
Investigation on the α/δ Crystal Transition of Poly(l-lactic Acid) with Different Molecular Weights



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
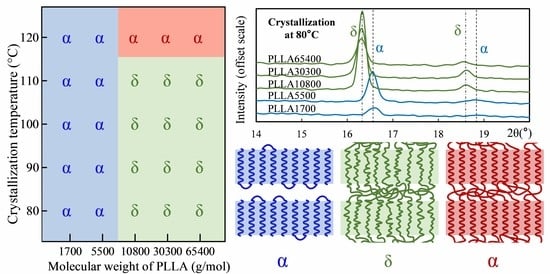
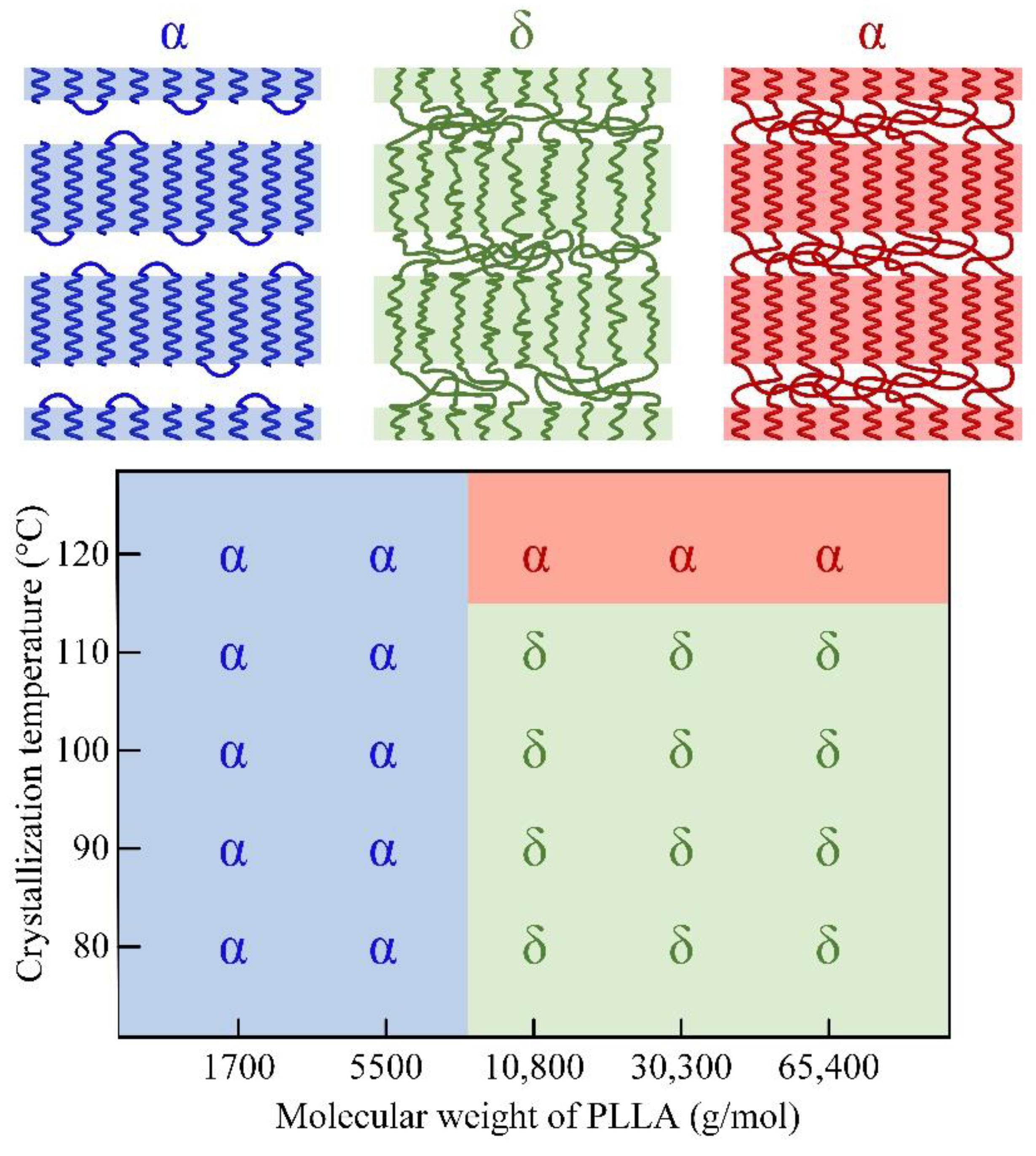
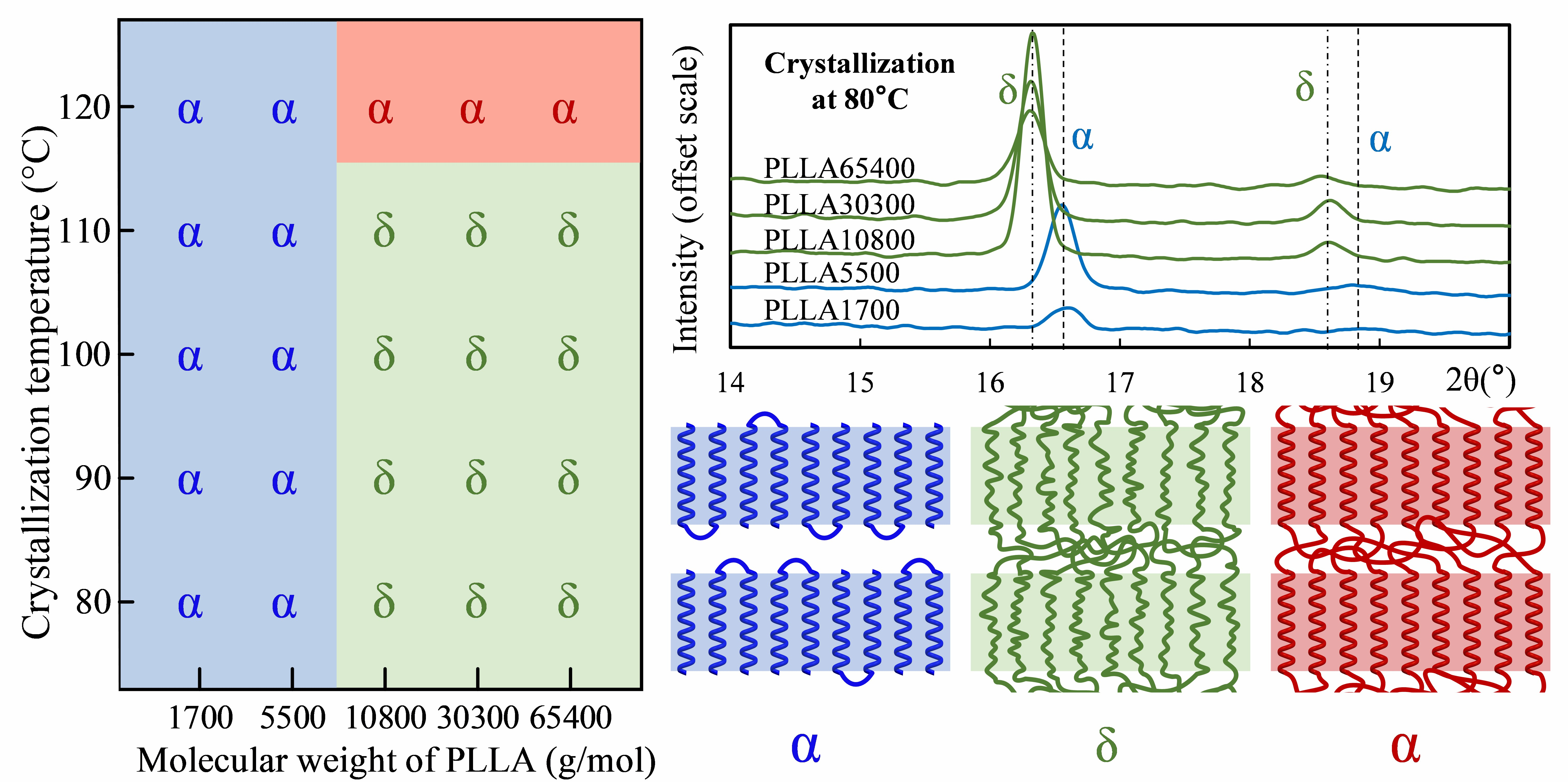
3.1. Crystal Morphology of PLLA with Different Molecular Weights
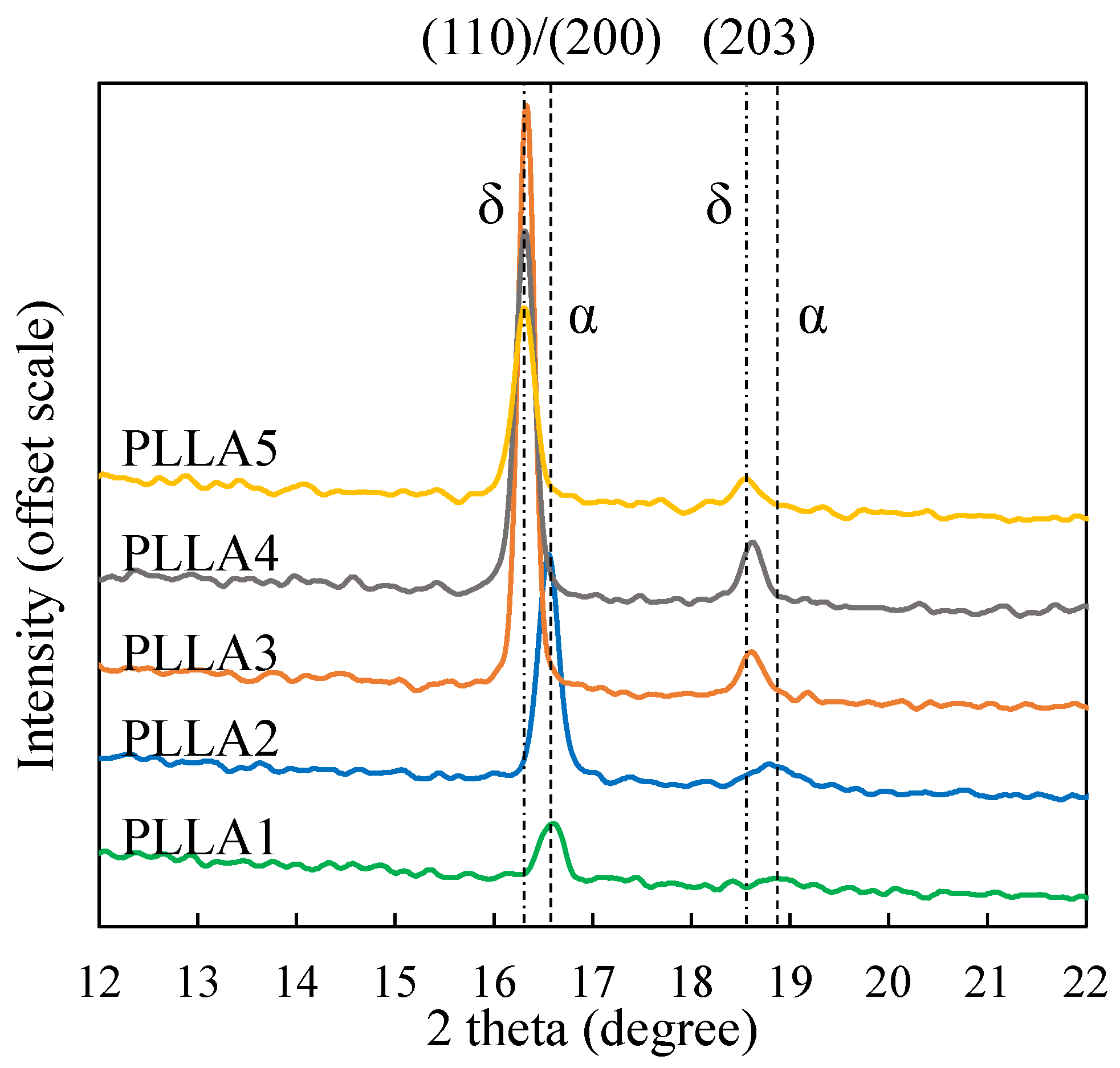
3.2. Form Transformation of PLLA Crystals
3.3. Calorimetric Properties of α and δ Crystal
3.4. SAXS Analysis of PLLA Samples
3.5. Rheology Properties of PLLA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Di Lorenzo, M.L.; Androsch, R. Synthesis, Structure and Properties of Poly(lactic acid); Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Kakroodi, A.R.; Kazemi, Y.; Ding, W.D.; Ameli, A.; Park, C.B. poly(lactic acid)-based In Situ Microfibrillar Composites with Enhanced Crystallization Kinetics, Mechanical Properties, Rheological Behavior, and Foaming Ability. Biomacromolecules 2015, 16, 3925–3935. [Google Scholar] [CrossRef]
- Bai, H.; Huang, C.; Xiu, H.; Zhang, Q.; Deng, H.; Wang, K.; Chen, F.; Fu, Q. Significantly Improving Oxygen Barrier Properties of Polylactide Via Constructing Parallel-Aligned Shish-Kebab-Like Crystals with Well-Interlocked Boundaries. Biomacromolecules 2014, 15, 1507–1514. [Google Scholar] [CrossRef]
- Aleman, C.; Lotz, B.; Puiggali, J. Crystal Structure of the α-form of Poly(L-lactide). Macromolecules 2001, 34, 4795–4801. [Google Scholar] [CrossRef]
- Wasanasuk, K.; Tashiro, K.; Hanesaka, M.; Ohhara, T.; Kurihara, K.; Kuroki, R.; Tamada, T.; Ozeki, T.; Kanamoto, T. Crystal Structure Analysis of poly(L-lactic Acid) α Form on the Basis of the 2-Dimensional Wide-Angle Synchrotron X-Ray and Neutron Diffraction Measurements. Macromolecules 2011, 44, 6441–6452. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, J.; Tashiro, K. Phase Transition Mechanism of Poly(L-lactic acid) among the α, δ, and β Forms on the Basis of the Reinvestigated Crystal Structure of the β Form. Macromolecules 2017, 50, 3285–3300. [Google Scholar] [CrossRef]
- Zhang, J.; Tashiro, K.; Tsuji, H.; Domb, A.J. Disorder-to-Order Phase Transition and Multiple Melting Behavior of Poly(L-lactide) Investigated by Simultaneous Measurements of WAXD and DSC. Macromolecules 2008, 41, 1352–1357. [Google Scholar] [CrossRef]
- Wasanasuk, K.; Tashiro, K. Crystal Structure and Disorder in Poly(L-lactic Acid) δ Form (α′ Form) and the Phase Transition Mechanism to the Ordered α Form. Polymer 2011, 52, 6097–6109. [Google Scholar] [CrossRef]
- Hoogsteen, W.; Postema, A.R.; Pennings, A.J.; Ten Brinke, G.; Zugenmaier, P. Crystal Structure, Conformation and Morphology of Solution-spun poly(L-lactide) Fibers. Macromolecules 1990, 23, 634–642. [Google Scholar] [CrossRef]
- Puiggali, J.; Ikada, Y.; Tsuji, H.; Cartier, L.; Okihara, T.; Lotz, B. The Frustrated Structure of Poly(L-lactide). Polymer 2000, 41, 8921–8930. [Google Scholar] [CrossRef]
- Sawai, D.; Takahashi, K.; Imamura, T.; Nakamura, K.; Kanamoto, T.; Hyon, S.-H. Preparation of Oriented β-Form Poly(Llactic Acid) by Solid-State Extrusion. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 95–104. [Google Scholar] [CrossRef]
- Eling, B.; Gogolewski, S.; Pennings, A.J. Biodegradable Materials of Poly(L-Lactic Acid): 1. Melt-spun and Solution-spun Fibres. Polymer 1982, 23, 1587–1593. [Google Scholar] [CrossRef]
- Cartier, L.; Okihara, T.; Ikada, Y.; Tsuji, H.; Puiggali, J.; Lotz, B. Epitaxial Crystallization and Crystalline Polymorphism of Polylactides. Polymer 2000, 41, 8909–8919. [Google Scholar] [CrossRef]
- Sasaki, S.; Asakura, T. Helix Distortion and Crystal Structure of the α-Form of poly(L-lactide). Macromolecules 2003, 36, 8385–8390. [Google Scholar] [CrossRef]
- Wasanasuk, K.; Tashiro, K. Structural Regularization in the Crystallization Process from the Glass or Melt of Poly(L-lactic Acid) Viewed from the Temperature-dependent and Time-resolved Measurements of FTIR and Wide-angle/Small-angle X-ray Scatterings. Macromolecules 2011, 44, 9650–9660. [Google Scholar] [CrossRef]
- Zhang, J.; Tashiro, K.; Domb, A.J.; Tsuji, H. Confirmation of Disorder α Form of Poly(L-lactic acid) by the X-ray Fiber Pattern and Polarized IR/Raman Spectra Measured for Uniaxially-oriented Samples. Macromol. Symp. 2006, 242, 274–278. [Google Scholar] [CrossRef]
- Zhang, J.; Tashiro, K.; Tsuji, H.; Domb, A.J. Investigation of Phase Transitional Behavior of Poly(L-lactide)/Poly(D-lactide) Blend Used to Prepare the Highly-oriented stereocomplex. Macromolecules 2007, 40, 1049–1054. [Google Scholar] [CrossRef]
- Zhang, J.; Duan, Y.; Sato, H.; Tsuji, H.; Noda, I.; Yan, S.; Ozaki, Y. Crystal modifications and thermal behavior of poly(L-lactic acid) revealed by infrared spectroscopy. Macromolecules 2005, 38, 8012–8021. [Google Scholar] [CrossRef]
- Pan, P.; Kai, W.; Zhu, B.; Dong, T.; Inoue, Y. Polymorphous Crystallization and Multiple Melting Behavior of Poly(L-lactide): Molecular Weight Dependence. Macromolecules 2007, 40, 6898–6905. [Google Scholar] [CrossRef]
- Pan, P.; Zhu, B.; Kai, W.; Dong, T.; Inoue, Y. Polymorphic transition in disordered poly(L-lactide) crystals induced by annealing at elevated temperatures. Macromolecules 2008, 41, 4296–4304. [Google Scholar] [CrossRef]
- Pan, P.; Han, L.; Shan, G.; Bao, Y. Heating and annealing induced structural reorganization and embrittlement of solution-crystallized poly(L-lactic acid). Macromolecules 2014, 47, 8126–8130. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, G.; Wang, G. Investigation on the growth of snowflake-shaped poly(L-Lactic Acid) crystal by in-situ high-pressure microscope. Polymer 2019, 177, 25–34. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, G.; Wang, G. Investigation on the influence of fold conformation on PLLA lamellar splaying by film crystallization in supercritical CO2. CrystEngComm 2020, 22, 1459–1472. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, G.; Wang, G. Investigation of the Influence of Pressurized CO2 on Crystal Growth of poly(L-Lactic Acid) by Using In-situ High-pressure Optical System. Soft Matter 2019, 15, 5714–5727. [Google Scholar] [CrossRef]
- Chai, Y.; Raegen, A.N.; Zhu, S.; Forrest, J.A. Crystallization of low molecular weight atactic polystyrene. Soft Matter 2018, 14, 6883–6891. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Li, Y. Mesophase-Mediated Crystallization of Poly(L-lactide): Deterministic Pathways to Nanostructured Morphology and Superstructure Control. Macromolecules 2016, 49, 7387–7399. [Google Scholar] [CrossRef]
- Lan, Q.; Li, Y.; Chi, H. Highly enhanced mesophase formation in glassy poly(L-lactide) at low temperatures by low-pressure CO2 that provides moderately increased molecular mobility. Macromolecules 2016, 49, 2262–2271. [Google Scholar] [CrossRef]
- Zhang, J.; Sato, H.; Tsuji, H.; Noda, I.; Ozaki, Y. Infrared Spectroscopic Study of CH3•••O=C Interaction during Poly(L-lactide)/Poly(D-lactide) Stereocomplex Formation. Macromolecules 2005, 38, 1822–1829. [Google Scholar] [CrossRef]
- Nyquist, R. The Infrared Spectra Atlas of Monomers and Polymers; Amazon: Seattle, WA, USA, 1980. [Google Scholar]
- Qiao, Y.; Yang, F.; Lu, Y.; Liu, P.; Li, Y.; Men, Y. Spontaneous form II to I transition in low molar mass polybutene-1 at crystallization temperature reveals stabilization role of intercrystalline links and entanglements for metastable form II crystals. Macromolecules 2018, 51, 8298–8305. [Google Scholar] [CrossRef]
- Shieh, Y.T.; Lin, Y.G.; Chen, H.L. Effect of Supercritical CO2 on Phase Structure of PEO/PVAc Blends Evaluated from SAXS Absolute Intensity Measurement. Polymer 2002, 43, 3691–3698. [Google Scholar] [CrossRef]
- Shieh, Y.T.; Liu, K.H.; Lin, T.L. Effect of Supercritical CO2 on Morphology of Compatible Crystalline/Amorphous PEO/PMMA blends. J. Supercrit. Fluid 2004, 28, 101–112. [Google Scholar] [CrossRef]
- Shieh, Y.T.; Hsiao, T.T.; Chang, S.K. CO2 Pressure Effects on Melting, Crystallization, and Morphology of Poly(vinylidene fluoride). Polymer 2006, 47, 5929–5937. [Google Scholar] [CrossRef]
- Strobl, G.R.; Schneider, M. Direct Evaluation of the Electron Density Correlation Function of Partially Crystalline Polymers. J. Polym. Sci. Part B Polym. Phys. 1980, 18, 1343–1359. [Google Scholar] [CrossRef]
- Wang, X.; Liu, R.; Wu, M.; Wang, Z.; Huang, Y. Effect of chain disentanglement on melt crystallization behavior of isotactic polypropylene. Polymer 2009, 50, 5824–5827. [Google Scholar] [CrossRef]
- Kurz, R.; Schulz, M.; Scheliga, F.; Men, Y.; Seidlitz, A.; Thurn-Albrecht, T.; Saalwächter, K. Interplay between Crystallization and Entanglements in the Amorphous Phase of the Crystal-fixed Polymer Poly(ϵ-caprolactone). Macromolecules 2018, 51, 5831–5841. [Google Scholar] [CrossRef]
- Dorgan, J.R.; Janzen, J.; Clayton, M.P.; Hait, S.B.; Knauss, D.M. Melt Rheology of Variable L-content Poly(lactic acid). J. Rheol. 2005, 49, 607–619. [Google Scholar] [CrossRef]
- Cooper-White, J.J.; Mackay, M.E. Rheological Properties of Poly(lactides). Effect of Molecular Weight and Temperature on the Viscoelasticity of Poly(l-lactic acid). J. Polym. Sci. Part B Polym. Phys. 1999, 37, 1803–1814. [Google Scholar] [CrossRef]
- Hu, W. The Physics of Polymer Chain-Folding. Phys. Rep. 2018, 747, 1–50. [Google Scholar] [CrossRef]
- Hu, W.; Frenkel, D.; Mathot, V.B.F. Intramolecular Nucleation Model for Polymer Crystallization. Macromolecules 2003, 36, 8178–8183. [Google Scholar] [CrossRef] [Green Version]
- Wunderlich, B.; Mehta, A. Macromolecular Nucleation. J. Polym. Sci. Part B Polym. Phys. 1974, 12, 255–263. [Google Scholar] [CrossRef]
- Mehta, A.; Wunderlich, B. A Study of Molecular Fractionation during the Crystallization of Polymers. Colloid Polym. Sci. 1975, 253, 193–205. [Google Scholar] [CrossRef]
- Wunderlich, B. Molecular Nucleation and Segregation. Faraday Discuss. Chem. Soc. 1979, 68, 239–243. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Wunderlich, B. Molecular Segregation and Nucleation of poly(ethylene oxide) Crystallized from the Melt. I. Calorimetric Study. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 577–594. [Google Scholar] [CrossRef]
- Hong, Y.; Koga, T.; Miyoshi, T. Chain Trajectory and Crystallization Mechanism of a Semicrystalline Polymer in Melt-And Solution-Grown Crystals as Studied Using 13C-13C Double-Quantum NMR. Macromolecules 2015, 48, 3282–3293. [Google Scholar] [CrossRef]
- Hong, Y.; Yuan, S.; Li, Z.; Ke, Y.; Nozaki, K.; Miyoshi, T. Three-Dimensional Conformation of Folded Polymers in Single Crystals. Phys. Rev. Lett. 2015, 115, 168301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Chen, W.; Yuan, S.; Kang, J.; Miyoshi, T. Chain Trajectory of Semicrystalline Polymers as Revealed by Solid-State NMR Spectroscopy. ACS Macro Lett. 2016, 5, 355–358. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, S.; Chen, W.; He, Q.; Hong, Y.; Miyoshi, T. Solid-State NMR Study of the Chain Trajectory and Crystallization Mechanism of poly(l-lactic acid) in Dilute Solution. Macromolecules 2017, 50, 6404–6414. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, S.; Chen, W.; Zhou, Y.; Hong, Y.; Miyoshi, T. Structural Unit of Polymer Crystallization in Dilute Solution as Studied by Solid-State NMR and 13C Isotope Labeling. Macromolecules 2018, 51, 8729–8737. [Google Scholar] [CrossRef]
- Ungar, G.; Stejny, J.; Keller, A.; Bidd, I.; Whiting, M.C. The Crystallization of Ultralong Normal Paraffins: The Onset of Chain Folding. Science 1985, 229, 386–389. [Google Scholar] [CrossRef]
- Ungar, G.; Keller, A. Time-resolved Synchrotron X-ray Study of Chain-folded Crystallization of Long Paraffins. Polymer 1986, 27, 1835–1844. [Google Scholar] [CrossRef]
- Fritzsching, K.J.; Mao, K.; Schmidt-Rohr, K. Avoidance of Density Anomalies as a Structural Principle for Semicrystalline Polymers: The Importance of Chain Ends and Chain Tilt. Macromolecules 2017, 50, 1521–1540. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikada, Y. Stereocomplex formation between enantiomeric poly(lactic acid)s. 6. Binary blends from copolymers. Macromolecules 1992, 25, 5719–5723. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikada, Y. Crystallization from the melt of poly(lactide) s with different optical purities and their blends. Macromol. Chem. Phys. 1996, 197, 3483–3499. [Google Scholar] [CrossRef]
- Tsuji, H.; Sobue, T. Stereocomplexation of quaternary or ternary monomer units and dual stereo-complexation in enantiomeric binary and quaternary polymer blends of poly(2-hydroxybutanoic acid)s, poly(2-hydroxybutanoic acid-co-lactic acid)s, and poly(lactic acid)s. RSC Adv. 2015, 5, 83331–83342. [Google Scholar] [CrossRef]
- Tsuji, H.; Osanai, K.; Arakawa, Y. Stereocomplex crystallization between L-and D-configured staggered asymmetric random copolymers based on 2-hydroxyalkanoic acids. Cryst. Growth Des. 2018, 18, 6009–6019. [Google Scholar] [CrossRef]
- Tsuji, H.; Yamasaki, M.; Arakawa, Y. Stereocomplex Formation between Enantiomeric Alternating Lactic Acid-Based Copolymers as a Versatile Method for the Preparation of High Performance Biobased Biodegradable Materials. ACS Appl. Polym. Mater. 2019, 1, 1476–1484. [Google Scholar] [CrossRef]
- Tsuji, H.; Sato, S.; Masaki, N.; Arakawa, Y.; Kuzuya, A.; Ohya, Y. Synthesis, stereo-complex crystallization and homo-crystallization of enantiomeric poly(lactic acid-co-alanine) s with ester and amide linkages. Polym. Chem. 2018, 9, 565–575. [Google Scholar] [CrossRef]
- Tsuji, H.; Sato, S.; Masaki, N.; Arakawa, Y.; Yoshizaki, Y.; Kuzuya, A.; Ohya, Y. Stereocomplex crystallization, homocrystallization, and polymorphism of enantiomeric copolyesteramides poly(lactic acid-co-alanine)s from the melt. Polym. Cryst. 2020, 3, e10094. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | [α]purified (°) | ||||
|---|---|---|---|---|---|
| PLLA1 | 1700 | 1.18 | * | 124.9 | −151.3 |
| PLLA2 | 5500 | 1.22 | 48.0 | 152.2 | −153.2 |
| PLLA3 | 10,800 | 1.14 | 55.2 | 166.9 | −154.6 |
| PLLA4 | 30,300 | 1.23 | 58.1 | 173.4 | −155.4 |
| PLLA5 | 65,400 | 1.26 | 58.2 | 174.2 | −155.7 |
| Sample Name | |||||
|---|---|---|---|---|---|
| PLLA1 | 10.83 | 11.41 | 5.15 | 6.26 | 1.26 |
| PLLA2 | 16.11 | 15.61 | 6.78 | 8.83 | 1.96 |
| PLLA3 | 14.61 | 15.25 | 6.98 | 8.27 | 2.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Zhao, G.; Wang, G. Investigation on the α/δ Crystal Transition of Poly(l-lactic Acid) with Different Molecular Weights. Polymers 2021, 13, 3280. https://doi.org/10.3390/polym13193280
Zhang L, Zhao G, Wang G. Investigation on the α/δ Crystal Transition of Poly(l-lactic Acid) with Different Molecular Weights. Polymers. 2021; 13(19):3280. https://doi.org/10.3390/polym13193280
Chicago/Turabian StyleZhang, Lei, Guoqun Zhao, and Guilong Wang. 2021. "Investigation on the α/δ Crystal Transition of Poly(l-lactic Acid) with Different Molecular Weights" Polymers 13, no. 19: 3280. https://doi.org/10.3390/polym13193280
APA StyleZhang, L., Zhao, G., & Wang, G. (2021). Investigation on the α/δ Crystal Transition of Poly(l-lactic Acid) with Different Molecular Weights. Polymers, 13(19), 3280. https://doi.org/10.3390/polym13193280