
Influence of the Filler Particles’ Surface Morphology on the Polyurethane Matrix’s Structure Formation in the Composite

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Polymer Matrix
2.1.2. Filler
2.2. Sample Preparation
2.3. Measurements
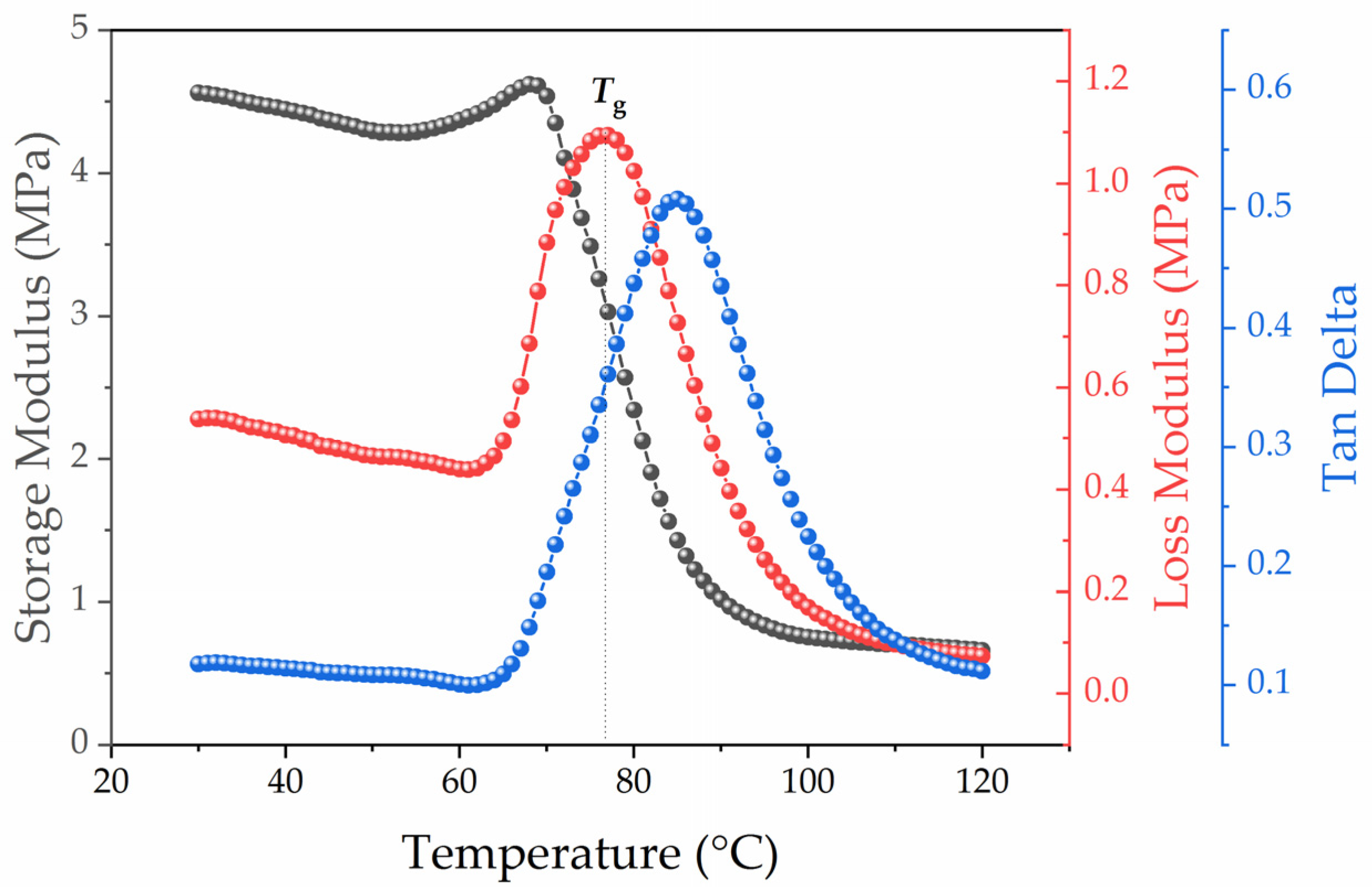
Dynamic Mechanical Analysis (DMA)
3. Results and Discussion
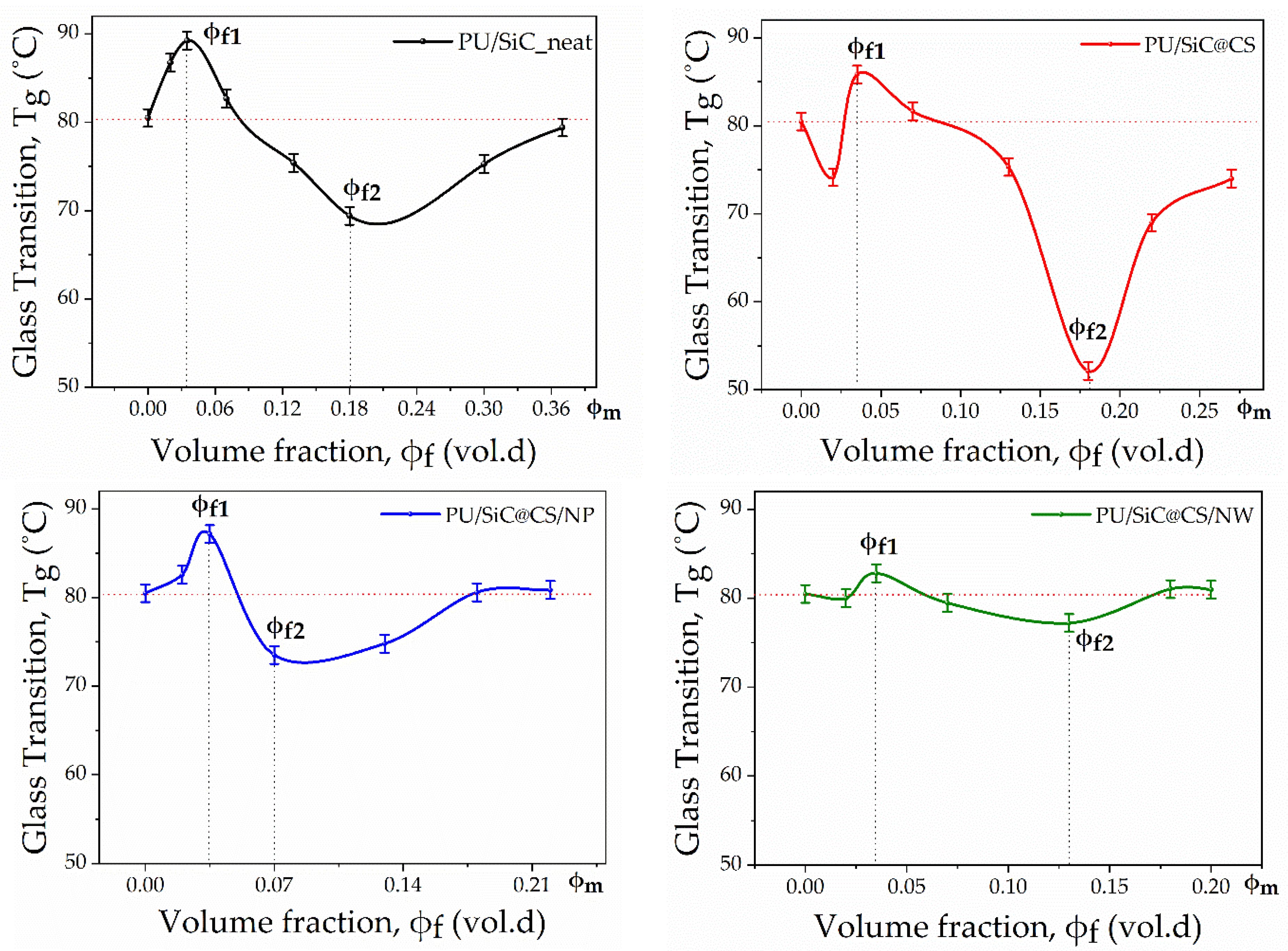
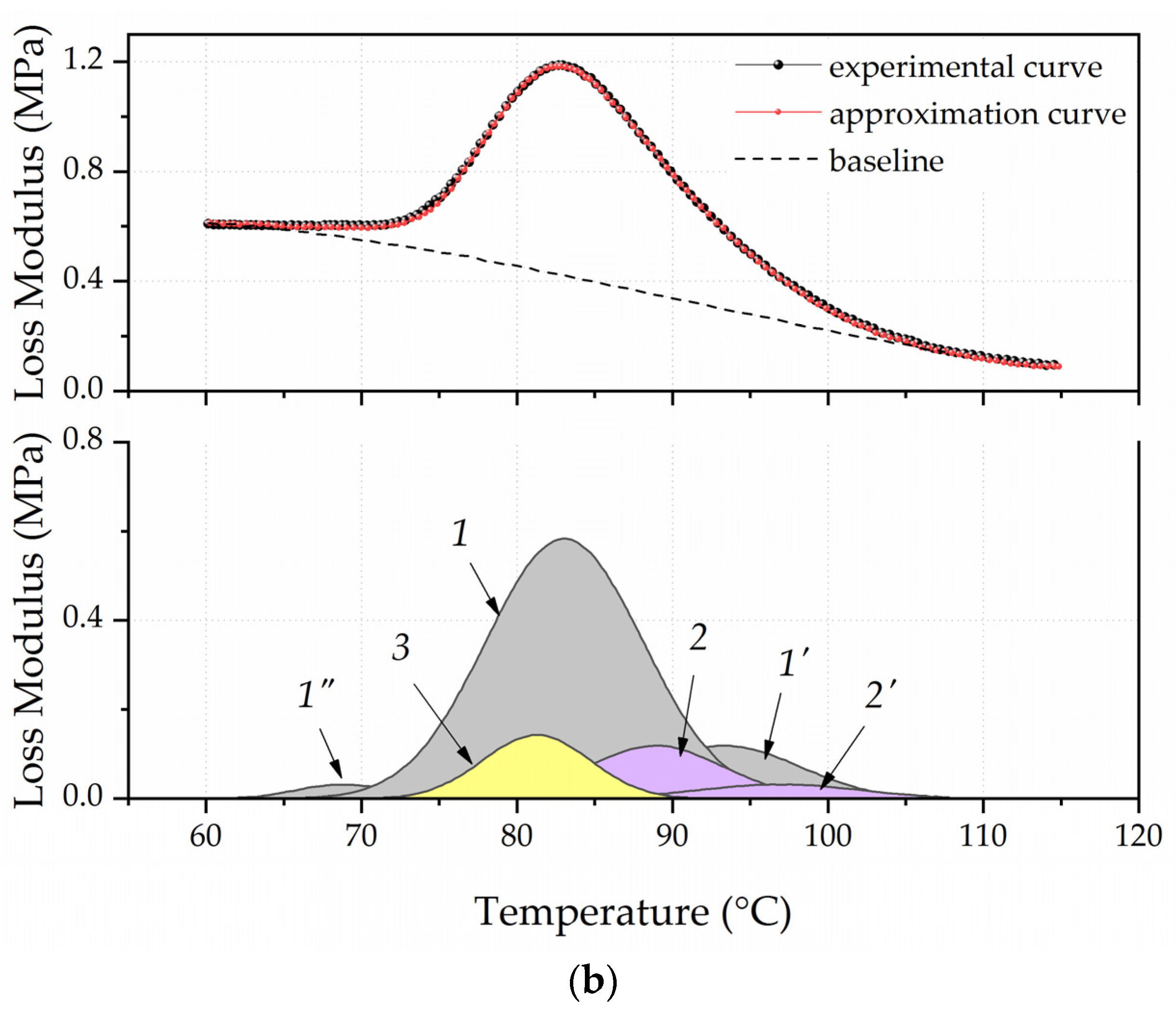
3.1. Dependence of the Composite’s Relaxation Properties from the Filler Particle Surface Morphology and Its Concentration
3.2. Determination of the Polymer’s Proportion Forming the Boundary, Transition and Bulk Matrix Layer
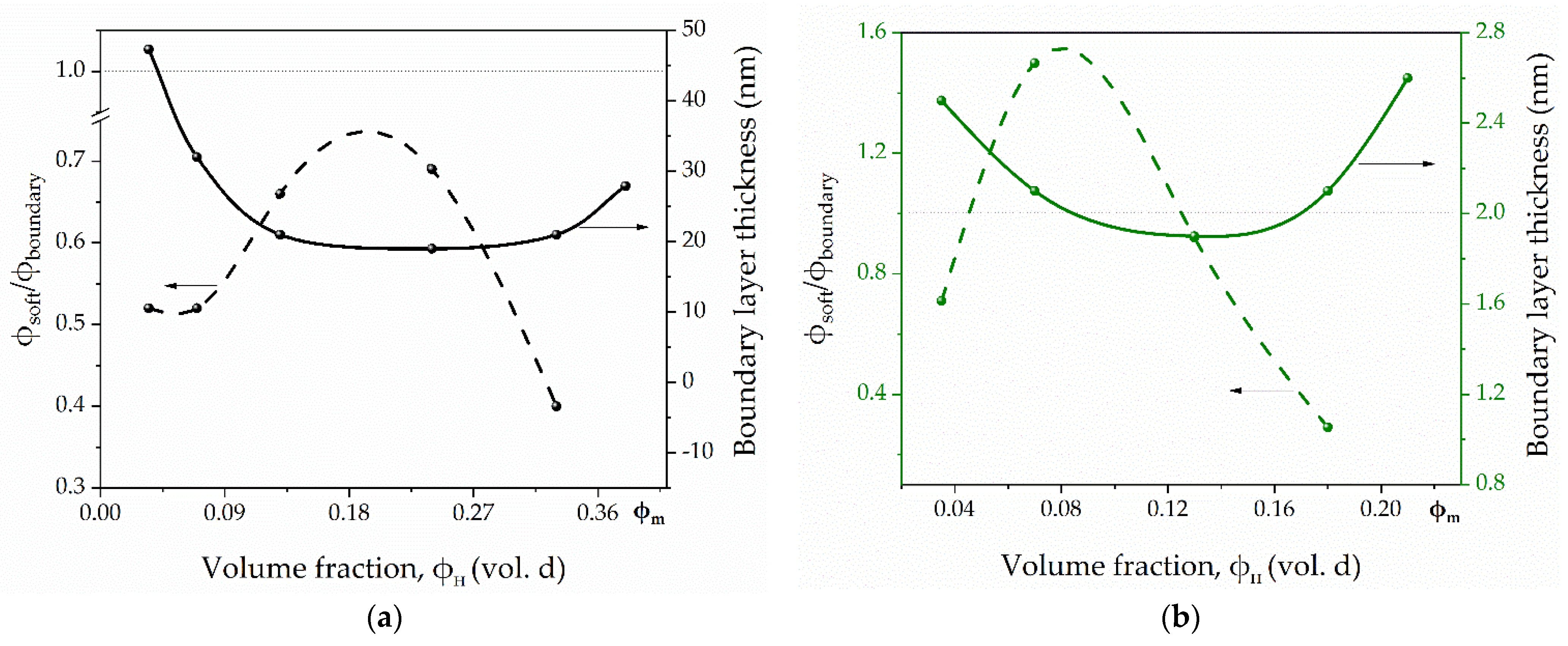
3.3. The Boundary Layer Thickness Determination
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kausar, A.; Rafique, I.; Bakhtiar, M. Aerospace Application of Polymer Nanocomposite with Carbon Nanotube, Graphite, Graphene Oxide, and Nanoclay. Polym.-Plast. Technol. Eng. 2017, 56, 1438–1456. [Google Scholar] [CrossRef]
- Krishnakumar, S.; Senthilvelan, T. Polymer composites in dentistry and orthopedic applications-a review. Mater. Today Proc. 2021, 46, 9707–9713. [Google Scholar] [CrossRef]
- Supova, M.; Martynkova, G.S.; Barabasyova, K. Effect of nanofillers dispersion in polymer matrices: A review. Sci. Adv. Mater. 2011, 3, 1–25. [Google Scholar] [CrossRef]
- Lepcio, P.; Ondreas, F.; Zarybnicka, K.; Svatik, J.; Jancar, J. Phase diagram of bare particles in polymer nanocomposites: Uniting solution and melt blending. Polymer 2021, 230, 124033. [Google Scholar] [CrossRef]
- Jouault, N.; Dalmas, F.; Boue, F.; Jestin, J. Multiscale characterization of filler dispersion and origins of mechanical reinforcement in model nanocomposites. Polymer 2012, 53, 761–775. [Google Scholar] [CrossRef]
- Forintos, N.; Czigany, T. Multifunctional application of carbon fiber reinforced polymer composites: Electrical properties of the reinforcing carbon fibers—A short review. Compos. Part B Eng. 2019, 162, 331–343. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Zhdanov, A.A. Role of various factors in the restriction of molecular mobility of polymer chains in filled polymers. Polym. Sci. USSR 1987, 30, 1000–1006. [Google Scholar] [CrossRef]
- Li, Y.; Abberton, B.C.; Kröger, M.; Liu, W.K. Challenges in multiscale modeling of polymer dynamics. Polymers 2013, 5, 751–832. [Google Scholar] [CrossRef] [Green Version]
- Arrighi, V.; Higgins, J.S.; Burgess, A.N.; Floudas, G. Local dynamics of poly(dimethyl siloxane) in the presence of reinforcing filler particles. Polymer 1998, 39, 6369–6376. [Google Scholar] [CrossRef]
- Jancar, J.; Hoy, R.S.; Jancarova, E.; Zidek, J. Effect of temperature, strain rate and particle size on the yield stresses and post-yield strain softening of PMMA and its composites. Polymer 2015, 63, 196–207. [Google Scholar] [CrossRef]
- Pakhomov, K.S.; Simonov-Emel’yanov, I.D.; Antipov, Y.V.; Kul’kov, A.A. The compaction of fibrous fillers under pressure and the structure formation in reinforced organoplastics during processing. Int. Polym. Sci. Technol. 2015, 42, 11–16. [Google Scholar] [CrossRef]
- Burroughs, M.J.; Napolitano, S.; Cangialosi, D.; Priestley, R.D. Direct measurement of glass transition temperature in exposed and buried adsorbed polymer nanolayers. Macromolecules 2016, 49, 4647–4655. [Google Scholar] [CrossRef]
- Napolitano, S.; Glynos, E.; Tito, N.B. Glass transition of polymers in bulk, confined geometries, and near interfaces. Rep. Prog. Phys. 2017, 80, 036602. [Google Scholar] [CrossRef]
- Karatrantos, A.V.; Clarke, N. Polymer Dynamics in Polymer-Nanoparticle Interface. Theory Modeling Polym. Nanocomposites 2021, 310, 81–100. [Google Scholar] [CrossRef]
- Zuo, B.; Zhou, H.; Davis, M.J.; Wang, X.; Priestley, R.D. Effect of Local Chain Conformation in Adsorbed Nanolayers on Confined Polymer Molecular Mobility. Phys. Rev. Lett. 2019, 122, 217801. [Google Scholar] [CrossRef] [PubMed]
- Campoy-Quiles, M.; Sims, M.; Etchegoin, P.G.; Bradley, D.D.C. Thickness- dependent thermal transition temperatures in thin conjugated polymer films. Macromolecules 2006, 39, 7673–7680. [Google Scholar] [CrossRef]
- Yang, S.; Liu, S.; Narayanan, S.; Zhang, C.; Akcora, P. Chemical heterogeneity in interfacial layers of polymer nanocomposites. Soft Matter 2018, 14, 4784–4791. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.J.; Winey, K.I. Dynamics of polymer segments, polymer chains, and nanoparticles in polymer nanocomposite melts: A review. Prog. Polym. Sci. 2020, 105, 101242. [Google Scholar] [CrossRef]
- Țălu, Ș. Micro and Nanoscale Characterization of Three Dimensional Surfaces. Basics and Applications; Napoca Star Publ House: Cluj-Napoca, Romania, 2005; pp. 21–27. [Google Scholar]
- Hong, Y.; Li, Y.; Wang, F.; Zuo, B.; Wang, X.; Zhang, L.; Kawaguchi, D.; Tanaka, K. Enhanced thermal stability of polystyrene by interfacial noncovalent interactions. Macromolecules 2018, 51, 5620–5627. [Google Scholar] [CrossRef]
- Ma, H.; Gao, B.; Wang, M. Strategies for enhancing thermal conductivity of polymer-based thermal interface materials: A review. J. Mater. Sci. 2021, 56, 1064–1086. [Google Scholar] [CrossRef]
- Georgakilas, V.; Otyepka, M.; Bourlinos, A.B.; Chandra, V.; Kim, N.; Kemp, K.C.; Hobza, P.; Zboril, R.; Kim, K.S. Functionalization of Graphene: Covalent and Non-Covalent Approaches, Derivatives and Applications. Chem. Rev. 2012, 112, 6156–6214. [Google Scholar] [CrossRef]
- Yuan, B.; Zhao, B.; Cong, Z.; Cheng, Z.; Wang, Q.; Lu, Y.; Han, X. A Flexible, fireproof, composite polymer electrolyte reinforced by electrospun polyimide for room-temperature solid-state batteries. Polymers 2021, 13, 3622. [Google Scholar] [CrossRef]
- Lisuzzo, L.; Caruso, M.R.; Cavallaro, G.; Milioto, S.; Lazzara, G. Hydroxypropyl Cellulose Films Filled with Halloysite Nanotubes/Wax Hybrid Microspheres. Ind. Eng. Chem. Res. 2021, 60, 1656–1665. [Google Scholar] [CrossRef]
- Lisuzzo, L.; Cavallaro, G.; Milioto, S.; Lazzara, G. Effects of halloysite content on the thermo-mechanical performances ofcomposite bioplastics. Appl. Clay Sci. 2020, 185, 105416. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Venna, S.R.; Hopkinson, D.P. Interactions at the interface of polymer matrix-filler particle composites. Polymer 2016, 103, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Simonov-Emel’yanov, I.D. The structure and calculation of compositions of disperse-filled polymer adhesives and sealants in mass and volume units. Polym. Sci. Ser. D 2020, 13, 169–171. [Google Scholar] [CrossRef]
- Simonov-Emel’yanov, I.D.; Apeksimov, N.V.; Zubkov, S.B.; Trofimov, A.N. The structure formation in polymer composites with hollow glass microspheres. Int. Polym. Sci. Technol. 2014, 41, 21–26. [Google Scholar] [CrossRef]
- Simonov-Emelyanov, I.D. Structure and calculation of filler content in particle-filled polymer composites in mass and volume units. Plast. Massy 2019, 5, 9–10. [Google Scholar] [CrossRef] [Green Version]
- Lipatov, Y.S. Physicochemical Bases of Polymer Filling; Chemistry: Moscow, Russia, 1991; p. 259. [Google Scholar]
- Startsev, O.V.; Perepechko, I.I.; Startseva, L.T.; Mashinskaya, G.P. Structural Changes in Plasticized Network Epoxy Polymers. Vysokomol. Soedin. Ser. B 1983, 25, 457–461. [Google Scholar]
- Arrighi, V.; McEwen, I.J.; Qian, H.; Serrano Prieto, M.B. The glass transition and interfacial layer in styrene-butadiene rubber containing silica nanofiller. Polymer 2003, 44, 6259–6266. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SiC_neat | SiC@C | SiC@C/NP | SiC@C/NW | |
|---|---|---|---|---|
| specific surface area (Sg), m2 g−l | 3 | 15 | 38.8 | 45 |
| maximum packing volume fraction (φm) | 0.39 | 0.28 | 0.23 | 0.21 |
| Percent in Weight of Filler, wt. % | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 5 | 10 | 20 | 40 | 60 | 65 | 70 | 75 | 99 | |
| φf (SiC_neat) | 0.02 | 0.035 | 0.08 | 0.15 | 0.19 | 0.21 | 0.22 | 0.28 | 0.38 |
| φf (SiC@C) | 0.02 | 0.035 | 0.08 | 0.15 | 0.19 | 0.21 | 0.22 | 0.28 | |
| φf (SiC@C/NP) | 0.02 | 0.035 | 0.08 | 0.15 | 0.19 | 0.21 | 0.22 | ||
| φf (SiC@C/NW) | 0.02 | 0.035 | 0.08 | 0.15 | 0.19 | 0.21 | |||
| φf, vol.d | PU/SiC_neat | PU/SiC@C/NW | ||||
|---|---|---|---|---|---|---|
| φb, vol.d | φt, vol.d | φv, vol.d | φb, vol.d | φt, vol.d | φv, vol.d | |
| 0.035 | 0.17 | 0.09 | 0.74 | 0.14 | 0.10 | 0.76 |
| 0.08 | 0.23 | 0.12 | 0.65 | 0.22 | 0.30 | 0.48 |
| 0.15 | 0.30 | 0.20 | 0.50 | 0.41 | 0.38 | 0.21 |
| 0.19 | 0.43 | 0.30 | 0.27 | 0.68 | 0.20 | 0.12 |
| 0.36 | 0.62 | 0.25 | 0.13 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalygina, T.A.; Rudenko, M.S.; Nemtsev, I.V.; Parfenov, V.A.; Voronina, S.Y.; Simonov-Emelyanov, I.D.; Borisova, P.E. Influence of the Filler Particles’ Surface Morphology on the Polyurethane Matrix’s Structure Formation in the Composite. Polymers 2021, 13, 3864. https://doi.org/10.3390/polym13223864
Shalygina TA, Rudenko MS, Nemtsev IV, Parfenov VA, Voronina SY, Simonov-Emelyanov ID, Borisova PE. Influence of the Filler Particles’ Surface Morphology on the Polyurethane Matrix’s Structure Formation in the Composite. Polymers. 2021; 13(22):3864. https://doi.org/10.3390/polym13223864
Chicago/Turabian StyleShalygina, Taisiya A., Mikhail S. Rudenko, Ivan V. Nemtsev, Vladimir A. Parfenov, Svetlana Y. Voronina, Igor D. Simonov-Emelyanov, and Polina E. Borisova. 2021. "Influence of the Filler Particles’ Surface Morphology on the Polyurethane Matrix’s Structure Formation in the Composite" Polymers 13, no. 22: 3864. https://doi.org/10.3390/polym13223864
APA StyleShalygina, T. A., Rudenko, M. S., Nemtsev, I. V., Parfenov, V. A., Voronina, S. Y., Simonov-Emelyanov, I. D., & Borisova, P. E. (2021). Influence of the Filler Particles’ Surface Morphology on the Polyurethane Matrix’s Structure Formation in the Composite. Polymers, 13(22), 3864. https://doi.org/10.3390/polym13223864