polymers. Polym. Chem. 2014, 5, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Lu, Y.; Chia, A.; Zhang, M.; Rupar, P.A.; Gunari, N.; Walker, G.C.; Cambridge, G.; He, F.; Guerin, G.; et al. Self-Seeding in One Dimension: A Route to Uniform Fiber-like Nanostructures from Block Copolymers with a Crystallizable Core-Forming Block. ACS Nano 2013, 7, 3754–3766. [Google Scholar] [CrossRef]
- Hsiao, M.-S.; Yusoff, S.F.M.; Winnik, M.A.; Manners, I. Crystallization-Driven Self-Assembly of Block Copolymers with a Short Crystallizable Core-Forming Segment: Controlling Micelle Morphology through the Influence of Molar Mass and Solvent Selectivity. Macromolecules 2014, 47, 2361–2372. [Google Scholar] [CrossRef]
- Sun, L.; Petzetakis, N.; Pitto-Barry, A.; Schiller, T.L.; Kirby, N.; Keddie, D.J.; Boyd, B.J.; O’Reilly, R.K.; Dove, A.P. Tuning the Size of Cylindrical Micelles from Poly(l-lactide)-b-poly(acrylic acid) Diblock Copolymers Based on Crystallization-Driven Self-Assembly. Macromolecules 2013, 46, 9074–9082. [Google Scholar] [CrossRef]
- Presa Soto, A.; Gilroy, J.B.; Winnik, M.A.; Manners, I. Pointed-Oval-Shaped Micelles from Crystalline-Coil Block Copolymers by Crystallization-Driven Living Self-Assembly. Angew. Chem. Int. Ed. 2010, 49, 8220–8223. [Google Scholar] [CrossRef]
- Petzetakis, N.; Dove, A.P.; O’Reilly, R.K. Cylindrical micelles from the living crystallization-driven self-assembly of poly(lactide)-containing block copolymers. Chem. Sci. 2011, 2, 955–960. [Google Scholar] [CrossRef]
- Hudson, Z.M.; Boott, C.E.; Robinson, M.E.; Rupar, P.A.; Winnik, M.A.; Manners, I. Tailored hierarchical micelle architectures using living crystallization-driven self-assembly in two dimensions. Nat. Chem. 2014, 6, 893–898. [Google Scholar] [CrossRef]
- Finnegan, J.R.; Lunn, D.J.; Gould, O.E.C.; Hudson, Z.M.; Whittell, G.R.; Winnik, M.A.; Manners, I. Gradient Crystallization-Driven Self-Assembly: Cylindrical Micelles with “Patchy” Segmented Coronas via the Coassembly of Linear and Brush Block Copolymers. J. Am. Chem. Soc. 2014, 136, 13835–13844. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K.; Ahmed, R.; Whittell, G.R.; Lunn, D.J.; Dunphy, E.L.; Winnik, M.A.; Manners, I. Cylindrical Micelles of Controlled Length with a π-Conjugated Polythiophene Core via Crystallization-Driven Self-Assembly. J. Am. Chem. Soc. 2011, 133, 8842–8845. [Google Scholar] [CrossRef]
- Qiu, H.; Gao, Y.; Du, V.A.; Harniman, R.; Winnik, M.A.; Manners, I. Branched Micelles by Living Crystallization-Driven Block Copolymer Self-Assembly under Kinetic Control. J. Am. Chem. Soc. 2015, 137, 2375–2385. [Google Scholar] [CrossRef]
- Qian, J.; Li, X.; Lunn, D.J.; Gwyther, J.; Hudson, Z.M.; Kynaston, E.; Rupar, P.A.; Winnik, M.A.; Manners, I. Uniform, High Aspect Ratio Fiber-like Micelles and Block Co-micelles with a Crystalline π-Conjugated Polythiophene Core by Self-Seeding. J. Am. Chem. Soc. 2014, 136, 4121–4124. [Google Scholar] [CrossRef]
- Tritschler, U.; Gwyther, J.; Harniman, R.L.; Whittell, G.R.; Winnik, M.A.; Manners, I. Toward Uniform Nanofibers with a π-Conjugated Core: Optimizing the “Living” Crystallization-Driven Self-Assembly of Diblock Copolymers with a Poly(3-octylthiophene) Core-Forming Block. Macromolecules 2018, 51, 5101–5113. [Google Scholar] [CrossRef]
- Geng, Y.; Dalhaimer, P.; Cai, S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D.E. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2, 249–255. [Google Scholar] [CrossRef]
- Kim, Y.; Dalhaimer, P.; Christian, D.A.; Discher, D.E. Polymeric worm micelles as nano-carriers for drug delivery. Nanotechnology 2005, 16, S484–S491. [Google Scholar] [CrossRef]
- Hartgerink, J.D.; Beniash, E.; Stupp, S.I. Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers. Science 2001, 294, 1684–1688. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tekobo, S.; Tu, Y.; Zhou, Q.; Jin, X.; Dergunov, S.A.; Pinkhassik, E.; Yan, B. Permission to enter cell by shape: Nanodisk vs nanosphere. ACS Appl. Mater. Interfaces 2012, 4, 4099–4105. [Google Scholar] [CrossRef] [PubMed]
- Rizis, G.; van de Ven, T.G.M.; Eisenberg, A. “Raft” Formation by Two-Dimensional Self-Assembly of Block Copolymer Rod Micelles in Aqueous Solution. Angew. Chem. Int. Ed. 2014, 53, 9000–9003. [Google Scholar] [CrossRef]
- Li, X.; Jin, B.; Gao, Y.; Hayward, D.W.; Winnik, M.A.; Luo, Y.; Manners, I. Monodisperse Cylindrical Micelles of Controlled Length with a Liquid-Crystalline Perfluorinated Core by 1D “Self-Seeding”. Angew. Chem. Int. Ed. 2016, 55, 11392–11396. [Google Scholar] [CrossRef] [Green Version]
- Jin, B.; Sano, K.; Aya, S.; Ishida, Y.; Gianneschi, N.; Luo, Y.; Li, X. One-pot universal initiation-growth methods from a liquid crystalline block copolymer. Nat. Commun. 2019, 10, 2397. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gao, H.; Lin, J.; Wang, L.; Wang, X.-S.; Yang, C.; Lin, S. Growth and Termination of Cylindrical Micelles via Liquid-Crystallization-Driven Self-Assembly. Macromolecules 2020, 53, 8992–8999. [Google Scholar] [CrossRef]
- Rider, D.A.; Manners, I. Synthesis, Self-Assembly, and Applications of Polyferrocenylsilane Block Copolymers. Polym. Rev. 2007, 47, 165–195. [Google Scholar] [CrossRef]
- Hailes, R.L.N.; Oliver, A.M.; Gwyther, J.; Whittell, G.R.; Manners, I. Polyferrocenylsilanes: Synthesis, properties, and applications. Chem. Soc. Rev. 2016, 45, 5358–5407. [Google Scholar] [CrossRef] [Green Version]
- Kirshenbaum, K.; Barron, A.E.; Goldsmith, R.A.; Armand, P.; Bradley, E.K.; Truong, K.T.V.; Dill, K.A.; Cohen, F.E.; Zuckermann, R.N. Sequence-specific polypeptoids: A diverse family of heteropolymers with stable secondary structure. Proc. Natl. Acad. Sci. USA 1998, 95, 4303–4308. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lahasky, S.H.; Guo, L.; Lee, C.-U.; Lavan, M. Polypeptoid Materials: Current Status and Future Perspectives. Macromolecules 2012, 45, 5833–5841. [Google Scholar] [CrossRef]
- Sun, J.; Zuckermann, R.N. Peptoid Polymers: A Highly Designable Bioinspired Material. ACS Nano 2013, 7, 4715–4732. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, N.; Ulbricht, J.; Lorson, T.; Schlaad, H.; Luxenhofer, R. Peptoids and Polypeptoids at the Frontier of Supra- and Macromolecular Engineering. Chem. Rev. 2016, 116, 1753–1802. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.A.; Xuan, S.; Li, A.; Simpson, J.M.; Sternhagen, G.L.; Yu, T.; Darvish, O.A.; Jiang, N.; Zhang, D. Polypeptoid polymers: Synthesis, characterization, and properties. Biopolymers 2017, 109, e23070. [Google Scholar]
- Xuan, S.; Zuckermann, R.N. Diblock copolypeptoids: A review of phase separation, crystallization, self-assembly and biological applications. J. Mater. Chem. B 2020, 8, 5380–5394. [Google Scholar] [CrossRef] [PubMed]
- Statz, A.R.; Meagher, R.J.; Barron, A.E.; Messersmith, P.B. New Peptidomimetic Polymers for Antifouling Surfaces. J. Am. Chem. Soc. 2005, 127, 7972–7973. [Google Scholar] [CrossRef] [PubMed]
- Statz, A.R.; Barron, A.E.; Messersmith, P.B. Protein, cell and bacterial fouling resistance of polypeptoid-modified surfaces: Effect of side-chain chemistry. Soft Matter 2008, 4, 131–139. [Google Scholar] [CrossRef]
- Lau, K.H.A.; Ren, C.; Sileika, T.S.; Park, S.H.; Szleifer, I.; Messersmith, P.B. Surface-Grafted Polysarcosine as a Peptoid Antifouling Polymer Brush. Langmuir 2012, 28, 16099–16107. [Google Scholar] [CrossRef] [Green Version]
- Leng, C.; Buss, H.G.; Segalman, R.A.; Chen, Z. Surface Structure and Hydration of Sequence-Specific Amphiphilic Polypeptoids for Antifouling/Fouling Release Applications. Langmuir 2015, 31, 9306–9311. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.L.; Wenning, B.; Rizis, G.; Calabrese, D.R.; Finlay, J.A.; Franco, S.C.; Zuckermann, R.N.; Clare, A.S.; Kramer, E.J.; Ober, C.K.; et al. Role of Backbone Chemistry and Monomer Sequence in Amphiphilic Oligopeptide- and Oligopeptoid-Functionalized PDMS- and PEO-Based Block Copolymers for Marine Antifouling and Fouling Release Coatings. Macromolecules 2017, 50, 2656–2667. [Google Scholar] [CrossRef]
- Gao, Q.; Li, P.; Zhao, H.; Chen, Y.; Jiang, L.; Ma, P.X. Methacrylate-ended polypeptides and polypeptoids for antimicrobial and antifouling coatings. Polym. Chem. 2017, 8, 6386–6397. [Google Scholar] [CrossRef]
- Li, A.; Zhang, D. Synthesis and Characterization of Cleavable Core-Cross-Linked Micelles Based on Amphiphilic Block Copolypeptoids as Smart Drug Carriers. Biomacromolecules 2016, 17, 852–861. [Google Scholar] [CrossRef]
- Zhu, L.P.; Simpson, J.M.; Xu, X.; He, H.; Zhang, D.H.; Yin, L.C. Cationic Polypeptoids with Optimized Molecular Characteristics toward Efficient Nonviral Gene Delivery. ACS Appl. Mater. Interfaces 2017, 9, 23476–23486. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Chen, H.; Tao, X.; Cao, F.; Trépout, S.; Ling, J.; Li, M.-H. Oxidation-Sensitive Polymersomes Based on Amphiphilic Diblock Copolypeptoids. Biomacromolecules 2019, 20, 3435–3444. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, M.; Li, S.; Jin, H.; Cai, X.; Du, D.; Li, H.; Chen, C.-L.; Lin, Y. Efficient Cytosolic Delivery Using Crystalline Nanoflowers Assembled from Fluorinated Peptoids. Small 2018, 14, 1803544. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Song, Y.; Wang, M.; Jian, T.; Ding, S.; Mu, P.; Liao, Z.; Shi, Q.; Cai, X.; Jin, H.; et al. Bioinspired Peptoid Nanotubes for Targeted Tumor Cell Imaging and Chemo-Photodynamic Therapy. Small 2019, 15, 1902485. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.J.; Kim, J.H.; Grzincic, E.M.; Kim, S.C.; Abate, A.R.; Zuckermann, R.N. Uniform, Large-Area, Highly Ordered Peptoid Monolayer and Bilayer Films for Sensing Applications. Langmuir 2019, 35, 13671–13680. [Google Scholar] [CrossRef]
- Tao, X.; Chen, H.; Trépout, S.; Cen, J.; Ling, J.; Li, M.-H. Polymersomes with aggregation-induced emission based on amphiphilic block copolypeptoids. Chem. Commun. 2019, 55, 13530–13533. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, D. Cyclic Poly(α-peptoid)s and Their Block Copolymers from N-Heterocyclic Carbene-Mediated Ring-Opening Polymerizations of N-Substituted N-Carboxylanhydrides. J. Am. Chem. Soc. 2009, 131, 18072–18074. [Google Scholar] [CrossRef]
- Lee, C.-U.; Smart, T.P.; Guo, L.; Epps, T.H.; Zhang, D. Synthesis and Characterization of Amphiphilic Cyclic Diblock Copolypeptoids from N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-Carboxyanhydride. Macromolecules 2011, 44, 9574–9585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fetsch, C.; Grossmann, A.; Holz, L.; Nawroth, J.F.; Luxenhofer, R. Polypeptoids from N-Substituted Glycine N-Carboxyanhydrides: Hydrophilic, Hydrophobic, and Amphiphilic Polymers with Poisson Distribution. Macromolecules 2011, 44, 6746–6758. [Google Scholar] [CrossRef]
- Fetsch, C.; Luxenhofer, R. Highly Defined Multiblock Copolypeptoids: Pushing the Limits of Living Nucleophilic Ring-Opening Polymerization. Macromol. Rapid Commun. 2012, 33, 1708–1713. [Google Scholar] [CrossRef]
- Guo, L.; Lahasky, S.H.; Ghale, K.; Zhang, D. N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-Carboxyanhydrides toward Poly(α-peptoid)s: Kinetic, Mechanism, and Architectural Control. J. Am. Chem. Soc. 2012, 134, 9163–9171. [Google Scholar] [CrossRef]
- Robinson, J.W.; Schlaad, H. A versatile polypeptoid platform based on N-allyl glycine. Chem. Commun. 2012, 48, 7835–7837. [Google Scholar] [CrossRef]
- Tao, X.; Du, J.; Wang, Y.; Ling, J. Polypeptoids with tunable cloud point temperatures synthesized from N-substituted glycine N-thiocarboxyanhydrides. Polym. Chem. 2015, 6, 3164–3174. [Google Scholar] [CrossRef]
- Tao, X.; Deng, Y.; Shen, Z.; Ling, J. Controlled Polymerization of N-Substituted Glycine N-Thiocarboxyanhydrides Initiated by Rare Earth Borohydrides toward Hydrophilic and Hydrophobic Polypeptoids. Macromolecules 2014, 47, 6173–6180. [Google Scholar] [CrossRef]
- Xuan, S.; Lee, C.-U.; Chen, C.; Doyle, A.B.; Zhang, Y.; Guo, L.; John, V.T.; Hayes, D.; Zhang, D. Thermoreversible and Injectable ABC Polypeptoid Hydrogels: Controlling the Hydrogel Properties through Molecular Design. Chem. Mater. 2016, 28, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternhagen, G.L.; Gupta, S.; Zhang, Y.; John, V.; Schneider, G.J.; Zhang, D. Solution Self-Assemblies of Sequence-Defined Ionic Peptoid Block Copolymers. J. Am. Chem. Soc. 2018, 140, 4100–4109. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; Shelby, S.A.; Choi, P.H.; Marciel, A.B.; Chen, R.; Tan, L.; Chu, T.K.; Mesch, R.A.; Lee, B.C.; Connolly, M.D.; et al. Free-floating ultrathin two-dimensional crystals from sequence-specific peptoid polymers. Nat. Mater. 2010, 9, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-U.; Lu, L.; Chen, J.; Garno, J.C.; Zhang, D. Crystallization-Driven Thermoreversible Gelation of Coil-Crystalline Cyclic and Linear Diblock Copolypeptoids. ACS Macro Lett. 2013, 2, 436–440. [Google Scholar] [CrossRef]
- Sanii, B.; Haxton, T.K.; Olivier, G.K.; Cho, A.; Barton, B.; Proulx, C.; Whitelam, S.; Zuckermann, R.N. Structure-Determining Step in the Hierarchical Assembly of Peptoid Nanosheets. ACS Nano 2014, 8, 11674–11684. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Jiang, X.; Lund, R.; Downing, K.H.; Balsara, N.P.; Zuckermann, R.N. Self-assembly of crystalline nanotubes from monodisperse amphiphilic diblock copolypeptoid tiles. Proc. Natl. Acad. Sci. USA 2016, 113, 3954–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Jiao, F.; Daily, M.D.; Chen, Y.; Yan, F.; Ding, Y.-H.; Zhang, X.; Robertson, E.J.; Baer, M.D.; Chen, C.-L. Highly stable and self-repairing membrane-mimetic 2D nanomaterials assembled from lipid-like peptoids. Nat. Commun. 2016, 7, 12252. [Google Scholar] [CrossRef] [PubMed]
- Fetsch, C.; Gaitzsch, J.; Messager, L.; Battaglia, G.; Luxenhofer, R. Self-Assembly of Amphiphilic Block Copolypeptoids—Micelles, Worms and Polymersomes. Sci. Rep. 2016, 6, 33491. [Google Scholar] [CrossRef] [Green Version]
- Robertson, E.J.; Battigelli, A.; Proulx, C.; Mannige, R.V.; Haxton, T.K.; Yun, L.S.; Whitelam, S.; Zuckermann, R.N. Design, Synthesis, Assembly, and Engineering of Peptoid Nanosheets. Acc. Chem. Res. 2016, 49, 379–389. [Google Scholar] [CrossRef]
- Ni, Y.; Sun, J.; Wei, Y.; Fu, X.; Zhu, C.; Li, Z. Two-Dimensional Supramolecular Assemblies from pH-Responsive Poly(ethyl glycol)-b-poly(l-glutamic acid)-b-poly(N-octylglycine) Triblock Copolymer. Biomacromolecules 2017, 18, 3367–3374. [Google Scholar] [CrossRef]
- Shi, Z.; Wei, Y.; Zhu, C.; Sun, J.; Li, Z. Crystallization-Driven Two-Dimensional Nanosheet from Hierarchical Self-Assembly of Polypeptoid-Based Diblock Copolymers. Macromolecules 2018, 51, 6344–6351. [Google Scholar] [CrossRef]
- Wei, Y.; Tian, J.; Zhang, Z.; Zhu, C.; Sun, J.; Li, Z. Supramolecular Nanosheets Assembled from Poly(ethylene glycol)-b-poly(N-(2-phenylethyl)glycine) Diblock Copolymer Containing Crystallizable Hydrophobic Polypeptoid: Crystallization Driven Assembly Transition from Filaments to Nanosheets. Macromolecules 2019, 52, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Yu, T.; Darvish, O.A.; Qian, S.; Mkam Tsengam, I.K.; John, V.; Zhang, D. Crystallization-Driven Self-Assembly of Coil–Comb-Shaped Polypeptoid Block Copolymers: Solution Morphology and Self-Assembly Pathways. Macromolecules 2019, 52, 8867–8877. [Google Scholar] [CrossRef]
- Sun, J.; Wang, Z.; Zhu, C.; Wang, M.; Shi, Z.; Wei, Y.; Fu, X.; Chen, X.; Zuckermann, R.N. Hierarchical supramolecular assembly of a single peptoid polymer into a planar nanobrush with two distinct molecular packing motifs. Proc. Natl. Acad. Sci. USA 2020, 117, 31639–31647. [Google Scholar] [CrossRef]
- Gangloff, N.; Höferth, M.; Stepanenko, V.; Sochor, B.; Schummer, B.; Nickel, J.; Walles, H.; Hanke, R.; Würthner, F.; Zuckermann, R.N.; et al. Linking two worlds in polymer chemistry: The influence of block uniformity and dispersity in amphiphilic block copolypeptoids on their self-assembly. Biopolymers 2019, 110, e23259. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lin, M.; Bonduelle, C.; Li, R.; Shi, Z.; Zhu, C.; Lecommandoux, S.; Li, Z.; Sun, J. Thermoinduced Crystallization-Driven Self-Assembly of Bioinspired Block Copolymers in Aqueous Solution. Biomacromolecules 2020, 21, 3411–3419. [Google Scholar] [CrossRef]
- Xuan, S.; Jiang, X.; Balsara, N.P.; Zuckermann, R.N. Crystallization and self-assembly of shape-complementary sequence-defined peptoids. Polym. Chem. 2021, 12, 4770–4777. [Google Scholar] [CrossRef]
- Kang, L.; Chao, A.; Zhang, M.; Yu, T.; Wang, J.; Wang, Q.; Yu, H.; Jiang, N.; Zhang, D. Modulating the Molecular Geometry and Solution Self-Assembly of Amphiphilic Polypeptoid Block Copolymers by Side Chain Branching Pattern. J. Am. Chem. Soc. 2021, 143, 5890–5902. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liu, F.; Li, M.; Li, Z.; Sun, J. Dimension control on self-assembly of a crystalline core-forming polypeptoid block copolymer: 1D nanofibers versus 2D nanosheets. Polym. Chem. 2021, 12, 1147–1154. [Google Scholar] [CrossRef]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K.; et al. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Qi, J.H.; Tao, J.H.; Zuckermann, R.N.; DeYoreo, J.J. Tuning calcite morphology and growth acceleration by a rational design of highly stable protein-mimetics. Sci. Rep. 2014, 4, 6266. [Google Scholar] [CrossRef] [Green Version]
- Rosales, A.M.; Murnen, H.K.; Zuckermann, R.N.; Segalman, R.A. Control of Crystallization and Melting Behavior in Sequence Specific Polypeptoids. Macromolecules 2010, 43, 5627–5636. [Google Scholar] [CrossRef]
- Sun, J.; Teran, A.A.; Liao, X.X.; Balsara, N.P.; Zuckermann, R.N. Crystallization in Sequence-Defined Peptoid Diblock Copolymers Induced by Microphase Separation. J. Am. Chem. Soc. 2014, 136, 2070–2077. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Teran, A.A.; Liao, X.X.; Balsara, N.P.; Zuckermann, R.N. Nanoscale Phase Separation in Sequence-Defined Peptoid Diblock Copolymers. J. Am. Chem. Soc. 2013, 135, 14119–14124. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Jiang, X.; Siegmund, A.; Connolly, M.D.; Downing, K.H.; Balsara, N.P.; Zuckermann, R.N. Morphology and Proton Transport in Humidified Phosphonated Peptoid Block Copolymers. Macromolecules 2016, 49, 3083–3090. [Google Scholar] [CrossRef]
- Chen, C.L.; Zuckermann, R.N.; DeYoreo, J.J. Surface-Directed Assembly of Sequence Defined Synthetic Polymers into Networks of Hexagonally Patterned Nanoribbons with Controlled Functionalities. ACS Nano 2016, 10, 5314–5320. [Google Scholar] [CrossRef]
- Guo, L.; Li, J.; Brown, Z.; Ghale, K.; Zhang, D. Synthesis and characterization of cyclic and linear helical poly(α-peptoid)s by N-heterocyclic carbene-mediated ring-opening polymerizations of N-substituted N-carboxyanhydrides. Peptide Sci. 2011, 96, 596–603. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, D. Synthesis and Characterization of Helix-Coil Block Copoly(α-peptoid)s. In Non-Conventional Functional Block Copolymers; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2011; Volume 1066, pp. 71–79. [Google Scholar]
- Tao, X.; Zheng, B.; Bai, T.; Li, M.-H.; Ling, J. Polymerization of N-Substituted Glycine N-Thiocarboxyanhydride through Regioselective Initiation of Cysteamine: A Direct Way toward Thiol-Capped Polypeptoids. Macromolecules 2018, 51, 4494–4501. [Google Scholar] [CrossRef]
- Tao, X.; Zheng, B.; Kricheldorf, H.R.; Ling, J. Are N-substituted glycine N-thiocarboxyanhydride monomers really hard to polymerize? J. Polym. Sci. A Polym. Chem. 2017, 55, 404–410. [Google Scholar] [CrossRef]
- Lee, C.-U.; Li, A.; Ghale, K.; Zhang, D. Crystallization and Melting Behaviors of Cyclic and Linear Polypeptoids with Alkyl Side Chains. Macromolecules 2013, 46, 8213–8223. [Google Scholar] [CrossRef]
- Li, A.; Lu, L.; Li, X.; He, L.; Do, C.; Garno, J.C.; Zhang, D. Amidine-Mediated Zwitterionic Ring-Opening Polymerization of N-Alkyl N-Carboxyanhydride: Mechanism, Kinetics, and Architecture Elucidation. Macromolecules 2016, 49, 1163–1171. [Google Scholar] [CrossRef]
- Du, P.; Li, A.; Li, X.; Zhang, Y.; Do, C.; He, L.; Rick, S.W.; John, V.T.; Kumar, R.; Zhang, D. Aggregation of cyclic polypeptoids bearing zwitterionic end-groups with attractive dipole-dipole and solvophobic interactions: A study by small-angle neutron scattering and molecular dynamics simulation. Phys. Chem. Chem. Phys. 2017, 19, 14388–14400. [Google Scholar] [CrossRef] [PubMed]
- Fessler, J.H.; Ogston, A.G. Studies of the sedimentation, diffusion and viscosity of some sarcosine polymers in aqueous solution. Trans. Faraday Soc. 1951, 47, 667–679. [Google Scholar] [CrossRef]
- Bovey, F.A.; Ryan, J.J.; Hood, F.P. Polymer Nuclear Magnetic Resonance Spectroscopy. XV. The Conformation of Polysarcosine. Macromolecules 1968, 1, 305–307. [Google Scholar] [CrossRef]
- Birke, A.; Ling, J.; Barz, M. Polysarcosine-containing copolymers: Synthesis, characterization, self-assembly, and applications. Prog. Polym. Sci. 2018, 81, 163–208. [Google Scholar] [CrossRef]
- Ueda, M.; Uesaka, A.; Kimura, S. Selective disruption of each part of Janus molecular assemblies by lateral diffusion of stimuli-responsive amphiphilic peptides. Chem. Commun. 2015, 51, 1601–1604. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, Z.; Zhu, D.; Tao, X.; Gao, Y.; Zhu, H.; Mao, Z.; Ling, J. Gold nanoparticles coated with polysarcosine brushes to enhance their colloidal stability and circulation time in vivo. J. Colloid Interface Sci. 2016, 483, 201–210. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, Y.; Yan, F.J.; Chen, J.; Tao, X.F.; Ling, J.; Yang, B.; He, Q.J.; Mao, Z.W. Polysarcosine brush stabilized gold nanorods for in vivo near-infrared photothermal tumor therapy. Acta Biomater. 2017, 50, 534–545. [Google Scholar] [CrossRef]
- Hu, Y.; Hou, Y.; Wang, H.; Lu, H. Polysarcosine as an Alternative to PEG for Therapeutic Protein Conjugation. Bioconjug. Chem. 2018, 29, 2232–2238. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Birke, A.; Fischer, K.; Schmidt, M.; Barz, M. Solution Properties of Polysarcosine: From Absolute and Relative Molar Mass Determinations to Complement Activation. Macromolecules 2018, 51, 2653–2661. [Google Scholar] [CrossRef]
- Greer, D.R.; Stolberg, M.A.; Kundu, J.; Spencer, R.K.; Pascal, T.; Prendergast, D.; Balsara, N.P.; Zuckermann, R.N. Universal Relationship between Molecular Structure and Crystal Structure in Peptoid Polymers and Prevalence of the cis Backbone Conformation. J. Am. Chem. Soc. 2018, 140, 827–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fetsch, C.; Luxenhofer, R. Thermal Properties of Aliphatic Polypeptoids. Polymers 2013, 5, 112–127. [Google Scholar] [CrossRef] [Green Version]
- Heijboer, J. Physics of Non-Crystalline Solids; Prins, J.A., Ed.; North-Holland Publishing Co.: Amsterdam, The Netherlands, 1965; pp. 231–235. [Google Scholar]
- Greer, D.R.; Stolberg, M.A.; Xuan, S.; Jiang, X.; Balsara, N.P.; Zuckermann, R.N. Liquid-Crystalline Phase Behavior in Polypeptoid Diblock Copolymers. Macromolecules 2018, 51, 9519–9525. [Google Scholar] [CrossRef]
- Jiang, N.; Chen, J.; Yu, T.; Chao, A.; Kang, L.; Wu, Y.; Niu, K.; Li, R.; Fukuto, M.; Zhang, D. Cyclic Topology Enhancing Structural Ordering and Stability of Comb-Shaped Polypeptoid Thin Films against Melt-Induced Dewetting. Macromolecules 2020, 53, 7601–7612. [Google Scholar] [CrossRef]
- Platé, N.A.f.; Shibaev, V.P. Comb-Shaped Polymers and Liquid Crystals; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Robinson, J.W.; Secker, C.; Weidner, S.; Schlaad, H. Thermoresponsive Poly(N-C3 glycine)s. Macromolecules 2013, 46, 580–587. [Google Scholar] [CrossRef]
- Roe, R.J. Methods of X-ray and Neutron Scattering in Polymer Science; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Chu, B.; Hsiao, B.S. Small-Angle X-ray Scattering of Polymers. Chem. Rev. 2001, 101, 1727–1762. [Google Scholar] [CrossRef] [PubMed]
- Broennimann, C.; Eikenberry, E.F.; Henrich, B.; Horisberger, R.; Huelsen, G.; Pohl, E.; Schmitt, B.; Schulze-Briese, C.; Suzuki, M.; Tomizaki, T.; et al. The PILATUS 1M detector. J. Synchrotron Radiat. 2006, 13, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchet, C.E.; Spilotros, A.; Schwemmer, F.; Graewert, M.A.; Kikhney, A.; Jeffries, C.M.; Franke, D.; Mark, D.; Zengerle, R.; Cipriani, F.; et al. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystallogr. 2015, 48, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portale, G.; Hermida-Merino, D.; Bras, W. Polymer research and synchrotron radiation perspectives. Eur. Polym. J. 2016, 81, 415–432. [Google Scholar] [CrossRef]
- Trebbin, M.; Steinhauser, D.; Perlich, J.; Buffet, A.; Roth, S.V.; Zimmermann, W.; Thiele, J.; Förster, S. Anisotropic particles align perpendicular to the flow direction in narrow microchannels. Proc. Natl. Acad. Sci. USA 2013, 110, 6706–6711. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Altunbas, A.; Yucel, T.; Nagarkar, R.P.; Schneider, J.P.; Pochan, D.J. Injectable solid hydrogel: Mechanism of shear-thinning and immediate recovery of injectable β-hairpin peptide hydrogels. Soft Matter 2010, 6, 5143–5156. [Google Scholar] [CrossRef] [Green Version]
- Butler, P. Shear induced structures and transformations in complex fluids. Curr. Opin. Colloid Interface Sci. 1999, 4, 214–221. [Google Scholar] [CrossRef]
- Pedersen, J.S.; Gerstenberg, M.C. Scattering Form Factor of Block Copolymer Micelles. Macromolecules 1996, 29, 1363–1365. [Google Scholar] [CrossRef]
- Pedersen, J.S. Form factors of block copolymer micelles with spherical, ellipsoidal and cylindrical cores. J. Appl. Crystallogr. 2000, 33, 637–640. [Google Scholar] [CrossRef]
- Xu, J.; Ma, Y.; Hu, W.; Rehahn, M.; Reiter, G. Cloning polymer single crystals through self-seeding. Nat. Mater. 2009, 8, 348–353. [Google Scholar] [CrossRef]
- Qian, J.; Guerin, G.; Lu, Y.; Cambridge, G.; Manners, I.; Winnik, M.A. Self-Seeding in One Dimension: An Approach to Control the Length of Fiberlike Polyisoprene–Polyferrocenylsilane Block Copolymer Micelles. Angew. Chem. Int. Ed. 2011, 50, 1622–1625. [Google Scholar] [CrossRef]
- Jiang, X.; Greer, D.R.; Kundu, J.; Ophus, C.; Minor, A.M.; Prendergast, D.; Zuckermann, R.N.; Balsara, N.P.; Downing, K.H. Imaging Unstained Synthetic Polymer Crystals and Defects on Atomic Length Scales Using Cryogenic Electron Microscopy. Macromolecules 2018, 51, 7794–7799. [Google Scholar] [CrossRef] [Green Version]
- Tschierske, C. Development of Structural Complexity by Liquid-Crystal Self-assembly. Angew. Chem. Int. Ed. 2013, 52, 8828–8878. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.J.; Keller, A. Solution crystallization of polyethylene at high temperatures. I: Lateral crystal habits. J. Mater. Sci. 1985, 20, 1571–1585. [Google Scholar] [CrossRef]
- Ganda, S.; Dulle, M.; Drechsler, M.; Förster, B.; Förster, S.; Stenzel, M.H. Two-Dimensional Self-Assembled Structures of Highly Ordered Bioactive Crystalline-Based Block Copolymers. Macromolecules 2017, 50, 8544–8553. [Google Scholar] [CrossRef]
- Cha, Y.; Jarrett-Wilkins, C.; Rahman, M.A.; Zhu, T.; Sha, Y.; Manners, I.; Tang, C. Crystallization-Driven Self-Assembly of Metallo-Polyelectrolyte Block Copolymers with a Polycaprolactone Core-Forming Segment. ACS Macro Lett. 2019, 8, 835–840. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, N.; Zhang, D. Solution Self-Assembly of Coil-Crystalline Diblock Copolypeptoids Bearing Alkyl Side Chains. Polymers 2021, 13, 3131. https://doi.org/10.3390/polym13183131
Jiang N, Zhang D. Solution Self-Assembly of Coil-Crystalline Diblock Copolypeptoids Bearing Alkyl Side Chains. Polymers. 2021; 13(18):3131. https://doi.org/10.3390/polym13183131
Chicago/Turabian StyleJiang, Naisheng, and Donghui Zhang. 2021. "Solution Self-Assembly of Coil-Crystalline Diblock Copolypeptoids Bearing Alkyl Side Chains" Polymers 13, no. 18: 3131. https://doi.org/10.3390/polym13183131