Poly(lactic acid) (PLA)/Poly(butylene succinate-co-adipate) (PBSA) Compatibilized Binary Biobased Blends: Melt Fluidity, Morphological, Thermo-Mechanical and Micromechanical Analysis

 ,
, 
 ,
,  and
and 
Abstract
:
1. Introduction
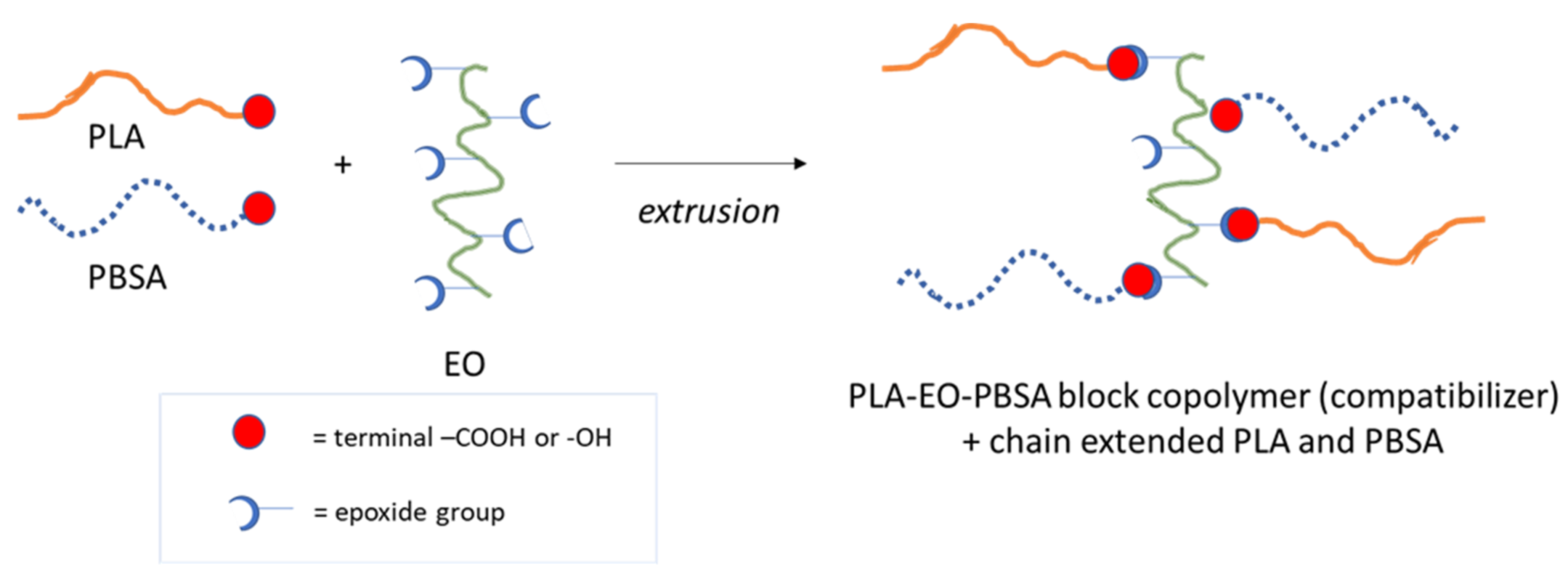
- Regarding binary blends, the compatibility/miscibility issues must be considered. It is noteworthy that the introduction of chain extenders that are able to reconnect cleaved chains, increases the molecular weight (consequently increasing the melt strength) [17,44,45]. Different types of chain extenders, also available on the market, have been extensively investigated and reported in literature such as: multi-functional epoxides [46], diisocyanate compounds [47], dianhydride [48], bis-oxazolines, tris(nonyl-phenyl) and phosphate (TNPP) [49]. The introduction of chain extenders also provides a better control of the polymer degradation [17,19,50,51,52] during the process and at the same time enhances the extrusion and injection foamability [52,53]. Moreover, the use of chain extenders can also improve the compatibility between the two phases constituting a binary blend because, especially in the interfacial region, the chain extender can react with both the polymers resulting in the formation of block copolymers acting as effective in situ generated compatibilizers. Chain extenders containing epoxy groups are the most suitable in this case, in fact they are able to react opening the epoxy group ring and creating covalent bonds [45] with both the hydroxyl and carboxylic groups of the polyester chain-ends. The high number of epoxy groups per macromolecule grants efficiency in limiting the decrease of viscosity during processing typical of polyesters that are generally affected by hydrolysis due to atmospheric humidity. For this reason, a multifunctional epoxy oligomer (EO) consisting of styrene, acrylic and glycidyl acrylate units, has been chosen for the binary PLA/PBSA polyester blends. Al-Itry et al. [52] and Wang et al. [54] studied the positive compatibilization effect of a similar EO in a PLA/PBAT system. Lascano et al. [55] explained that EO addition can be advantageous also in PLA/PBSA binary blends (thus very similar to the ones studied in this paper) because it reacts either with the hydroxyl terminal groups of PLA and PBSA, leading to a compatibilization effect and an effective toughening. However, the investigated blends contained up to 30% of PBSA and were not investigated in terms of their failure mechanism and melt fluidity.
2. Materials and Methods
2.1. Materials
- Poly(lactic) acid (PLA) Luminy LX175 produced by Total Corbion PLA. This biodegradable PLA is derived from natural resources, appears as white pellets and contains about 4% of D-lactic acid units. It is a general-purpose extrusion grade PLA that can be used alone or to produce formulated blends or composites; it can be easily processed on conventional equipment for film extrusion, thermoforming or fiber spinning (density: 1.24 g/cm3; melt flow index (MFI) (210 °C/2.16 kg): 6 g/10 min).
- Poly(butylene succinate-co-adipate) (PBSA), trade name BioPBS FD92PM, purchased from Mitsubishi Chemical Corporation, is a copolymer of succinic acid, adipic acid and butandiol. It is a soft and flexible semicrystalline polyester suitable for both blown and cast film extrusion (density of 1.24 g/cm3; MFI (190 °C, 2.16 kg): 4 g/10 min).
- The epoxy oligomer (EO) used in the work is Joncryl ADR 4468 produced by BASF. It is an oligomeric chain extender having about 20 epoxy groups per macromolecule that reacts with the terminal groups of polycondensates, increasing the melt viscosity (Mw: 7250; density: 1.08 g/cm3; epoxy equivalent weight: 310 g/mol).
2.2. Blends and Sample Preparation
2.3. Torque and Melt Flow Rate Analysis
2.4. Mechanical Characterization
2.5. Optical Analysis
2.6. Thermal Characterization
3. Results and Discussions
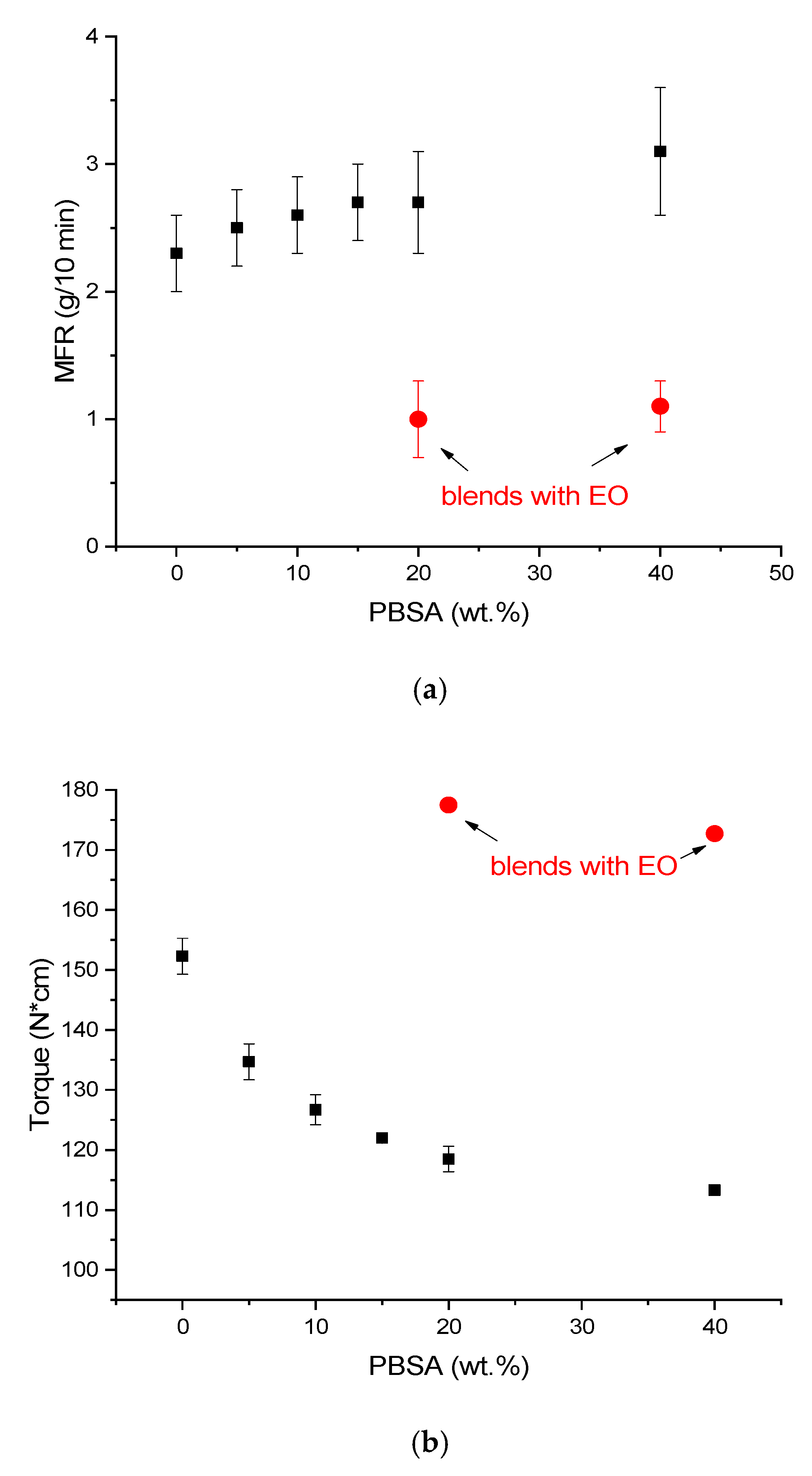
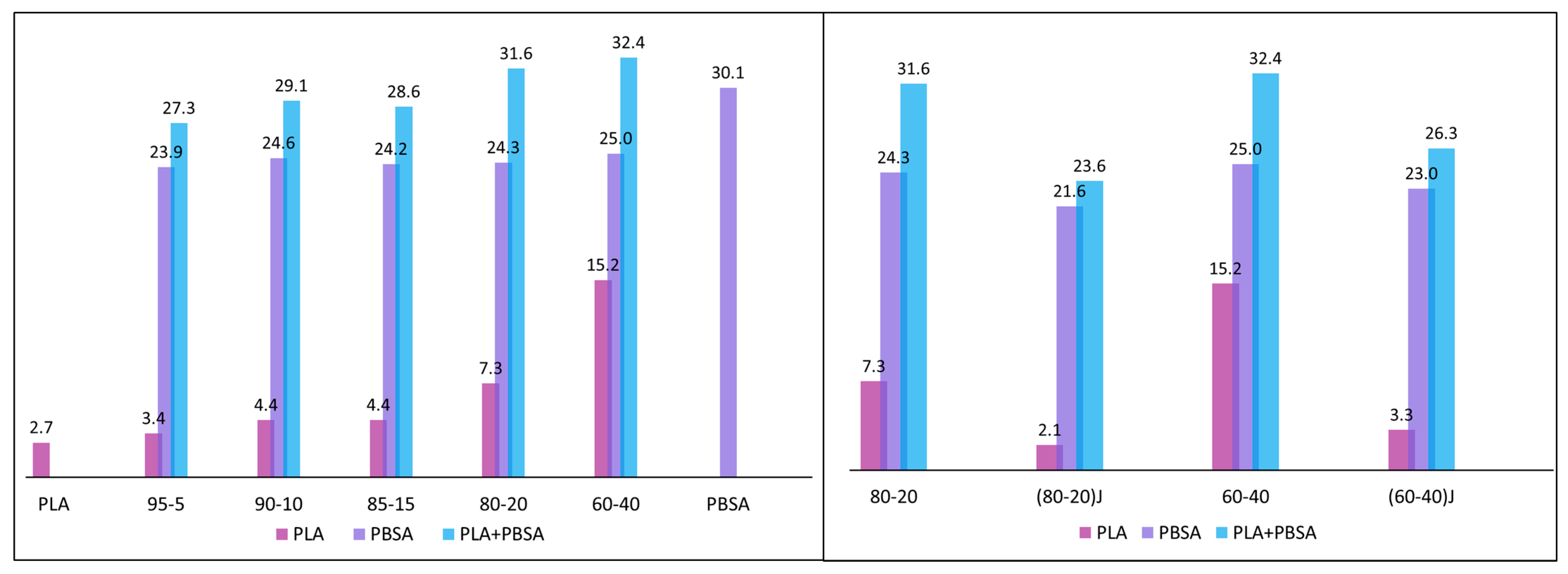
3.1. Melt Fluidity, Morphology and Thermal Properties of PLA/PBSA Blends
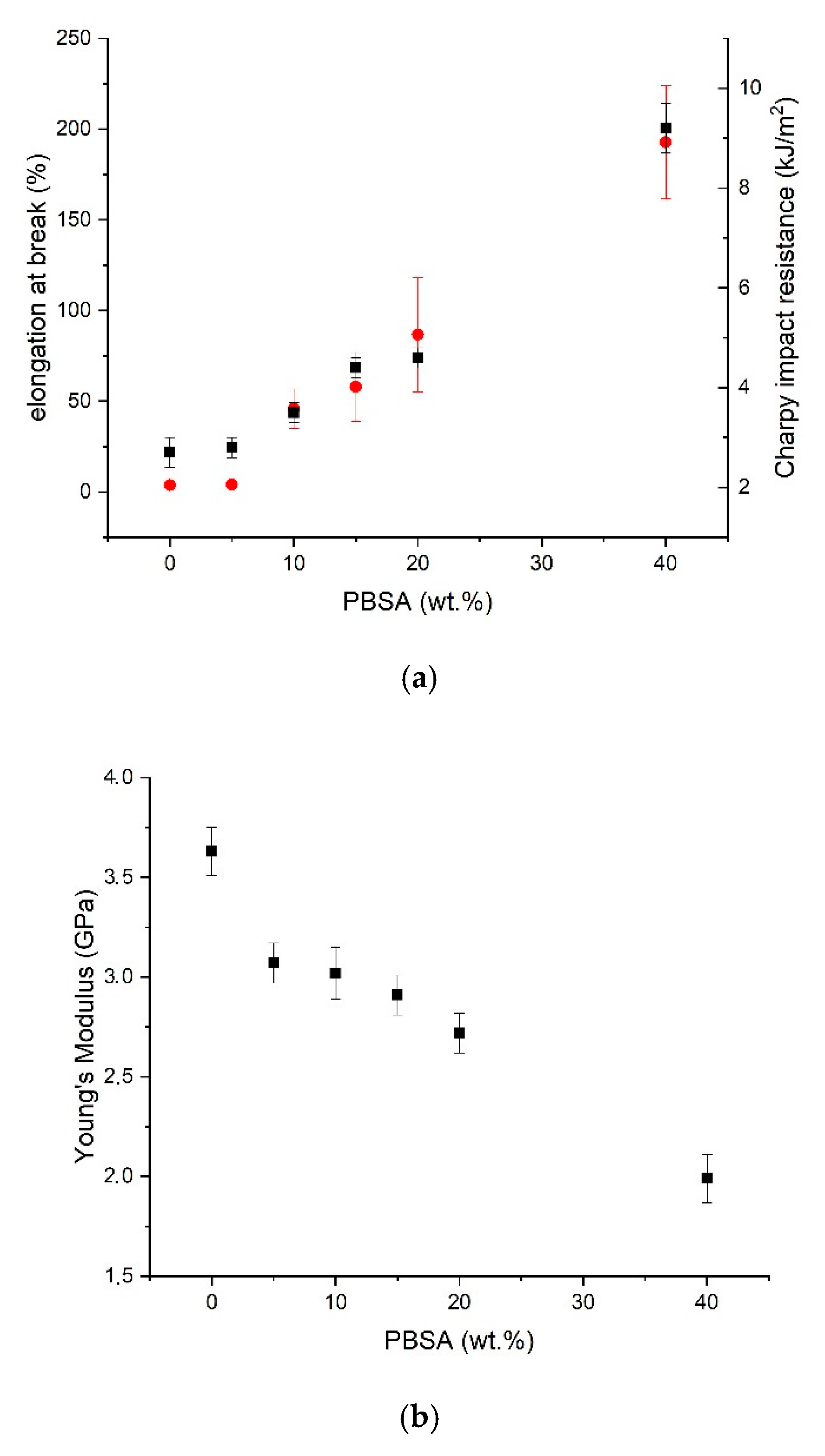
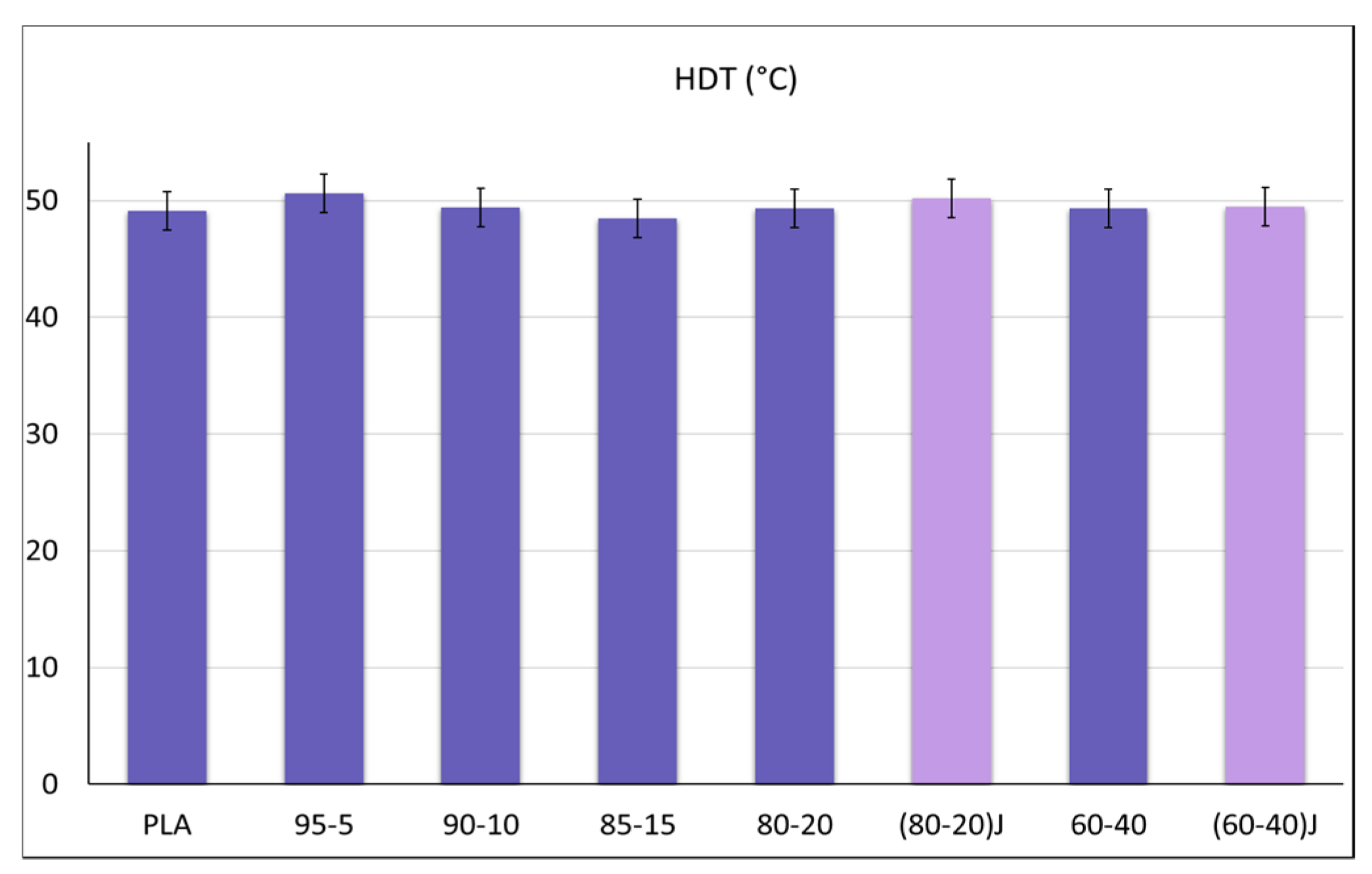
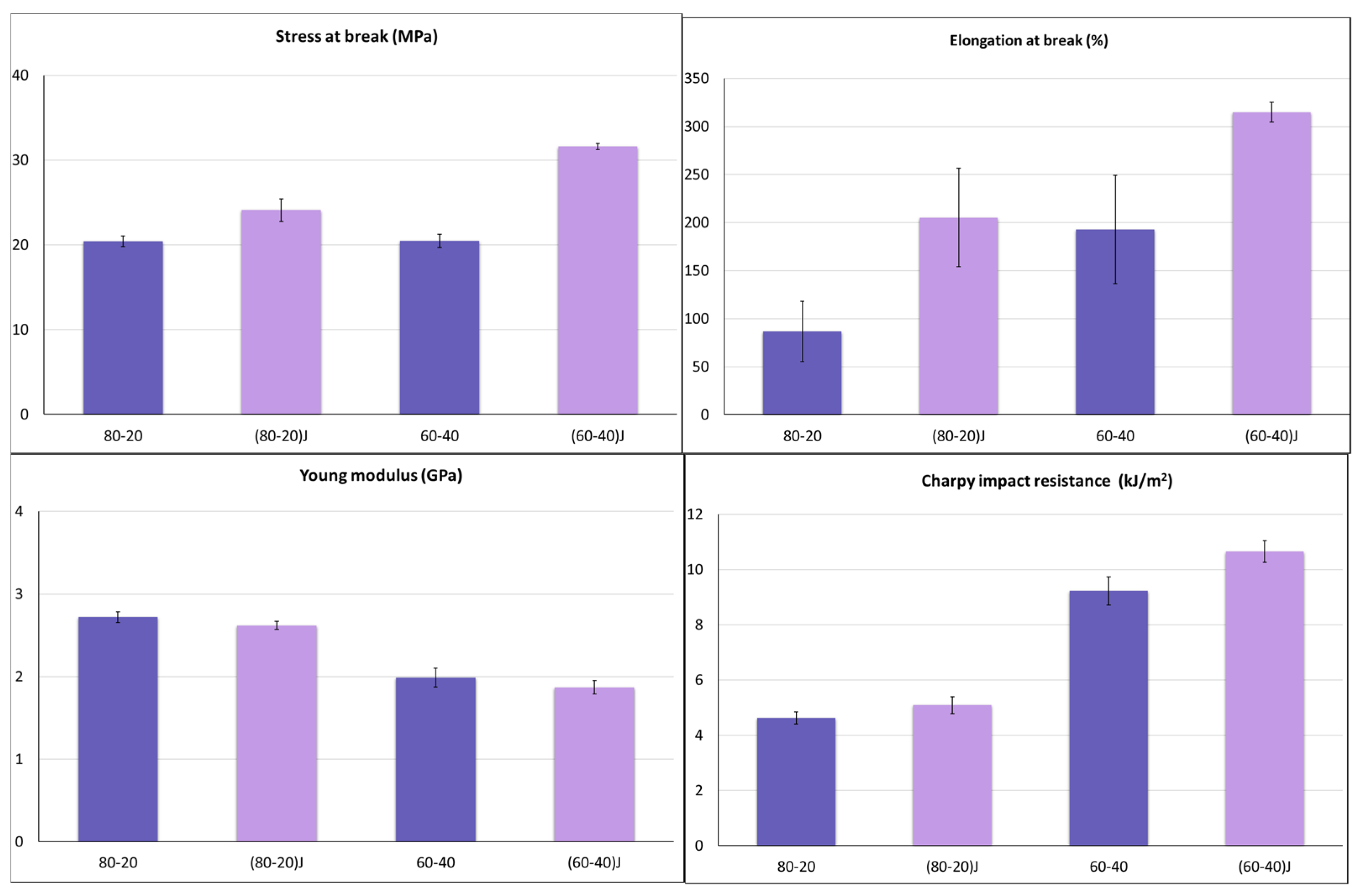
3.2. Effect of the EO on Mechanichal and Failure Behaviour of PLA/PBSA Blends
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vroman, I.; Tighzert, L. Biodegradable polymers. Materials 2009, 2, 307–344. [Google Scholar] [CrossRef]
- Luckachan, G.E.; Pillai, C.K.S. Biodegradable Polymers—A Review on Recent Trends and Emerging Perspectives. J. Polym. Environ. 2011, 19, 637–676. [Google Scholar] [CrossRef]
- La Mantia, F.P.; Morreale, M.; Botta, L.; Mistretta, M.C.; Ceraulo, M.; Scaffaro, R. Degradation of polymer blends: A brief review. Polym. Degrad. Stab. 2017, 145, 79–92. [Google Scholar] [CrossRef]
- Gross, R.A.; Kalra, B. Biodegradable Polymers for the Environment. Science 2002, 297, 803–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliotta, L.; Gigante, V.; Coltelli, M.; Cinelli, P.; Lazzeri, A.; Seggiani, M. Thermo-Mechanical Properties of PLA / Short Flax Fiber Biocomposites. Appl. Sci. 2019, 9, 3797. [Google Scholar]
- Harris, A.M.; Lee, E.C. Improving mechanical performance of injection molded PLA by controlling crystallinity. J. Appl. Polym. Sci. 2008, 107, 2246–2255. [Google Scholar] [CrossRef]
- Nofar, M.; Maani, A.; Sojoudi, H.; Heuzey, M.C.; Carreau, P.J. Interfacial and rheological properties of PLA/PBAT and PLA/PBSA blends and their morphological stability under shear flow. J. Rheol. 2015, 59, 317–333. [Google Scholar] [CrossRef]
- Bureepukdee, C.; Suttireungwong, S.; Seadan, M. A study on reactive blending of (polylactic acid) and poly(butylene succinate co adipate). IOP Conf. Ser. Mater. Sci. Eng. 2015, 87, 012070. [Google Scholar] [CrossRef] [Green Version]
- Nofar, M.; Tabatabaei, A.; Sojoudiasli, H.; Park, C.B.; Carreau, P.J.; Heuzey, M.-C.; Kamal, M.R. Mechanical and bead foaming behavior of PLA-PBAT and PLA-PBSA blends with different morphologies. Eur. Polym. J. 2017, 34, 231–244. [Google Scholar] [CrossRef]
- Yokohara, T.; Yamaguchi, M. Structure and properties for biomass-based polyester blends of PLA and PBS. Eur. Polym. J. 2008, 44, 677–685. [Google Scholar] [CrossRef]
- Hassan, E.; Wei, Y.; Jiao, H.; Yu, M. Dynamic Mechanical properties and Thermal stability of Poly(lactic acid) and poly(butylene succinate) blends composites. J. Fiber Bioeng. Inform. 2013, 6, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Pradeep, S.A.; Kharbas, H.; Turng, L.S.; Avalos, A.; Lawrence, J.G.; Pilla, S. Investigation of thermal and thermomechanical properties of biodegradable PLA/PBSA composites processed via supercritical fluid-assisted foam injection molding. Polymers 2017, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.W. Characterization and processing of Biodegradable polymer blends of poly(lactic acid) with poly(butylene succinate adipate). Korea Aust. Rheol. J. 2005, 17, 71–77. [Google Scholar]
- Takayama, T.; Todo, M.; Tsuji, H.; Arakawa, K. Improvement of fracture properties of PLA/PCL polymer blends by control of phase structures. Kobunshi Ronbunshu 2006, 63, 626–632. [Google Scholar] [CrossRef] [Green Version]
- Semba, T.; Kitagawa, K.; Ishiaku, U.S.; Kotaki, M.; Hamada, H. Effect of compounding procedure on mechanical properties and dispersed phase morphology of poly(lactic acid)/polycaprolactone blends containing peroxide. J. Appl. Polym. Sci. 2007, 103, 1066–1074. [Google Scholar] [CrossRef]
- Harada, M.; Lida, K.; Okamoto, K.; Hayashi, H.; Hirano, K. Reactive Compatibilization of Biodegrdable Poly(lactic acid)/Poly(caprolactone) Blends with reacive Processing Agents. Polym. Eng. Sci. 2008, 1359–1368. [Google Scholar] [CrossRef]
- Arruda, L.C.; Magaton, M.; Bretas, R.E.S.; Ueki, M.M. Influence of chain extender on mechanical, thermal and morphological properties of blown films of PLA/PBAT blends. Polym. Test. 2015. [Google Scholar] [CrossRef]
- Kumar, M.; Mohanty, S.; Nayak, S.K.; Rahail Parvaiz, M. Effect of glycidyl methacrylate (GMA) on the thermal, mechanical and morphological property of biodegradable PLA/PBAT blend and its nanocomposites. Bioresour. Technol. 2010, 101, 8406–8415. [Google Scholar] [CrossRef]
- Muthuraj, R.; Misra, M.; Mohanty, A.K. Biodegradable Poly(butylene succinate) and Poly(butylene adipate-co-terephthalate) Blends: Reactive Extrusion and Performance Evaluation. J. Polym. Environ. 2014, 22, 336–349. [Google Scholar] [CrossRef]
- Gigante, V.; Canesi, I.; Cinelli, P.; Coltelli, M.B.; Lazzeri, A. Rubber Toughening of Polylactic Acid (PLA) with Poly(butylene adipate-co-terephthalate) (PBAT): Mechanical Properties, Fracture Mechanics and Analysis of Ductile-to-Brittle Behavior while Varying Temperature and Test Speed. Eur. Polym. J. 2019, 115, 125–137. [Google Scholar] [CrossRef]
- Changwichan, K.; Silalertruksa, T.; Gheewala, S.H. Eco-efficiency assessment of bioplastics production systems and end-of-life options. Sustainability 2018, 10, 952. [Google Scholar] [CrossRef] [Green Version]
- European Bioplastics. Bioplastics Market Data. Available online: https://www.european-bioplastics.org/market/ (accessed on 25 September 2020).
- Gigante, V.; Coltelli, M.; Vannozzi, A.; Panariello, L.; Fusco, A.; Trombi, L.; Donnarumma, G.; Danti, S.; Lazzeri, A. Flat Die Extruded Biocompatible Poly( Lactic Acid ). Polymers 2019, 11, 1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucknall, C.B.; Heather, P.S.; Lazzeri, A. Rubber toughening of plastics. J. Mater. Sci. 2000, 24, 2255–2261. [Google Scholar] [CrossRef]
- Lazzeri, A.; Bucknall, C.B. Recent developments in the modeling of dilatational yielding in toughened plastics. Toughening Plast. Adv. Model. Exp. 2000, 264, 14–35. [Google Scholar]
- Nofar, M.; Salehiyan, R.; Ciftci, U.; Jalali, A.; Durmuş, A. Ductility improvements of PLA-based binary and ternary blends with controlled morphology using PBAT, PBSA, and nanoclay. Compos. Part B Eng. 2020, 182, 107661. [Google Scholar] [CrossRef]
- Meng, L.; Yu, L.; Khalid, S.; Liu, H.; Zhang, S.; Duan, Q.; Chen, L. Preparation, microstructure and performance of poly(lactic acid)-Poly(butylene succinate-co-butyleneadipate)-starch hybrid composites. Compos. Part B Eng. 2019, 177, 107384. [Google Scholar] [CrossRef]
- Garcia-Campo, M.; Quiles-Carrillo, L.; Sanchez-Nacher, L.; Balart, R.; Montanes, N. High toughness poly(lactic acid) (PLA) formulations obtained by ternary blends with poly(3-hydroxybutyrate) (PHB) and flexible polyesters from succinic acid. Polym. Bull. 2018, 76, 1839–1859. [Google Scholar] [CrossRef]
- Supthanyakula, R.; Kaabbuathongb, N.; Chirachanchai, S. Random poly(butylene succinate-co-lactic acid) as a multi-functional additive for miscibility, toughness, and clarity of PLA/PBS blends. Polymer 2016, 105, 1–9. [Google Scholar] [CrossRef]
- Su, S.; Kopitzky, R.; Tolga, S.; Kabasci, S. Polylactide (PLA) and Its Blends with Poly(butylene succinate) (PBS): A Brief Review. Polymers 2019, 11, 1193. [Google Scholar] [CrossRef] [Green Version]
- Hamad, K.; Kaseem, M.; Ayyoob, M.; Joo, J.; Deri, F. Polylactic acid blends: The future of green, light and tough. Prog. Polym. Sci. 2018, 85, 83–127. [Google Scholar] [CrossRef]
- Wang, R.; Wang, S.; Zhang, Y.; Wan, C.; Ma, P. Toughening modification of PLLA/PBS blends via in situ compatibilization. Polym. Eng. Sci. 2009, 49, 26–33. [Google Scholar] [CrossRef]
- Kfoury, G.; Raquez, J.-M.; Hassouna, F.; Odent, J.; Toniazzo, V.; Ruch, D.; Dubois, P. Recent advances in high performance poly(lactide): From “green” plasticization to super-tough materials via (reactive) compounding. Front. Chem. 2013, 1, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinaels, R.; Moldenaers, P. Morphology development in immiscible polymer blends. In Polymer Morphology: Priciples, Characterization and Properties; Guo, Q., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2016; Chapter 19; pp. 348–373. [Google Scholar]
- Thomas, S.; Grohens, Y.; Jyotishkumar, P. (Eds.) Characterization of Polymer Blends Miscibility, Morphology and Interfaces; Wiley-VCH Verlag GmbH & Co. KG: Weinheim, Germany, 2014. [Google Scholar]
- Harrats, C.; Thomas, S.; Groeninckx, G. (Eds.) Micro- and Nanostructured Multiphase Polymer Blend Systems: Phase Morphology and Interfaces; CRC Press, Taylor and Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Hu, L.; Vuillaume, P.Y. Chapter 7—Reactive Compatibilization of Polymer Blends by Coupling Agents and Interchange Catalysts. In Compatibilization of Polymer Blends; Ajitha, A.R., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 205–248. [Google Scholar]
- Homklin, R.; Hongsriphan, N. Mechanical and thermal properties of PLA/PBS cocontinuous blends adding nucleating agent. Energy Procedia 2013, 34, 871–879. [Google Scholar] [CrossRef] [Green Version]
- Dhibar, A.K.; Kim, J.K.; Khatua, B.B. Cocontinuous phase morphology of asymmetric compositions of polypropylene/high-density polyethylene blend by the addition of clay. J. Appl. Polym. Sci. 2011, 119, 3080–3092. [Google Scholar] [CrossRef]
- Ito, E.N.; Pessan, L.A.; Covas, J.A.; Hage, E., Jr. Analysis of the morphological development of PBT/ABS blends during the twin screw extrusion and injection molding processes. Int. Polym. Process. 2003, 18, 376–381. [Google Scholar] [CrossRef]
- Wu, D.; Yuan, L.; Laredo, E.; Zhang, M.; Zhou, W. Interfacial properties, viscoelasticity, and thermal behaviors of poly(butylene succinate)/polylactide blend. Ind. Eng. Chem. Res. 2012, 51, 2290–2298. [Google Scholar] [CrossRef]
- Macosko, C.W. Morphology Development and Control in Immiscible Polymer Blends. Macromol. Symp. 2000, 149, 171–184. [Google Scholar] [CrossRef]
- Ajitha, A.R.; Thomas, S. Chapter 1—Introduction: Polymer blends, thermodynamics, miscibility, phase separation, and compatibilization. In Compatibilization of Polymer Blends; Ajitha, A.R., Thomas, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–29. [Google Scholar]
- Coltelli, M.B.; Bronco, S.; Chinea, C. The effect of free radical reactions on structure and properties of poly(lactic acid) (PLA) based blends. Polym. Degrad. Stab. 2010, 95, 332–341. [Google Scholar] [CrossRef]
- Corre, Y.-M.; Duchet, J.; Reignier, J.; Maazouz, A. Melt strengthening of poly(lactic acid) through reactive extrusion with epoxy-functionalized chains. Rheol. Acta 2011, 50, 613–629. [Google Scholar] [CrossRef]
- Han, C.D. Rheology and Processing of Polymeric Materials: Volume 1: Polymer Rheology; Oxford University Press on Demand: Oxford, UK, 2007; Volume 1, ISBN 0195187822. [Google Scholar]
- Meng, Q.; Heuzey, M.-C.; Carreau, P.J. Control of thermal degradation of polylactide/clay nanocomposites during melt processing by chain extension reaction. Polym. Degrad. Stab. 2012, 97, 2010–2020. [Google Scholar] [CrossRef]
- Wu, C.-S. Utilization of peanut husks as a filler in aliphatic–aromatic polyesters: Preparation, characterization, and biodegradability. Polym. Degrad. Stab. 2012, 97, 2388–2395. [Google Scholar] [CrossRef]
- Bhatia, A.; Gupta, R.K.; Bhattacharya, S.N.; Choi, H.J. Compatibility of biodegradable poly(lactic acid) (PLA) and poly(butylene succinate) (PBS) blends for packaging application. Korea Aust. Rheol. J. 2007, 19, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Gu, S.Y.; Zhang, K.; Ren, J.; Zhan, H. Melt rheology of polylactide/poly(butylene adipate-co-terephthalate) blends. Carbohydr. Polym. 2008, 74, 79–85. [Google Scholar] [CrossRef]
- Lin, S.; Guo, W.; Chen, C.; Ma, J.; Wang, B. Mechanical properties and morphology of biodegradable poly(lactic acid)/poly(butylene adipate-co-terephthalate) blends compatibilized by transesterification. Mater. Des. 2012, 36, 604–608. [Google Scholar] [CrossRef]
- Al-Itry, R.; Lamnawar, K.; Maazouz, A. Improvement of thermal stability, rheological and mechanical properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy. Polym. Degrad. Stab. 2012, 97, 1898–1914. [Google Scholar] [CrossRef]
- Pilla, S.; Kim, S.G.; Auer, G.K.; Gong, S.; Park, C.B. Microcellular extrusion-foaming of polylactide with chain-extender. Polym. Eng. Sci. 2009, 49, 1653–1660. [Google Scholar] [CrossRef]
- Wang, X.; Peng, S.; Chen, H.; Yu, X.; Zhao, X. Mechanical properties, rheological behaviors, and phase morphologies of high-toughness PLA/PBAT blends by in-situ reactive compatibilization. Compos. Part B Eng. 2019, 173, 107028. [Google Scholar] [CrossRef]
- Lascano, D.; Quiles-Carrillo, L.; Balart, R.; Boronat, T.; Montanes, N. Toughened poly (lactic acid)-PLA formulations by binary blends with poly(butylene succinate-co-adipate)-PBSA and their shape memory behaviour. Materials 2019, 12, 622. [Google Scholar] [CrossRef] [Green Version]
- Chaiwutthinan, P.; Leejarkpai, T.; Kashima, D.P.; Chuayjuljit, S. Poly(Lactic Acid)/Poly(Butylene Succinate) Blends Filled with Epoxy Functionalised Polymeric Chain Extender. Adv. Mater. Res. 2013, 664, 644–648. [Google Scholar] [CrossRef]
- Elhassan, A.S.M.; Saeed, H.A.M.; Eltahir, Y.A.; Xia, Y.M.; Wang, Y.P. Modification of PLA with Chain Extender. Appl. Mech. Mater. 2014, 716–717, 44–47. [Google Scholar] [CrossRef]
- Nakayama, D.; Wu, F.; Mohanty, A.K.; Hirai, S.; Misra, M. Biodegradable Composites Developed from PBAT/PLA Binary Blends and Silk Powder: Compatibilization and Performance Evaluation. ACS Omega 2018, 3, 12412–12421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coltelli, M.-B.; Gigante, V.; Vannozzi, A.; Aliotta, L.; Danti, S.; Neri, S.; Gagliardini, A.; Morganti, P.; Panariello, L.; Lazzeri, A. Poly(lactic) acid (PLA) based nano-structured functional films for personal care applications. In Proceedings of the AUTEX 2019—19th World Textile Conference on Textiles at the Crossroads, Ghent, Belgium, 11–15 June 2019. [Google Scholar]
- Aliotta, L.; Cinelli, P.; Beatrice Coltelli, M.; Lazzeri, A. Rigid filler toughening in PLA-Calcium Carbonate composites: Effect of particle surface treatment and matrix plasticization. Eur. Polym. J. 2018, 113, 78–88. [Google Scholar] [CrossRef]
- Aliotta, L.; Gigante, V.; Acucella, O.; Signori, F.; Lazzeri, A. Thermal, Mechanical and Micromechanical Analysis of PLA/PBAT/POE-g-GMA Extruded Ternary Blends. Front. Mater. 2020. [Google Scholar] [CrossRef]
- Lazzeri, A.; Thio, Y.S.; Cohen, R.E. Volume strain measurements on CACO3/polypropylene particulate composites: The effect of particle size. J. Appl. Polym. Sci. 2004, 91, 925–935. [Google Scholar] [CrossRef]
- Bernal, C.R.; Montemartini, P.E.; Frontini, P.M. The use of load separation criterion and normalization method in ductile fracture characterization of thermoplastic polymers. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 1869–1880. [Google Scholar] [CrossRef]
- Baldi, F.; Agnelli, S.; Riccò, T. On the determination of the point of fracture initiation by the load separation criterion in J-testing of ductile polymers. Polym. Test. 2013, 32, 1326–1333. [Google Scholar] [CrossRef]
- Sharobeam, M.H.; Landes, J.D. The load separation criterion and methodology in ductile fracture mechanics. Int. J. Fract. 1991, 47, 81–104. [Google Scholar] [CrossRef]
- Baldi, F.; Agnelli, S.; Riccò, T. On the applicability of the load separation criterion in determining the fracture resistance (JIC) of ductile polymers at low and high loading rates. Int. J. Fract. 2010, 165, 105–119. [Google Scholar] [CrossRef]
- Agnelli, S.; Baldi, F.; Riccò, T. A tentative application of the energy separation principle to the determination of the fracture resistance (JIc) of rubbers. Eng. Fract. Mech. 2019, 90, 76–88. [Google Scholar] [CrossRef]
- Blackman, B.; Baldi, F.; Castellani, L.; Frontini, P.; Laiarinandrasana, L.; Pegoretti, A.; Rink, M.; Salazar, A.; Visser, H.; Agnelli, S. Application of the load separation criterion in J-testing of ductile polymers: A round-robin testing exercise. Polym. Test. 2015, 44, 72–81. [Google Scholar]
- Agnelli, S.; Baldi, F. A Testing Protocol for the Construction of the “Load SeparationParameter Curve” for Plastics; ESIS TC4 communication; ESIS: Delft, The Netherlands, 2013. [Google Scholar]
- Gui, Z.Y.; Wang, H.R.; Gao, Y.; Lu, C.; Cheng, S.J. Morphology and melt rheology of biodegradable poly(lactic acid)/poly(butylene succinate adipate) blends: Effect of blend compositions. Iran. Polym. J. 2012, 21, 81–89. [Google Scholar] [CrossRef]
- Fischer, E.W.; Sterzel, H.J.; Wegner, G. Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reactions. Kolloid Z. Z. Polym. 1973, 251, 980–990. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, C.; Liu, X.; Zhu, J. The crystallization behavior and mechanical properties of polylactic acid in the presence of a crystal nucleating agent. J. Appl. Polym. Sci. 2012, 125, 1108–1115. [Google Scholar] [CrossRef]
- Bhadane, P.A.; Champagne, M.F.; Huneault, M.A.; Tofan, F.; Favis, B.D. Erosion-dependant continuity development in high viscosity ratio blends of very low interfacial tension. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 1919–1929. [Google Scholar] [CrossRef]
- Xu, X.; Yan, X.; Zhu, T.; Zhang, C.; Sheng, J. Phase morphology development of polypropylene/ethylene-octene copolymer blends: Effects of blend composition and processing conditions. Polym. Bull. 2007, 58, 465–478. [Google Scholar] [CrossRef]
- Lazzeri, A.; Bucknall, C.B. Dilatational bands in rubber-toughened polymers. J. Mater. Sci. 1993, 28, 6799–6808. [Google Scholar] [CrossRef]
- Wu, S. Chain structure, phase morphology, and toughness relationships in polymers and blends. Polym. Eng. Sci. 1990, 30, 753–761. [Google Scholar] [CrossRef]
- Pötschke, P.; Paul, D.R. Formation of Co-continuous Structures in Melt-Mixed Immiscible Polymer Blends. J. Macromol. Sci. Part C 2003, 43, 87–141. [Google Scholar] [CrossRef]
- Harrats, C.; Coltelli, M.B.; Groeninckx, G. Features on the development and stability of phase morphology in complex multicomponent polymeric systems: Main focus on processing aspects. In Polymer Morphology: Principles, Characterization, and Processing, 1st ed.; Guo, Q., Ed.; Wiley: Hoboken, NJ, USA, 2016; pp. 418–438. [Google Scholar]
- Coltelli, M.-B.; Mallegni, N.; Rizzo, S.; Cinelli, P.; Lazzeri, A. Improved Impact Properties in Poly(lactic acid) (PLA) Blends Containing Cellulose Acetate (CA) Prepared by Reactive Extrusion. Materials 2019, 12, 270. [Google Scholar] [CrossRef] [Green Version]
- Coltelli, M.B.; Harrats, C.; Aglietto, M.; Groeninckx, G. Influence of compatibilizer precursor structure on the phase distribution of low density poly(ethylene) in a poly(ethylene terephthalate) matrix. Polym. Eng. Sci. 2008, 48, 1424–1433. [Google Scholar] [CrossRef]
- Coltelli, M.B.; Toncelli, C.; Ciardelli, F.; Bronco, S. Compatible blends of biorelated polyesters through catalytic transesterification in the melt. Polym. Degrad. Stab. 2011, 96, 982–990. [Google Scholar] [CrossRef]
- Quiles-Carrillo, L.; Montanes, N.; Lagaron, J.M.; Balart, R.; Torres-Giner, S. In Situ Compatibilization of Biopolymer Ternary Blends by Reactive Extrusion with Low-Functionality Epoxy-Based Styrene–Acrylic Oligomer. J. Polym. Environ. 2019, 27, 84–96. [Google Scholar] [CrossRef]
- Rasselet, D.; Caro-Bretelle, A.-S.; Taguet, A.; Lopez-Cuesta, J.-M. Reactive Compatibilization of PLA/PA11 Blends and Their Application in Additive Manufacturing. Materials 2019, 12, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de CD Nunes, E.; de Souza, A.G.; dos S. Rosa, D. Effect of the Joncryl® ADR Compatibilizing Agent in Blends of Poly(butylene adipate-co-terephthalate)/Poly(lactic acid). Macromol. Symp. 2019, 383, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bhattacharya, M.; Mano, J.F. Thermal analysis of the multiple melting behavior of poly(butylene succinate-co-adipate). J. Polym. Sci. Part B Polym. Phys. 2005, 43, 3077–3082. [Google Scholar] [CrossRef]
- Fenni, S.E.; Wang, J.; Haddaoui, N.; Favis, B.D.; Müller, A.J.; Cavallo, D. Crystallization and self-nucleation of PLA, PBS and PCL in their immiscible binary and ternary blends. Thermochim. Acta 2019, 677, 117–130. [Google Scholar] [CrossRef]
- Wunderlich, B. Macromolecular Physics; Elsevier: Amsterdam, The Netherlands, 2012; Volume 2, ISBN 0323148948. [Google Scholar]
- Di Lorenzo, M.L. Calorimetric analysis of the multiple melting behavior of poly(L-lactic acid). J. Appl. Polym. Sci. 2006, 100, 3145–3151. [Google Scholar] [CrossRef]
- Aliotta, L.; Cinelli, P.; Coltelli, M.B.; Righetti, M.C.; Gazzano, M.; Lazzeri, A. Effect of nucleating agents on crystallinity and properties of poly(lactic acid) (PLA). Eur. Polym. J. 2017, 93, 822–832. [Google Scholar] [CrossRef]
- Mallegni, N.; Phuong, T.V.; Coltelli, M.B.; Cinelli, P.; Lazzeri, A. Poly(lactic acid) (PLA) based tear resistant and biodegradable flexible films by blown film extrusion. Materials 2018, 11, 148. [Google Scholar] [CrossRef] [Green Version]
- Ostrowska, J.; Sadurski, W.; Paluch, M.; Tyński, P.; Bogusz, J. The effect of poly(butylene succinate) content on the structure and thermal and mechanical properties of its blends with polylactide. Polym. Int. 2019, 68, 1271–1279. [Google Scholar] [CrossRef]
- Eslami, H.; Kamal, M.R. Effect of a chain extender on the rheological and mechanical properties of biodegradable poly(lactic acid)/poly[(butylene succinate)-co-adipate] blends. J. Appl. Polym. Sci. 2013, 129, 2418–2428. [Google Scholar] [CrossRef]
- Supthanyakul, R.; Kaabbuathong, N.; Chirachanchai, S. Poly(l-lactide-b-butylene succinate-b-l-lactide) triblock copolymer: A multi-functional additive for PLA/PBS blend with a key performance on film clarity. Polym. Degrad. Stab. 2017, 142, 160–168. [Google Scholar] [CrossRef]
- Nascimento, L.; Gamez-Perez, J.; Santana, O.O.; Velasco, J.I.; Maspoch, M.L.; Franco-Urquiza, E. Effect of the Recycling and Annealing on the Mechanical and Fracture Properties of Poly(Lactic Acid). J. Polym. Environ. 2010, 18, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Todo, M.; Takayama, T. Fracture mechanisms of biodegradable PLA and PLA/PCL blends. Biomater. Chem. 2011. [Google Scholar]
- Naqui, S.I.; Robinson, I.M. Tensile dilatometric studies of deformation in polymeric materials and their composites. J. Mater. Sci. 1993, 28, 1421–1429. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blend Name | PLA–PBSA wt.% | EO wt.% |
|---|---|---|
| PLA | 100–0 | 0 |
| 95–5 | 95–5 | 0 |
| 90–10 | 90–10 | 0 |
| 85–15 | 85–15 | 0 |
| 80–20 | 80–20 | 0 |
| (80–20)J | 80–20 | 2 |
| 60–40 | 60–40 | 0 |
| (60–40)J | 60–40 | 2 |
| Main Injection Molding Parameters | PLA | 95–5 | 90–10 | 85–15 | 80–20 | (80–20)J | 60–40 | (60–40)J |
|---|---|---|---|---|---|---|---|---|
| Temperature profile (°C) | 180/185/190 | |||||||
| Mold temperature (°C) | 70 | 70 | 60 | 60 | 55 | 65 | 55 | 65 |
| Injection holding time (s) | 5 | |||||||
| Cooling time (s) | 15 | 25 | ||||||
| Injection pressure (bar) | 90 | 90 | 80 | 80 | 80 | 120 | 80 | 100 |
| Blend Name | Young’s Modulus (GPa) | Yield Stress (MPa) | Stress at Break (%) | Elongation at Break (%) | Charpy Impact Resistance (kJ/m2) |
|---|---|---|---|---|---|
| PLA | 3.63 ± 0.12 | / | 62.7 ± 1.7 | 3.7 ± 0.3 | 2.7 ± 0.3 |
| 95–5 | 3.07 ± 0.1 | / | 59.4 ± 0.1 | 4.0 ± 0.2 | 2.8 ± 0.3 |
| 90–10 | 3.02 ± 0.13 | 57.4 ± 0.7 | 23.3 ± 2.3 | 45.8 ± 10.9 | 3.5 ± 0.2 |
| 85–15 | 2.91 ± 0.10 | 55.3 ± 0.7 | 21.3 ± 1.1 | 57.9 ± 18.9 | 4.4 ± 0.2 |
| 80–20 | 2.72 ± 0.10 | 51.2 ± 0.6 | 19.7 ± 0.6 | 86.7 ± 31.5 | 4.6 ± 0.2 |
| 60–40 | 1.99 ± 0.12 | 39.9 ± 0.8 | 21.5 ± 0.8 | 192.8 ± 31.1 | 9.2 ± 0.5 |
| Blend Name | Rn (μm) | Rv (μm) | SD |
|---|---|---|---|
| 95–5 | 0.41 | 0.54 | 1.32 |
| 90–10 | 0.42 | 0.58 | 1.38 |
| 85–15 | 0.54 | 0.78 | 1.44 |
| 80–20 | 0.55 | 0.80 | 1.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliotta, L.; Vannozzi, A.; Canesi, I.; Cinelli, P.; Coltelli, M.-B.; Lazzeri, A. Poly(lactic acid) (PLA)/Poly(butylene succinate-co-adipate) (PBSA) Compatibilized Binary Biobased Blends: Melt Fluidity, Morphological, Thermo-Mechanical and Micromechanical Analysis. Polymers 2021, 13, 218. https://doi.org/10.3390/polym13020218
Aliotta L, Vannozzi A, Canesi I, Cinelli P, Coltelli M-B, Lazzeri A. Poly(lactic acid) (PLA)/Poly(butylene succinate-co-adipate) (PBSA) Compatibilized Binary Biobased Blends: Melt Fluidity, Morphological, Thermo-Mechanical and Micromechanical Analysis. Polymers. 2021; 13(2):218. https://doi.org/10.3390/polym13020218
Chicago/Turabian StyleAliotta, Laura, Alessandro Vannozzi, Ilaria Canesi, Patrizia Cinelli, Maria-Beatrice Coltelli, and Andrea Lazzeri. 2021. "Poly(lactic acid) (PLA)/Poly(butylene succinate-co-adipate) (PBSA) Compatibilized Binary Biobased Blends: Melt Fluidity, Morphological, Thermo-Mechanical and Micromechanical Analysis" Polymers 13, no. 2: 218. https://doi.org/10.3390/polym13020218
APA StyleAliotta, L., Vannozzi, A., Canesi, I., Cinelli, P., Coltelli, M.-B., & Lazzeri, A. (2021). Poly(lactic acid) (PLA)/Poly(butylene succinate-co-adipate) (PBSA) Compatibilized Binary Biobased Blends: Melt Fluidity, Morphological, Thermo-Mechanical and Micromechanical Analysis. Polymers, 13(2), 218. https://doi.org/10.3390/polym13020218