
Evaluating the Performance of a Semiaromatic/Aliphatic Polyamide Blend: The Case for Polyphthalamide (PPA) and Polyamide 4,10 (PA410)



Abstract
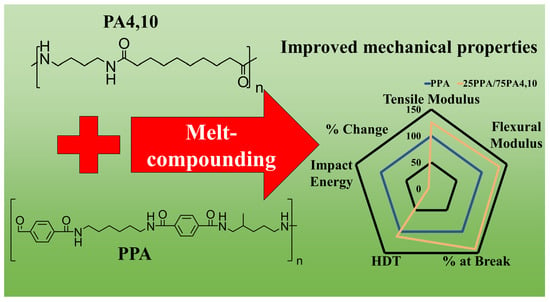
:
1. Introduction
2. Materials and Methods
2.1. Mechanical Analysis
2.2. Thermogravimetric Analysis (TGA)
2.3. Differential Scanning Calorimetry (DSC)
2.4. Scanning Electron Microscopy (SEM)
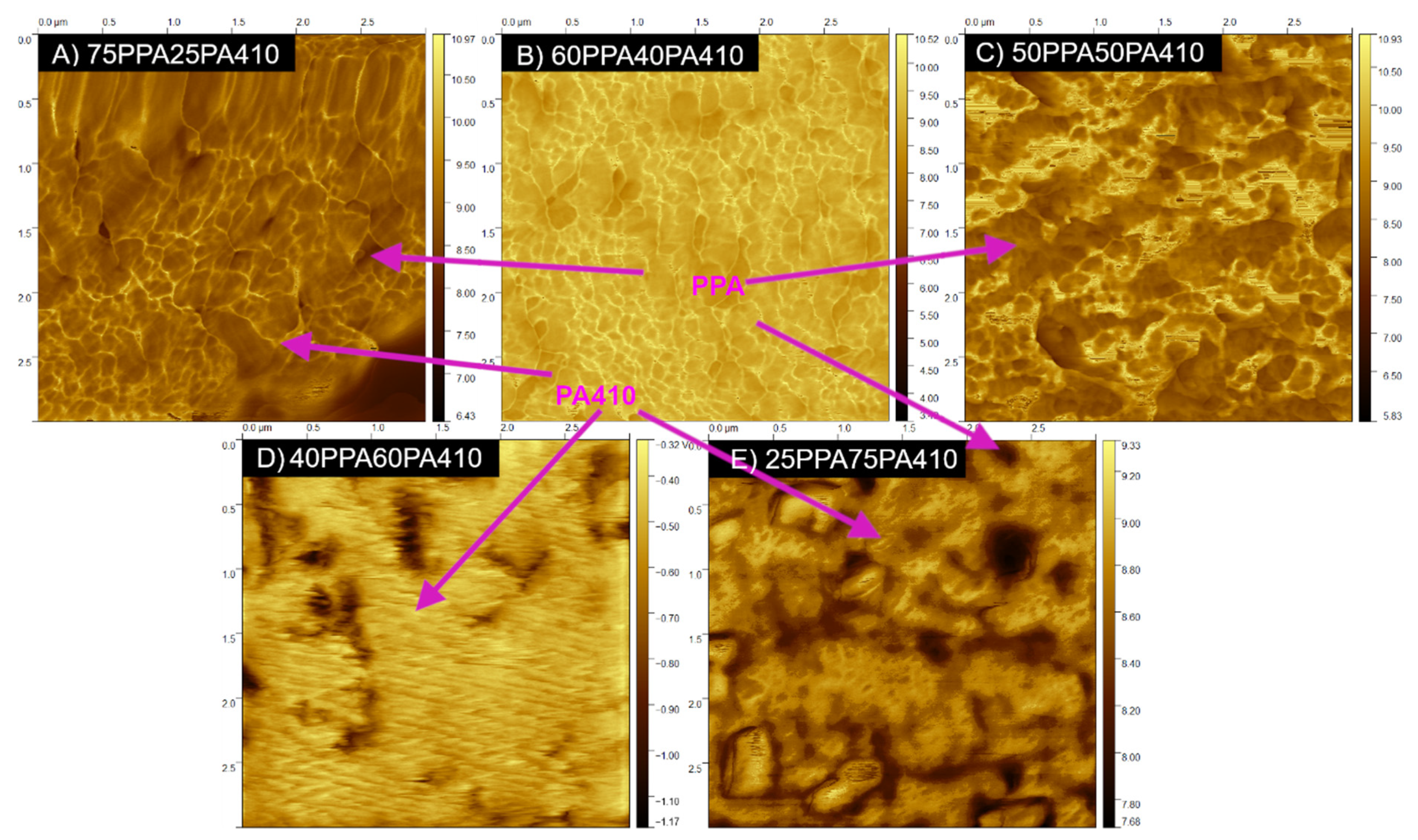
2.5. Atomic Force Microscopy (AFM)
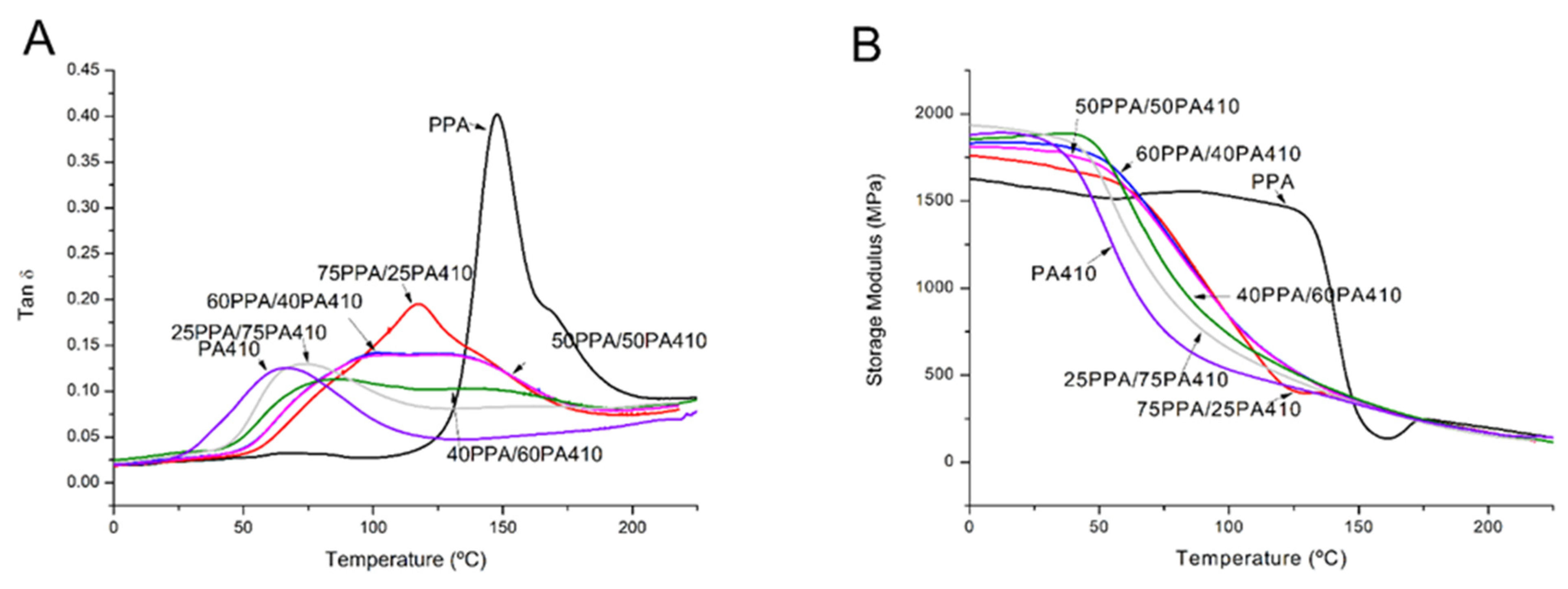
2.6. Dynamic Mechanical Analysis (DMA)
2.7. Polar Optical Microscopy (POM)
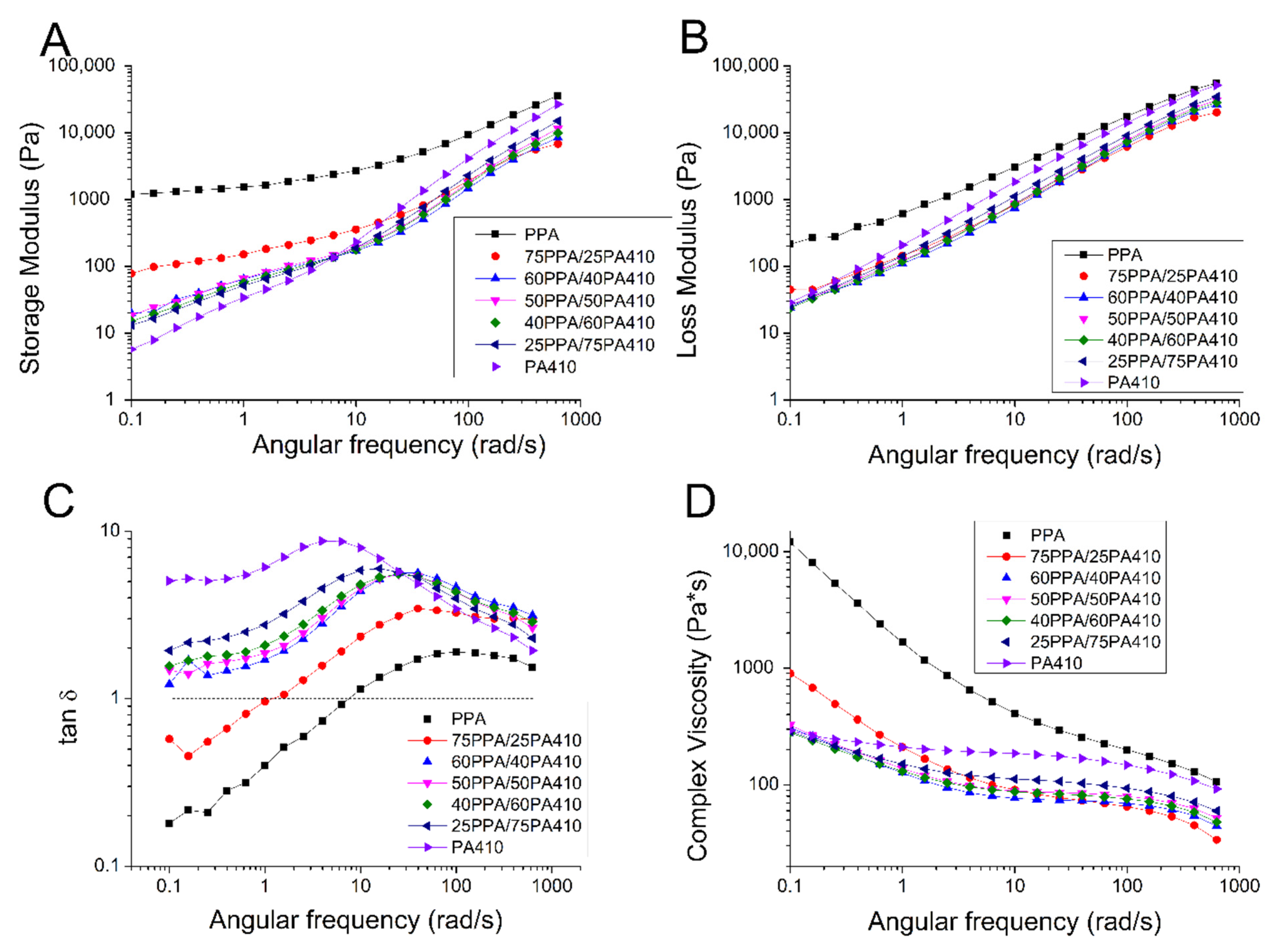
2.8. Rheology
2.9. Fourier-Transform Infrared Spectroscopy (FTIR)
3. Results and Discussion
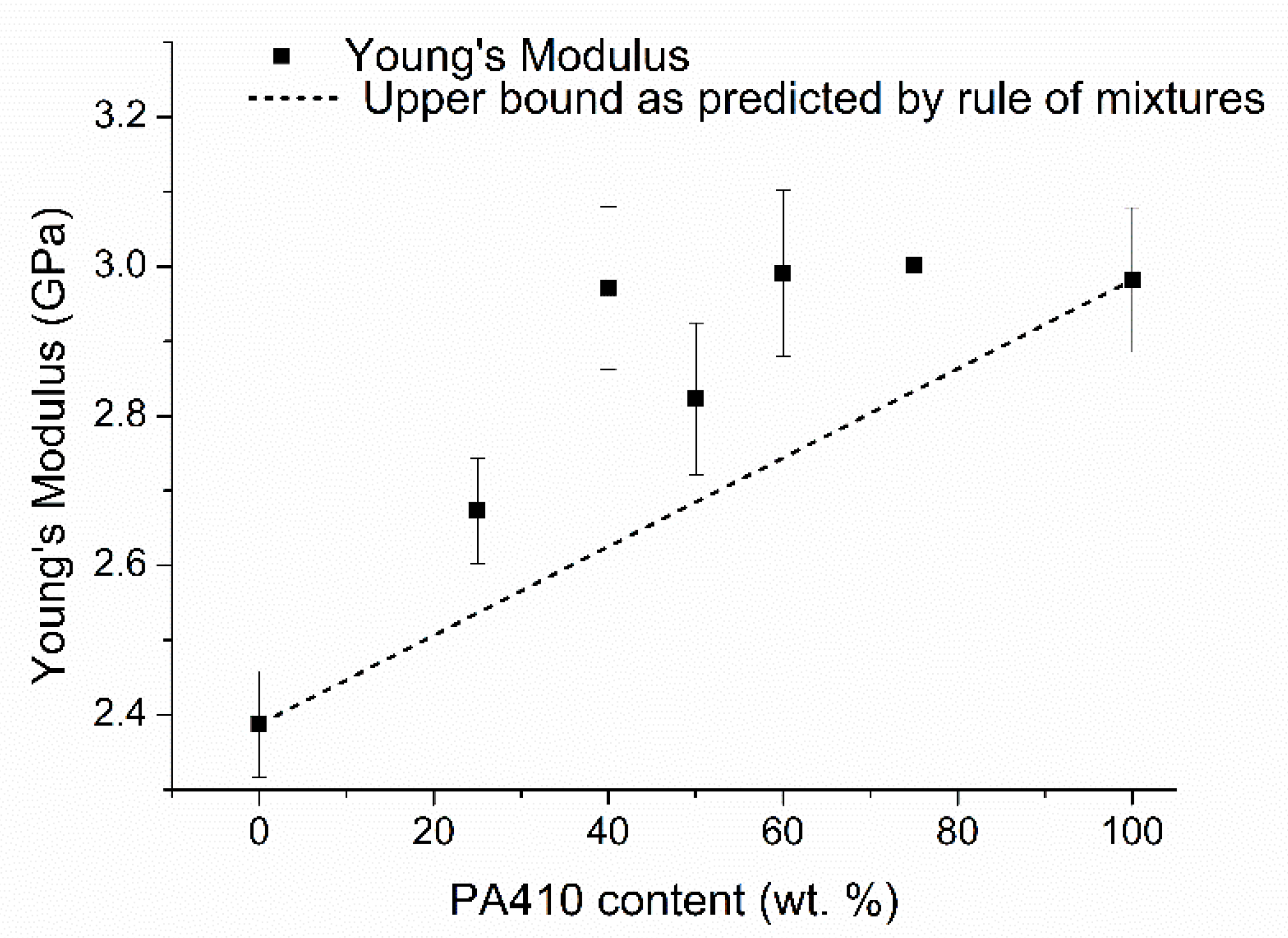
3.1. Mechanical Properties
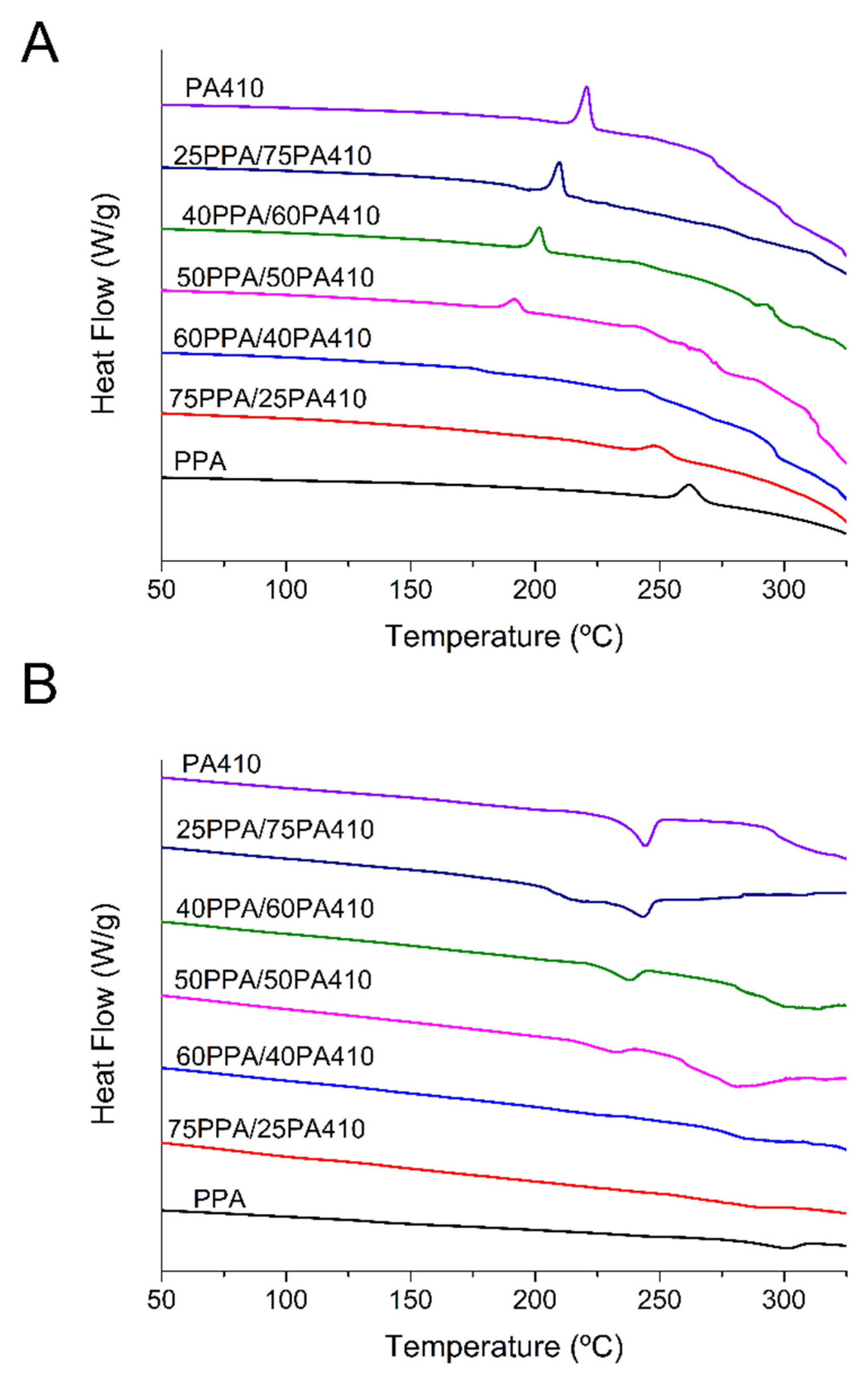
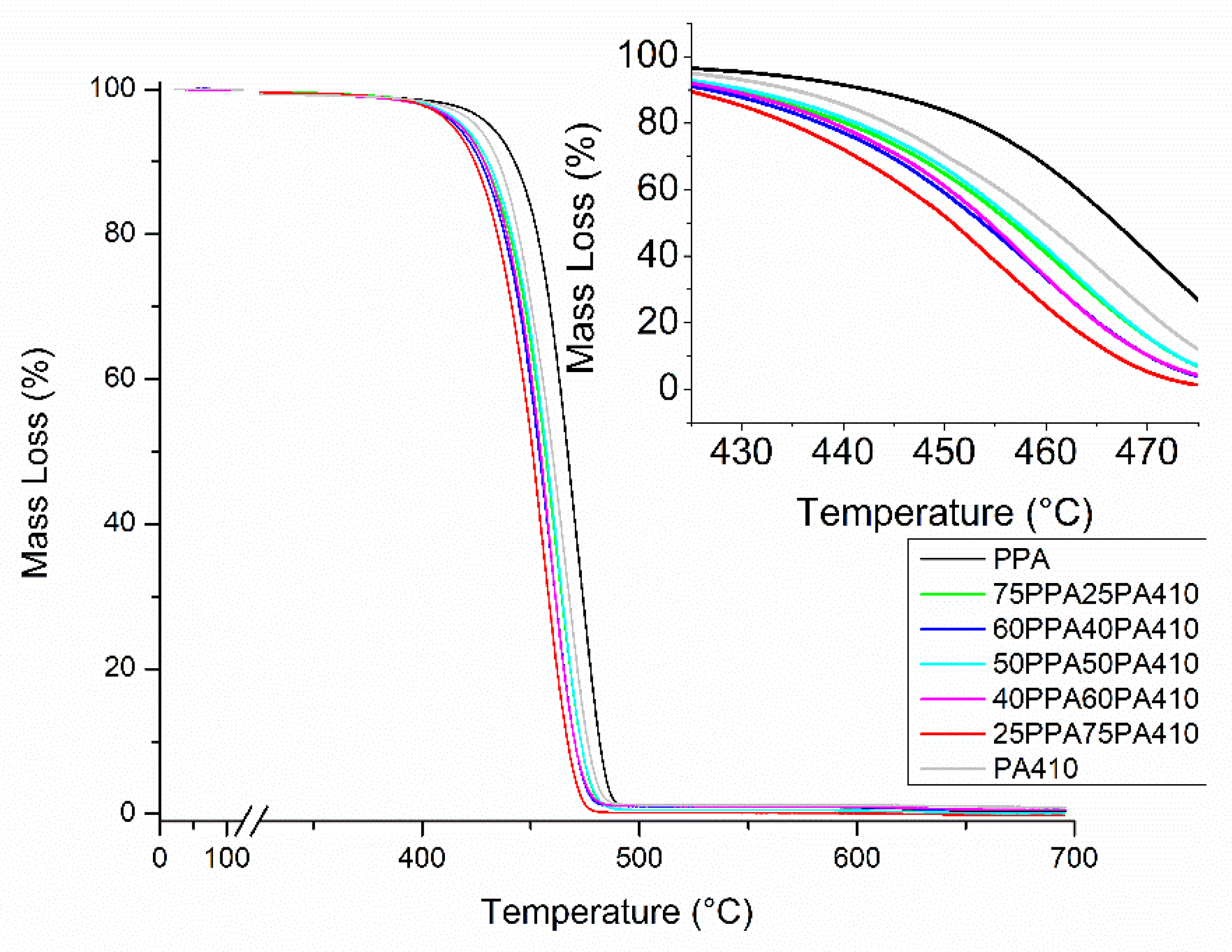
3.2. Thermal Analysis
3.3. Morphology Characterization
3.4. Viscoelastic Characterization
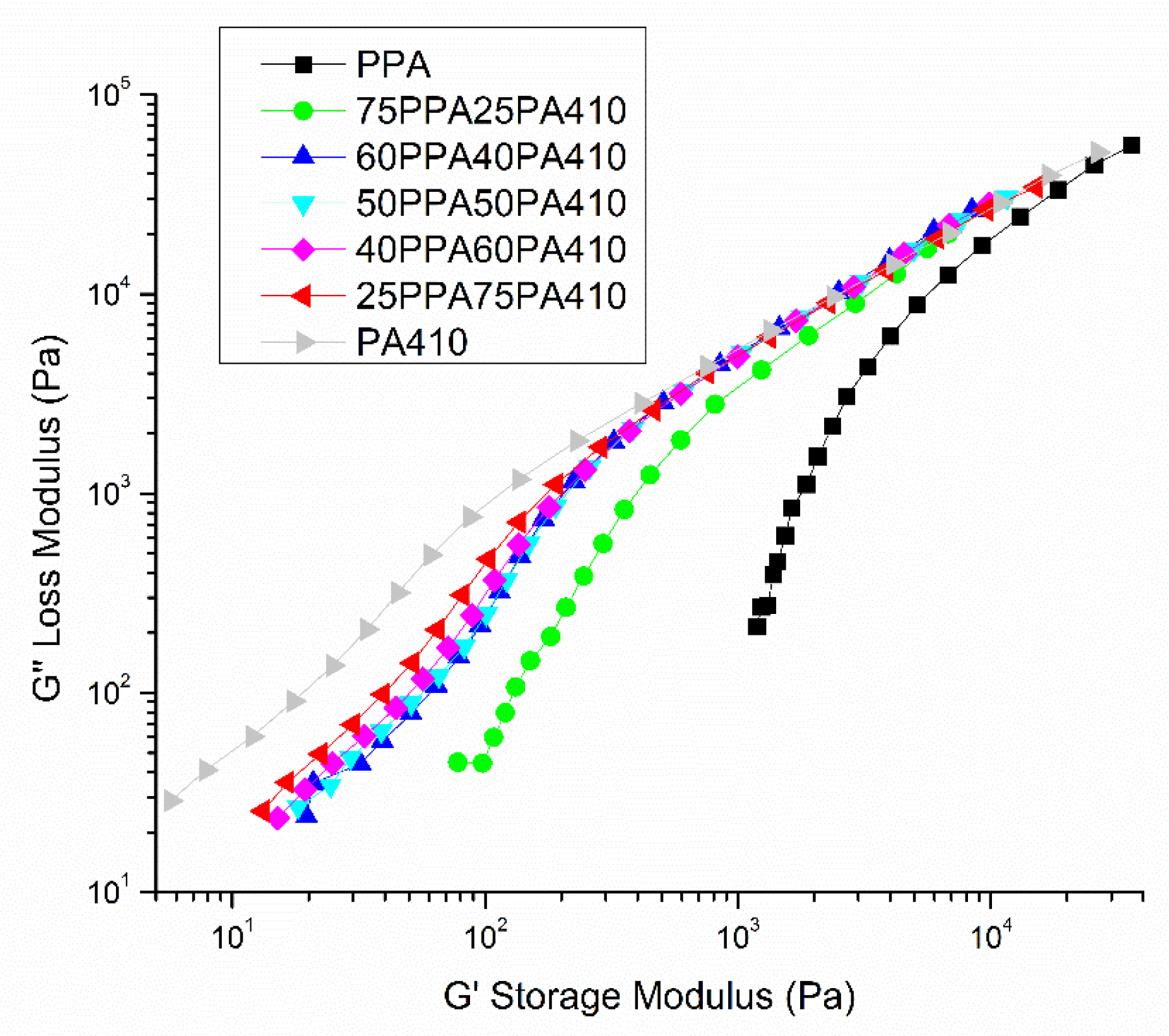
3.5. Rheological Properties
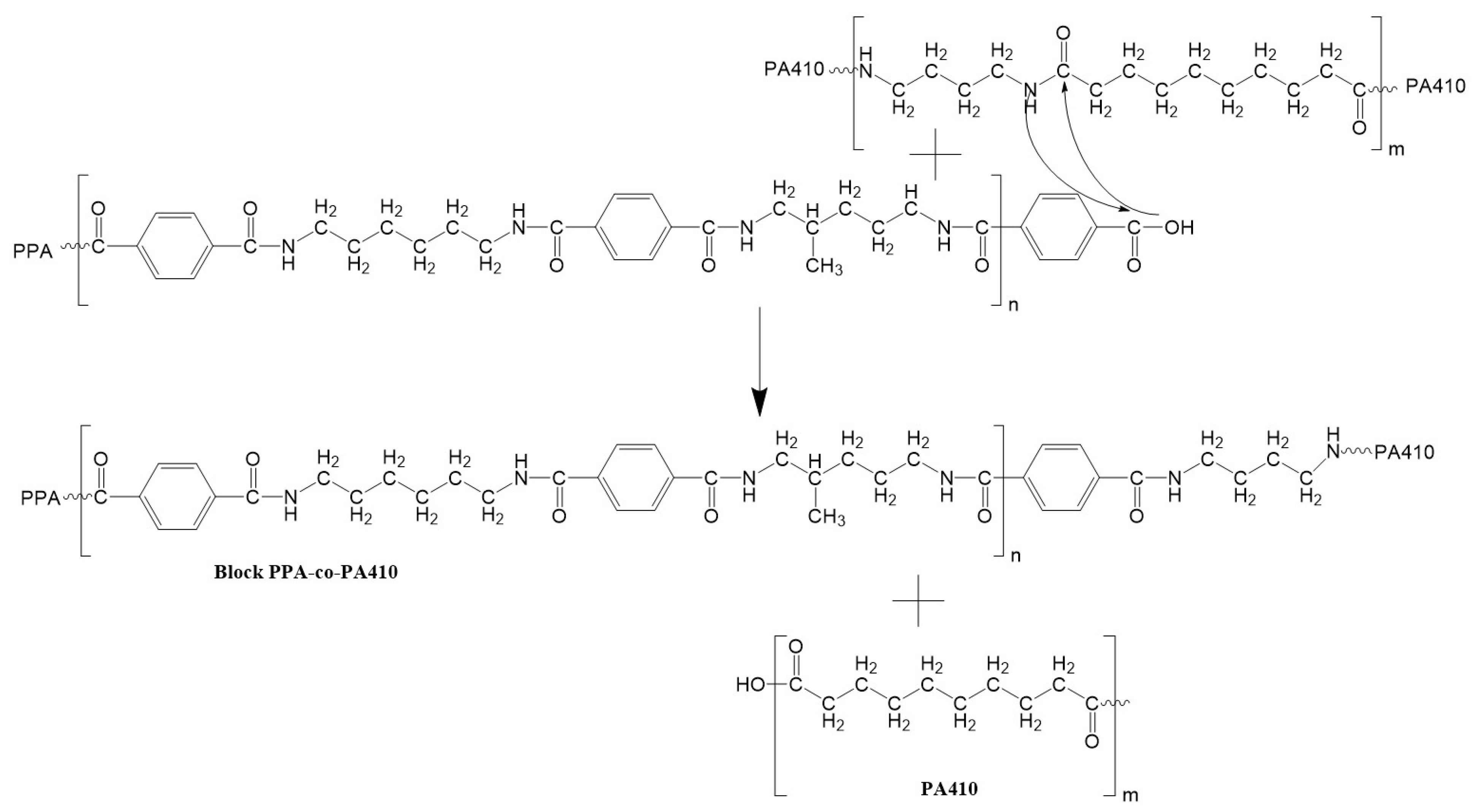
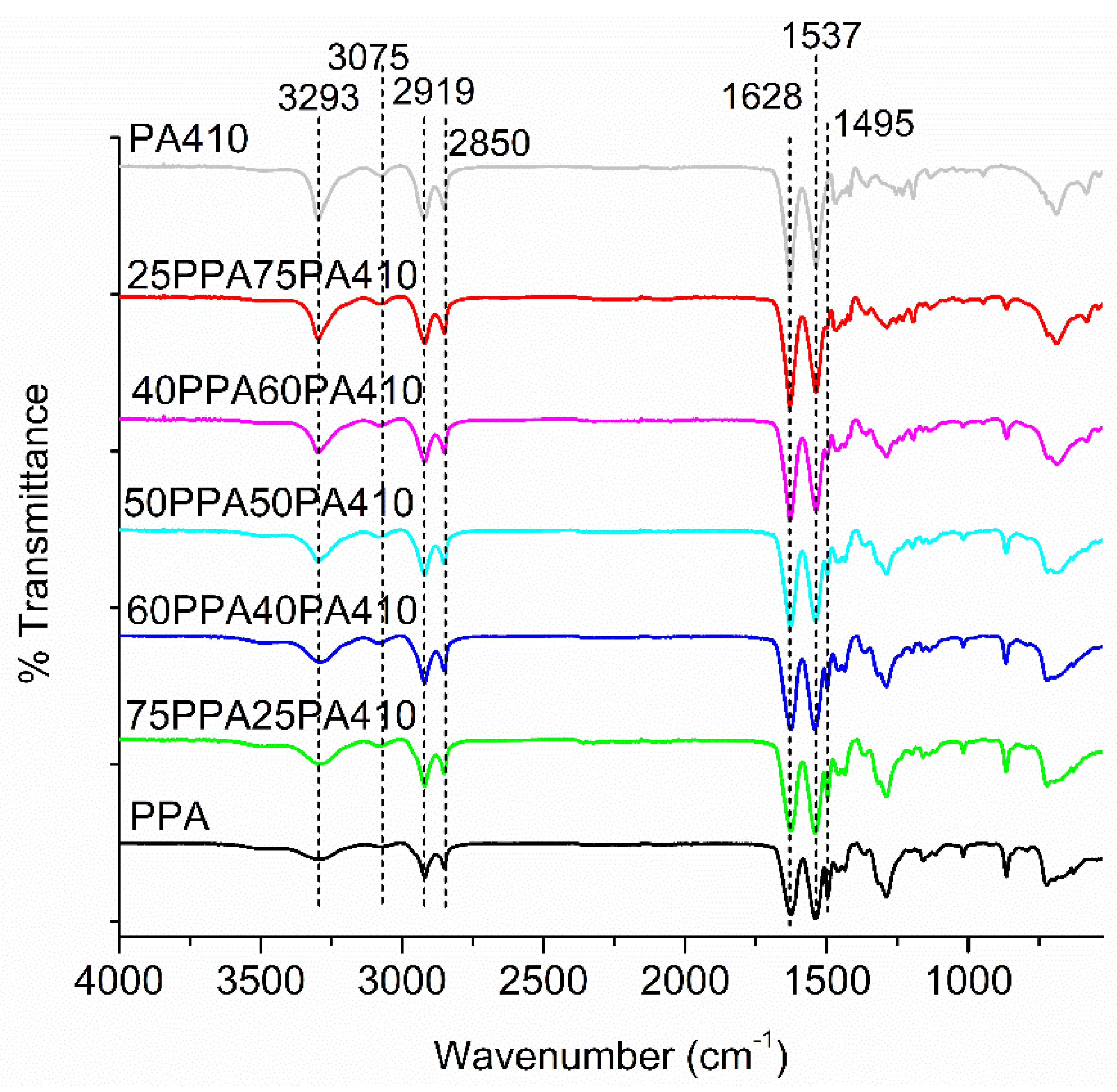
3.6. FTIR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Plastics—The Facts 2019; PlasticsEurope Deutschland e. V. and Messe Düsseldorf: Brussels, Belgium, 2019.
- Parker, D.; Bussink, J.; van de Grampel, H.T.; Wheatley, G.W.; Dorf, E.-U.; Ostlinning, E.; Reinking, K.; Schubert, F.; Jünger, O.; Wagener, R. Polymers, High-Temperature. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; ISBN 978-3-527-30673-2. [Google Scholar]
- Kemmish, D. 3 Semi-Aromatic Polyamides (Polyphthalamides). In Practical Guide to High Performance Engineering Plastics; Smithers Rapra: Shrewsbury, UK, 2011; pp. 47–55. ISBN 978-1-84735-576-8. [Google Scholar]
- D20 Committee Classification System and Basis for Specification for Polyphthalamide (PPA) Injection Molding Materials, D5336-15a; ASTM International: West Conshohocken, PA, USA, 2015.
- Hottle, T.A.; Bilec, M.M.; Landis, A.E. Biopolymer Production and End of Life Comparisons Using Life Cycle Assessment. Resour. Conserv. Recycl. 2017, 122, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Worrell, E.; Patel, M.K. Comparing Life Cycle Energy and GHG Emissions of Bio-Based PET, Recycled PET, PLA, and Man-Made Cellulosics: Modeling and Analysis: Comparing Life-Cycle Energy and GHG Emissions of Bioproducts. Biofuels Bioprod. Biorefin. 2012, 6, 625–639. [Google Scholar] [CrossRef]
- Arkema Rilsan® HT. Available online: https://www.extremematerials-arkema.com/en/product-families/rilsan-polyamide-11-family/rilsan-ht/ (accessed on 17 April 2020).
- No Contradiction: VESTAMID® HTplus Both Bio-Based and High-Performing. Available online: https://corporate.evonik.com/en/media/press_releases/corporate/no-contradiction-vestamid-htplus-both-bio-based-and-high-performing-103227.html (accessed on 17 April 2020).
- Moran, C.S.; Barthelon, A.; Pearsall, A.; Mittal, V.; Dorgan, J.R. Biorenewable Blends of Polyamide-4,10 and Polyamide-6,10. J. Appl. Polym. Sci. 2016, 133, 43626. [Google Scholar] [CrossRef]
- Leszczyńska, A.; Kiciliński, P.; Pielichowski, K. Biocomposites of Polyamide 4.10 and Surface Modified Microfibrillated Cellulose (MFC): Influence of Processing Parameters on Structure and Thermomechanical Properties. Cellulose (Dordrecht, The Netherlands) 2015, 22, 2551–2569. [Google Scholar] [CrossRef] [Green Version]
- Otaegi, I.; Aranburu, N.; Iturrondobeitia, M.; Ibarretxe, J.; Guerrica-Echevarría, G. The Effect of the Preparation Method and the Dispersion and Aspect Ratio of CNTs on the Mechanical and Electrical Properties of Bio-Based Polyamide-4,10/CNT Nanocomposites. Polymers 2019, 11, 2059. [Google Scholar] [CrossRef] [Green Version]
- Siciliano, A.; Severgnini, D.; Seves, A.; Pedrelli, T.; Vicini, L. Thermal and Mechanical Behavior of Polyamide 6/Polyamide 6I/6T Blends. J. Appl. Polym. Sci. 1996, 60, 1757–1764. [Google Scholar] [CrossRef]
- Persyn, O.; Miri, V.; Lefebvre, J.-M.; Ferreiro, V.; Brink, T.; Stroeks, A. Mechanical Behavior of Films of Miscible Polyamide 6/Polyamide 6I-6T Blends. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 1690–1701. [Google Scholar] [CrossRef]
- Eersels, K.L.L.; Groeninckx, G. Influence of Interchange Reactions on the Crystallization and Melting Behaviour of Polyamide Blends as Affected by the Processing Conditions. Polymer 1996, 37, 983–989. [Google Scholar] [CrossRef]
- Cretenoud, J.; Galland, S.; Plummer, C.J.G.; Michaud, V.; Bayer, A.; Lamberts, N.; Hoffmann, B.; Frauenrath, H. High-Temperature Copolyamides Obtained by the Efficient Transamidation of Crystalline-Crystalline Polyamide Blends. J. Appl. Polym. Sci. 2016, 134, 44349. [Google Scholar] [CrossRef]
- Voigt, W. Ueber die Beziehung zwischen den beiden Elasticitätsconstanten isotroper Körper. Ann. Phys. 1889, 274, 573–587. [Google Scholar] [CrossRef] [Green Version]
- Eersels, K.L.L.; Aerdts, A.M.; Groeninckx, G. Transamidation in Melt-Mixed Aliphatic and Aromatic Polyamides. 2. Molecular Characterization of PA 46/PA 6I Blends as a Function of the Extrusion Time, Extrusion Temperature, and Blend Composition. Macromolecules (Washington, DC, USA) 1996, 29, 1046–1050. [Google Scholar] [CrossRef]
- Ellis, T.S. Miscibility of Polyamide Blends: Effects of Configuration. Polymer 1995, 36, 3919–3926. [Google Scholar] [CrossRef]
- Utracki, L.A.; Wilkie, C.A. (Eds.) Polymer Blends Handbook; Springer: Dordrecht, The Netherlands, 2014; ISBN 978-94-007-6063-9. [Google Scholar]
- Eersels, K.L.L.; Groeninckx, G.; Koch, M.H.J.; Reynaers, H. Influence of Transreaction Processes on the Morphology of Semicrystalline Aliphatic/Aromatic Polyamide Blends. Polymer 1998, 39, 3893–3900. [Google Scholar] [CrossRef]
- Grätzl, T.; van Dijk, Y.; Schramm, N.; Kroll, L. Influence of the Automotive Paint Shop on Mechanical Properties of Continuous Fibre-Reinforced Thermoplastics. Compos. Struct. 2019, 208, 557–565. [Google Scholar] [CrossRef]
- Shashoua, V.E.; Eareckson, W.M. Interfacial Polycondensation. V. Polyterephthalamides from Short-Chain Aliphatic, Primary, and Secondary Diamines. J. Polym. Sci. (Hoboken, NJ, USA) 1959, 40, 343–358. [Google Scholar] [CrossRef]
- Edgar, O.B.; Hill, R. The P-Phenylene Linkage in Linear High Polymers: Some Structure–Property Relationships. J. Polym. Sci. (Hoboken, NJ, USA) 1952, 8, 1–22. [Google Scholar] [CrossRef]
- Zanjanijam, A.R.; Hakim, S.; Azizi, H. Rheological, Mechanical and Thermal Properties of the PA/PVB Blends and Their Nanocomposites: Structure-Property Relationships. Polym. Test. 2018, 66, 48–63. [Google Scholar] [CrossRef]
- La Mantia, F.P.; Morreale, M.; Botta, L.; Mistretta, M.C.; Ceraulo, M.; Scaffaro, R. Degradation of Polymer Blends: A Brief Review. Polym. Degrad. Stab. 2017, 145, 79–92. [Google Scholar] [CrossRef]
- Desio, G.P. Characterization and Properties of Polyphthalamide/Polyamide Blends and Polyphthalamide/Polyamide/Polyolefin Blends. J. Vinyl Addit. Technol. 1996, 2, 229–234. [Google Scholar] [CrossRef]
- Yao, Z.; Sun, J.; Wang, Q.; Cao, K. Study on Ester–Amide Exchange Reaction between PBS and PA6IcoT. Ind. Eng. Chem. Res. 2012, 51, 751–757. [Google Scholar] [CrossRef]
- Peng, W.-M.; Tong, X.; Zhang, M.-L.; Wang, X.-J.; Zhang, G.; Long, S.-R.; Yang, J. Semiaromatic Polyamide Poly(Hexamethylene Terephthalamide)-Co-Polycaprolactam: Thermal and Flame-Retardant Properties. J. Appl. Polym. Sci. 2018, 135, 46451. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, Q.; Du, L.; Yang, G. Influence of Preparation Methods on the Structures and Properties for the Blends between Polyamide 6co6T and Polyamide 6: Melt-Mixing Andin-Situ Blending. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 201–211. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, G.; Zhang, J.; Xu, J.; Shi, W. Non-Isothermal Crystallization Kinetics of Nylon 10T and Nylon 10T/1010 Copolymers: Effect of Sebacic Acid as a Third Comonomer. Chin. J. Chem. Eng. 2017, 25, 963–970. [Google Scholar] [CrossRef]
- Kaplan, D.S. Structure–Property Relationships in Copolymers to Composites: Molecular Interpretation of the Glass Transition Phenomenon. J. Appl. Polym. Sci. 1976, 20, 2615–2629. [Google Scholar] [CrossRef]
- Zytner, P.; Wu, F.; Misra, M.; Mohanty, A.K. Toughening of Biodegradable Poly(3-Hydroxybutyrate- Co -3-Hydroxyvalerate)/Poly(ε-Caprolactone) Blends by In Situ Reactive Compatibilization. ACS Omega 2020, 5, 14900–14910. [Google Scholar] [CrossRef]
- Wu, F.; Misra, M.; Mohanty, A.K. Super Toughened Poly(Lactic Acid)-Based Ternary Blends via Enhancing Interfacial Compatibility. ACS Omega 2019, 4, 1955–1968. [Google Scholar] [CrossRef]
- Zanjanijam, A.R.; Hakim, S.; Azizi, H. Morphological, Dynamic Mechanical, Rheological and Impact Strength Properties of the PP/PVB Blends: The Effect of Waste PVB as a Toughener. RSC Adv. 2016, 6, 44673–44686. [Google Scholar] [CrossRef]
- Liu, Y.; Yi, J.; Cai, X. Effect of a Novel Intumescent Retardant for ABS with Synergist Al(H2PO2)3. Polym. Bull. (Berl. Ger.) 2011, 67, 361–374. [Google Scholar] [CrossRef]
- Pagacz, J.; Raftopoulos, K.N.; Leszczyńska, A.; Pielichowski, K. Bio-Polyamides Based on Renewable Raw Materials: Glass Transition and Crystallinity Studies. J. Therm. Anal. Calorim. 2016, 123, 1225–1237. [Google Scholar] [CrossRef] [Green Version]
- Marchildon, K. Polyamides—Still Strong After Seventy Years. Macromol. React. Eng. 2011, 5, 22–54. [Google Scholar] [CrossRef]
- Zhang, C. Progress in Semicrystalline Heat-Resistant Polyamides. e-Polymers 2018, 18, 373–408. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | wt% of PPA | wt% of PA410 | Temperature of Melt Mixing [°C] |
|---|---|---|---|
| PPA | 100 | 0 | 325 |
| 75PPA/25PA410 | 75 | 25 | 305 |
| 60PPA/40PA410 | 60 | 40 | 305 |
| 50PPA/50PA410 | 50 | 50 | 305 |
| 40PPA/60PA410 | 40 | 60 | 305 |
| 25PPA/75PA410 | 25 | 75 | 305 |
| PA410 | 0 | 100 | 285 |
| Samples | Lowest Tm [°C] | ΔHm [J/m] | Tc [°C] | ΔHc [J/m] | χc [J/m] |
|---|---|---|---|---|---|
| PPA | 299.2 | 14.08 | 259.22 | 27.5 | N/A |
| 75PPA/25PA410 | 288.56 | 21.73 | 248.95 | 30.78 | 78.08 |
| 60PPA/40PA410 | 284.61 | 30.44 | 243.47 | 19.32 | 46.25 |
| 50PPA/50PA410 | 231.34 | 18.52 | 191.79 | 28.01 | 34.59 |
| 40PPA/60PA410 | 237.51 | 30.98 | 201.65 | 43.25 | 68.99 |
| 25PPA/75PA410 | 243.1 | 36.77 | 209.71 | 57.6 | 46.78 |
| PA410 | 244.21 | 52.93 | 220.72 | 69.07 | 45.35 |
| Samples | HDT (0.2% at 0.455 MPa) [°C] | 2% Weight Loss Temperature [°C] | 5% Weight Loss Temperature [°C] | 10% Weight Loss Temperature [°C] | Maximum Degradation Temperature [°C] |
|---|---|---|---|---|---|
| PPA | 122.30 ±17.97 | 404.86 ± 8.10 | 429.71 ± 0.33 | 441.72 ± 0.01 | 471.55 ± 1.22 |
| 75PPA/25PA410 | 109.91 ± 11.69 | 400.91 ± 0.99 | 417.71 ± 0.55 | 428.51 ± 0.36 | 461.47 ± 0.12 |
| 60PPA/40PA410 | 121.89 ± 8.65 | 398.42 ± 1.08 | 415.91 ± 0.18 | 427.01 ± 0.29 | 459.35 ± 0.49 |
| 50PPA/50PA410 | 121.20 ± 0.12 | 399.73 ± 1.68 | 418.29 ± 0.98 | 429.51 ± 1.17 | 461.50 ± 1.37 |
| 40PPA/60PA410 | 130.19 ± 2.18 | 397.58 ± 0.71 | 416.15 ± 1.70 | 427.26 ± 1.70 | 457.46 ± 1.08 |
| 25PPA/75PA410 | 135.99 ± 2.47 | 398.93 ± 0.66 | 414.51 ± 0.91 | 424.85 ± 0.93 | 455.75 ± 1.01 |
| PA410 | 160.03 ± 4.58 | 406.08 ± 0.25 | 424.17 ± 1.17 | 433.81 ± 1.68 | 463.02 ± 4.88 |
| Samples | Tg (from DMA) [°C] |
|---|---|
| PPA | 140.77 ± 9.84 |
| 75PPA/25PA410 | 117.00 ± 0.11 |
| 60PPA/40PA410 | 103.16 ± 2.64 |
| 50PPA/50PA410 | 101.39 ± 1.84 |
| 40PPA/60PA410 | 80.63 ± 2.52 |
| 25PPA/75PA410 | 70.32 ± 3.18 |
| PA410 | 66.44 ± 0.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez de Gortari, M.; Wu, F.; Mohanty, A.K.; Misra, M. Evaluating the Performance of a Semiaromatic/Aliphatic Polyamide Blend: The Case for Polyphthalamide (PPA) and Polyamide 4,10 (PA410). Polymers 2021, 13, 3391. https://doi.org/10.3390/polym13193391
Gonzalez de Gortari M, Wu F, Mohanty AK, Misra M. Evaluating the Performance of a Semiaromatic/Aliphatic Polyamide Blend: The Case for Polyphthalamide (PPA) and Polyamide 4,10 (PA410). Polymers. 2021; 13(19):3391. https://doi.org/10.3390/polym13193391
Chicago/Turabian StyleGonzalez de Gortari, Mateo, Feng Wu, Amar K. Mohanty, and Manjusri Misra. 2021. "Evaluating the Performance of a Semiaromatic/Aliphatic Polyamide Blend: The Case for Polyphthalamide (PPA) and Polyamide 4,10 (PA410)" Polymers 13, no. 19: 3391. https://doi.org/10.3390/polym13193391
APA StyleGonzalez de Gortari, M., Wu, F., Mohanty, A. K., & Misra, M. (2021). Evaluating the Performance of a Semiaromatic/Aliphatic Polyamide Blend: The Case for Polyphthalamide (PPA) and Polyamide 4,10 (PA410). Polymers, 13(19), 3391. https://doi.org/10.3390/polym13193391