1. Introduction
Shape memory polymers (SMPs) are smart materials with many potential biomedical applications. SMPs can be prepared in a primary/original shape, deformed into a temporary shape upon exposure to an external stimulus, and stored in this temporary shape once the stimulus is removed. The external stimulus can be temperature, light, pH, electrical stimulus, or a magnetic field. Upon re-exposure to the stimulus, the shape memory effect can be triggered to recover the material back to its original shape.
Based on the application, biomaterials require varying degrees of biodegradability, tissue integration, cell and blood interactions, nutrient transfer, space-filling ability, and clinical functionality. Polyurethane SMPs have been extensively employed as biomaterial scaffolds in vascular applications, [
1] drug delivery, [
2] and tissue engineering due to their excellent tunable mechanical properties, [
3] high cytocompatibility and biocompatibility, [
4,
5] and the ability to tune degradation rates to match application requirements [
6,
7].
Biodegradation affects cell infiltration, vascular in-growth, and neo-tissue formation to allow successful integration of host tissue with biomaterials at the implant location. Biodegradation can occur via three major mechanisms: oxidation, hydrolysis, and enzymatic degradation [
8]. Some applications, such as degradable sutures, require a fast degradation rate, while others require biostable scaffolds that remain in the body over long time frames. Polyurethane SMPs present an ideal system for controlling degradation rates by selectively incorporating oxidatively, hydrolytically, and/or enzymatically responsive groups. The ability to control architecture changes with shape memory properties while tuning degradation profiles presents several potential benefits for healing, and previous research in this area is rich [
9,
10,
11].
Within the large field of polyurethane SMPs, a crosslinked, amorphous polyurethane SMP foam system has been used for vascular occlusion applications, such as aneurysm filling [
12,
13], peripheral vascular disease [
14], and hemorrhage control [
15]. These materials are highly tunable, with prior work focusing on altering shape memory profiles, [
16] pore structure [
17], and/or toughness [
18] and on incorporation of functional motifs to enable in vivo imaging [
19,
20] or infection control [
21]. In vitro degradation characterization of SMP foams showed that they are hydrolytically stable, but that they degrade via oxidation [
7]. Degradation was attributed to tertiary amines in the polyol crosslinkers that are used to form the polyurethane network. In a rabbit elastase aneurysm model, Herting et al. found that the materials underwent ~97% mass loss by 90 days using cross-sectional histological images [
22].
Based on these findings, several subsequent studies have focused on improving the biostability of this valuable biomaterial system. Hasan et al. replaced the tertiary amine-containing monomers in the SMP foams with glycerol and hexanetriol. These foams were highly stable, with <10% mass loss over 45 days in accelerated hydrolytic and oxidation medias (0.1 M NaOH and 50% H
2O
2, respectively) [
23]. However, their shape recovery profiles were significantly slower than the original SMP foams, with 100% volume recovery achieved after ~40 min in 50 °C water vs. full recovery in <10 min in 37 °C water in the control foams. This property would limit their ability to be actuated upon implantation at body temperature in future applications.
Weems et al. focused on improving the biostability of shape memory polyurethanes by incorporating isocyanurate-containing alcohols [
24]. This approach resulted in increased biostability and delayed degradation. While tested in an accelerated oxidative degradation media (20% H
2O
2 catalyzed by 0.1 M CoCl
2), SMP films had more than 80% mass remaining after 100 days and porous SMP foams had close to 75% mass remaining after 40 days. This is a promising approach to significantly increasing biostability; however, the eventual degradation byproduct of these polymers may contain small molecules like cyclic isocyanurates, whose cytocompatibility has not been determined. Additionally, materials with intermediate degradation rates may be required.
In a separate approach, Weems et al. achieved a reduced degradation rate by incorporating small molecule antioxidants into the foams to form an SMP composite [
25]. The microparticles were physically mixed within the polymer solution; thus, this approach could result in the antioxidant-loaded microparticles leaching out of the polymer system to alter the scaffold biostability over time. In most of the composite formulations, the antioxidant payloads were released during the initial cleaning procedure. The composite that did have a well-retained antioxidant content after washing underwent complete release of the antioxidant during the first three days of the degradation study, and thus did not significantly alter the long-term degradation profile.
While these SMP foams have been widely employed in embolic applications, none of the prior studies characterized blood interactions following modifications, which are highly dependent on material chemistry. Additionally, there may be benefits to more moderate increases in biostability or in altering the physical erosion profiles of SMP foams, such as in load-bearing applications where bulk device failure may be detrimental to healing. To that end, we synthesized polyurethane SMP foams that were modified with ether linkages using diethylene glycol (DEG) or triethylene glycol (TEG) to extend their biostability relative to control foams. We characterized the ability to tune the rate of degradation while maintaining other properties, such as pore size and volume recovery rates, the physical erosion profiles, and cell and blood interactions in the resulting ether-containing foams. In the long-term, these foams could provide an option for biomaterial implants with controlled degradation after implantation to maintain scaffold properties over longer time frames and to eliminate the need for implant removal.
2. Materials and Methods
Materials: Hexamethylene diisocyante (HDI), diethylene glycol (DEG), triethylene glycol (TEG), N,N,N′,N′-tetrakis-(2-hydroxypropyl)-ethylene diamine (HPED), triethanol amine (TEA), hydrogen peroxide (H2O2, Certified ACS, 30%), sodium hydroxide (NaOH), and ethanol (reagent alcohol) were purchased as used as received from Fisher Scientific (Waltham, MA, USA). All chemicals were purchased at reagent grade unless specified. Catalysts (T-131 and BL-22) and surfactant (EP-H-190) were used as received from Evonik Corporation (Essen, Germany).
Foam Synthesis: Polyurethane foams were synthesized in a two-part process by first preparing an isocyanate (NCO) pre-mix that contained 35 equivalents of hydroxyl groups provided by varying ratios of HPED, TEA, DEG, and TEG, and 100 equivalents of isocyanates. The pre-polymer was formed by crosslinking the NCO pre-mix at 50 °C for 48 h. Surfactant (EP-H-190) was added to the pre-mix after 48 h. The NCO pre-mix was allowed to cool down to room temperature while the hydroxyl pre-mix was made. The hydroxyl (OH) mix contained the remaining hydroxyls to balance the NCO groups, deionized (DI) water as a chemical blowing agent, and catalysts (T-131-tin based gelling catalyst) and BL-22-amine based blowing catalyst). The hydroxyl components were mixed at 3500 rpm for 30 s. The required amount of catalysts were added to the hydroxyl contents and mixed at 3500 rpm for 30 s. The final hydroxyl mix was added to the isocyanate pre-mix and mixed at 1800 rpm for 5 s and poured into a large mold to form a gas-blown foam in an isothermal oven maintained at 50 °C. All mixing was carried out in a high-speed mixer (Flacktek, Landrum, SC, USA). The overall hydroxyl to isocyanate ratio in the foam was maintained at 1.04 to ensure the complete reaction of isocyanates during the synthesis. Synthesized foam compositions are shown in
Table 1.
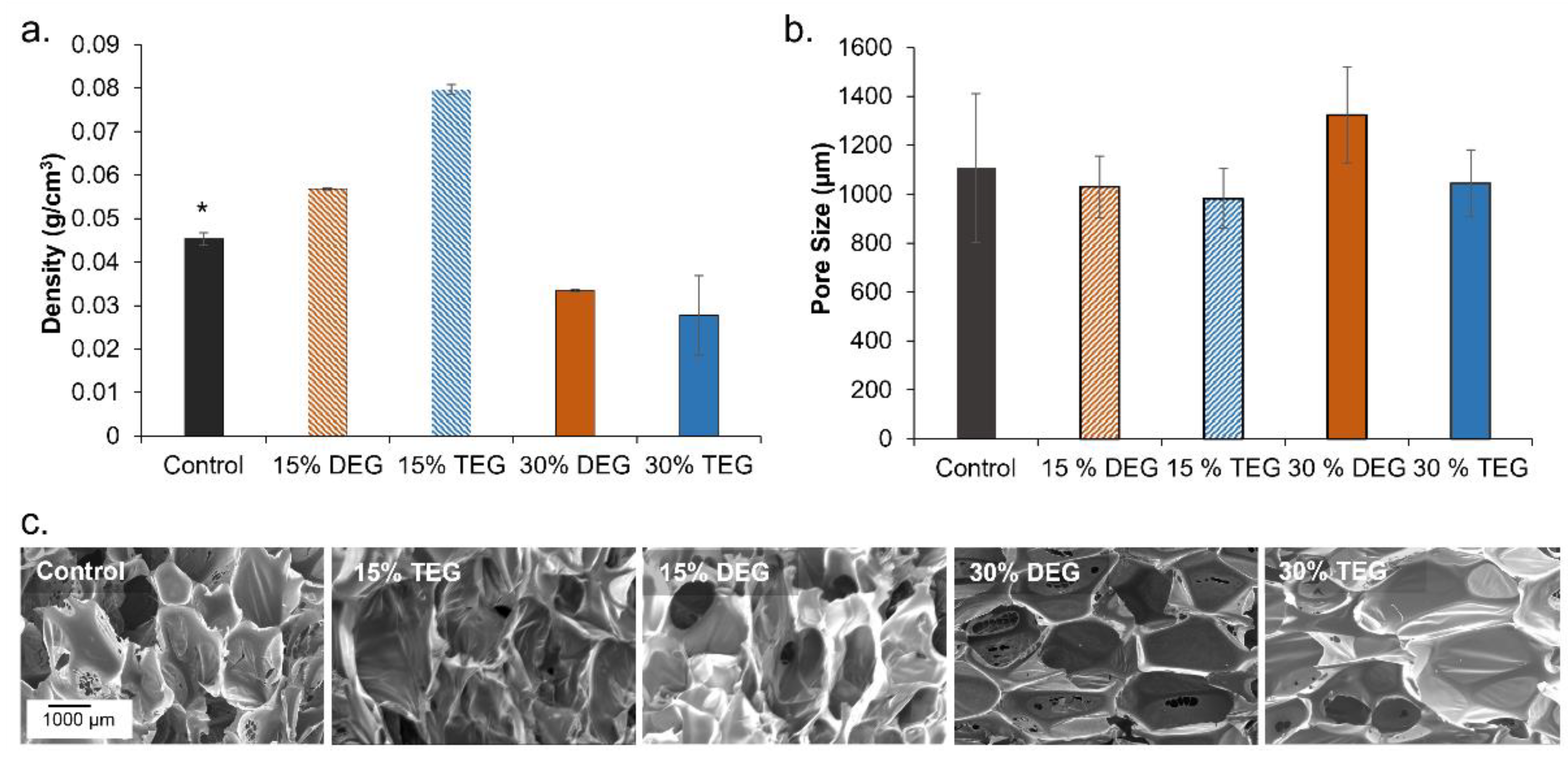
Foam Pore Analysis: Foam slices (n = 3, ~1 cm2) were cut parallel and perpendicular to the foam rise direction. Each piece was coated with gold using a high vacuum sputter coater (Denton, Moorestown, NJ, USA) at 100 mTorr for 45 s to form a consistent and a stable coating. Pore structures were characterized via a JEOL JSM 5600 scanning electron microscope (SEM; JEOL USA, Peabody, MA, USA) at 35× magnification under 10 kV high vacuum. The micrographs were analyzed via ImageJ (National Institutes of Health, Bethesda, MD, USA) to quantify pore diameters.
Density: Cube samples (n = 3, ~1 cm3) were cut via a hot wire cutter, (Proxxon Thermocut 115/E, Hickory, NC, USA). Dimensions and weights were measured to obtain foam densities.
Mechanical Testing: Dogbone punches were cut from each foam (n = 3) according to the ASTM D638 (scaled down by a factor of 4; length: 6.25 mm, width: ~1.5 mm). The thickness of each piece was measured prior to testing. Samples were tested in both dry and wet conditions. To test the samples in wet conditions, they were placed in DI water at 50 °C for 5 min and patted dry prior to analysis. Based on thermal and swelling analysis, this time frame/temperature provided equilibrium water absorption to ensure sample wetting. Samples (n = 3) were stretched in a tensile tester via a 24 N load cell at a rate of 2 mm/min until failure to measure elastic modulus, elongation at break, and ultimate tensile strength.
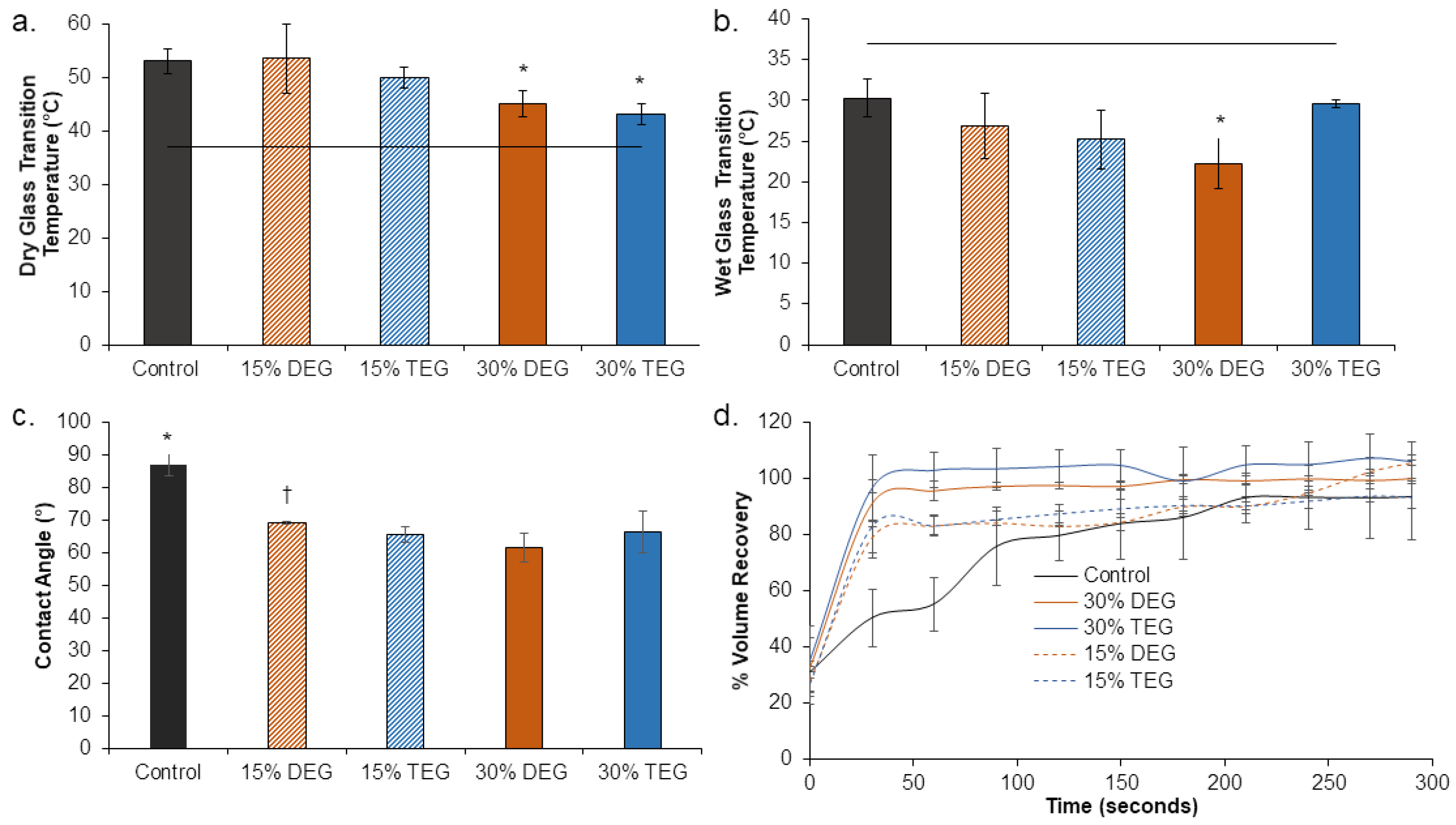
Thermal analysis: Glass transition temperature (Tg) was measured for each sample (n = 3, 3–5 mg) using a Q200 differential scanning calorimeter (DSC, TA instruments, New Castle, DE, USA) in both dry and wet (plasticized) conditions. Samples were placed in t-zero aluminum pans, equilibrated at −40 °C, heated to 120 °C at 10 °C/min, kept isothermally for 2 min, cooled to −40 °C at 10 °C/min, and heated back to 120 °C at 10 °C/min. Dry Tg was measured was measured as the half-height transition temperature during the second heating cycle. To measure wet Tg, samples were plasticized by placing in DI water at 50 °C for 10 min, pressed dry, and placed in t-zero aluminum pans with hermetic lids. A pin hole was pierced on the hermetic lid to allow water to escape during the heating cycle. Samples were equilibrated at −60 °C and heated to 80 °C at 10 °C/min. Wet Tg was measured as the half-height transition temperature during the single heating cycle.
Shape Memory Behavior: Volume expansion was used to quantify shape memory behavior. Cylindrical foam samples (1 cm long, 8 mm diameter) were cut, cleaned in DI water and 70% ethanol, and dried under vacuum for 24 h prior to testing. Each sample was heated to 100 °C for 10 min to allow softening, and the diameter was recorded using digital calipers prior to manual crimping in a radial compression crimper (Blockwise Engineering, Tempe, AZ, USA). After cooling to room temperature, the final crimped sample diameter and length were recorded, samples were placed in scintillation vial in a desiccator for 24 h and fixed on a 300 µm Nitinol wire to allow for complete shape setting and relaxation to occur. After 24 h, the foam’s initial diameter and length were measured, and samples were placed in a DI water bath set at 37 °C and allowed to expand for 5 min. Images were captured every 3 s to observe changes in diameter over time (t) and generate a volume recovery profile. Images were analyzed using ImageJ and volume recovery was measured as:
Change in volume vs. time was plotted over the expansion time frame.
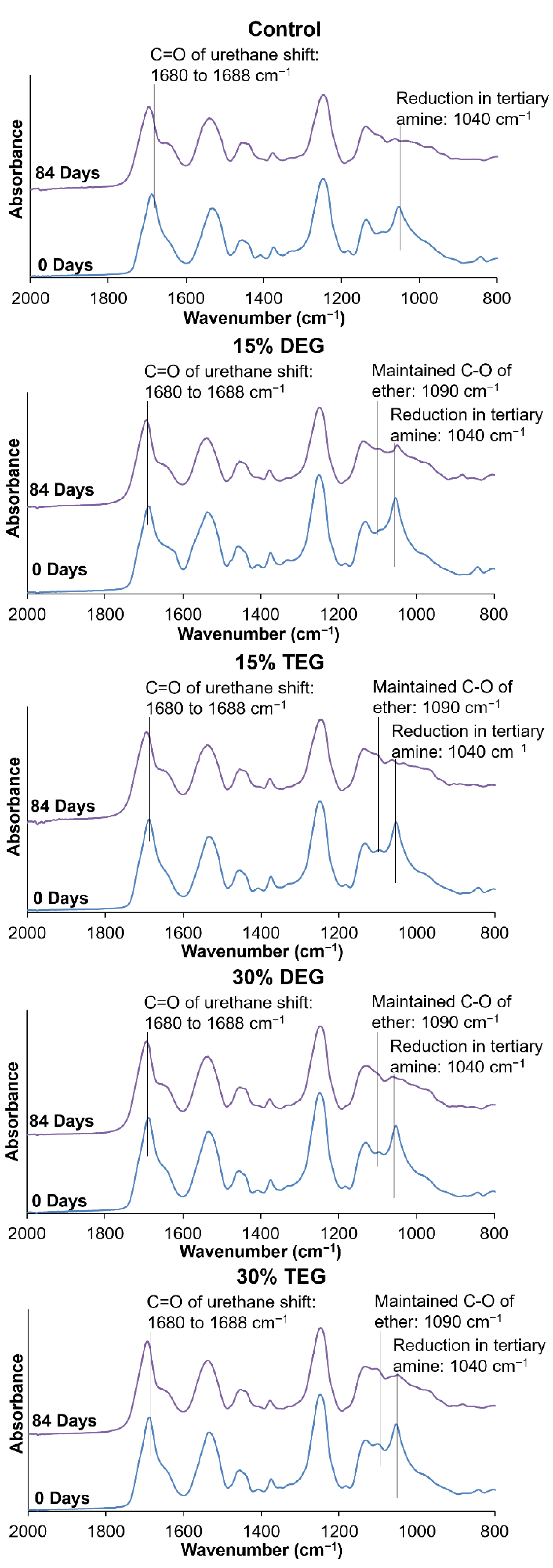
Spectroscopic Analysis: Surface chemistry was characterized on thin slices of cleaned foam pieces using a Nicolet i70 Attenuate total reflectance (ATR)-Fourier transform infrared (FTIR) Spectrometer (Fisher Scientific, Waltham, MA, USA) at 0.8 cm−1 resolution using OMNIC software (Fisher Scientific, Waltham, MA, USA). Incorporation of ethers into polyurethane foams was confirmed by the presence of peaks corresponding to the C-O of the ether group at ~1090 and ~1050 cm−1 and the carbonyl of urethane at ~1688 cm−1.
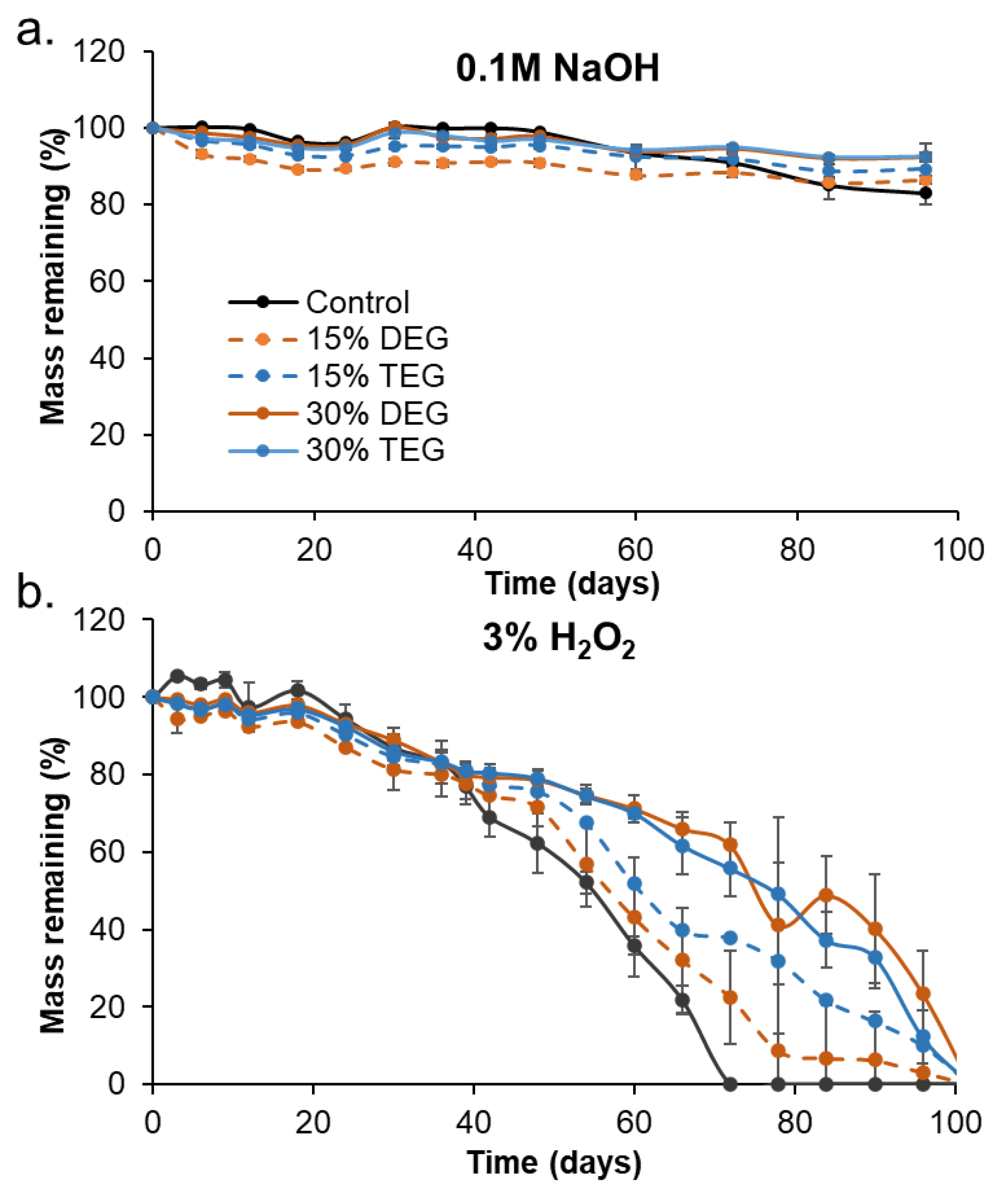
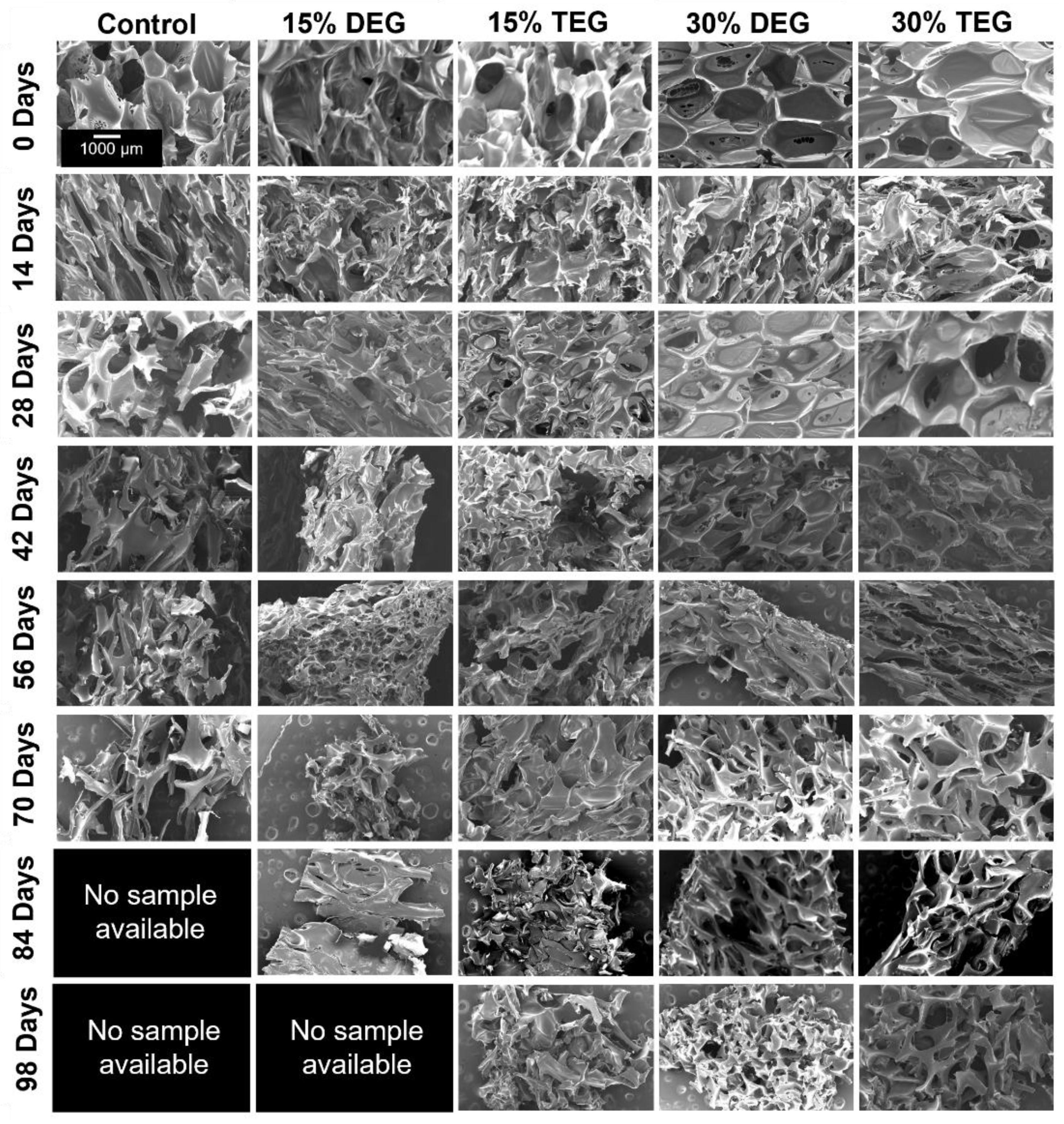
Degradation Analysis: Cylindrical foams (n = 8, 8 mm diameter, 1 cm height) were washed and dried, and initial masses were obtained using a gravimetric scale. Samples were placed in 3% H2O2 (real-time oxidative degradation media) or in 0.1 M NaOH (accelerated hydrolytic degradation media) at 37 °C with regular media changes. At selected time points, samples were washed with ethanol and dried under vacuum for 24 h. After drying, samples were imaged using a camera, and masses were measured (n = 5). A thin slice was cut from a sacrificial set of foams (n = 3) and used to measure pore morphology (SEM), Tg (DSC), and surface chemistry (FTIR) as described above.
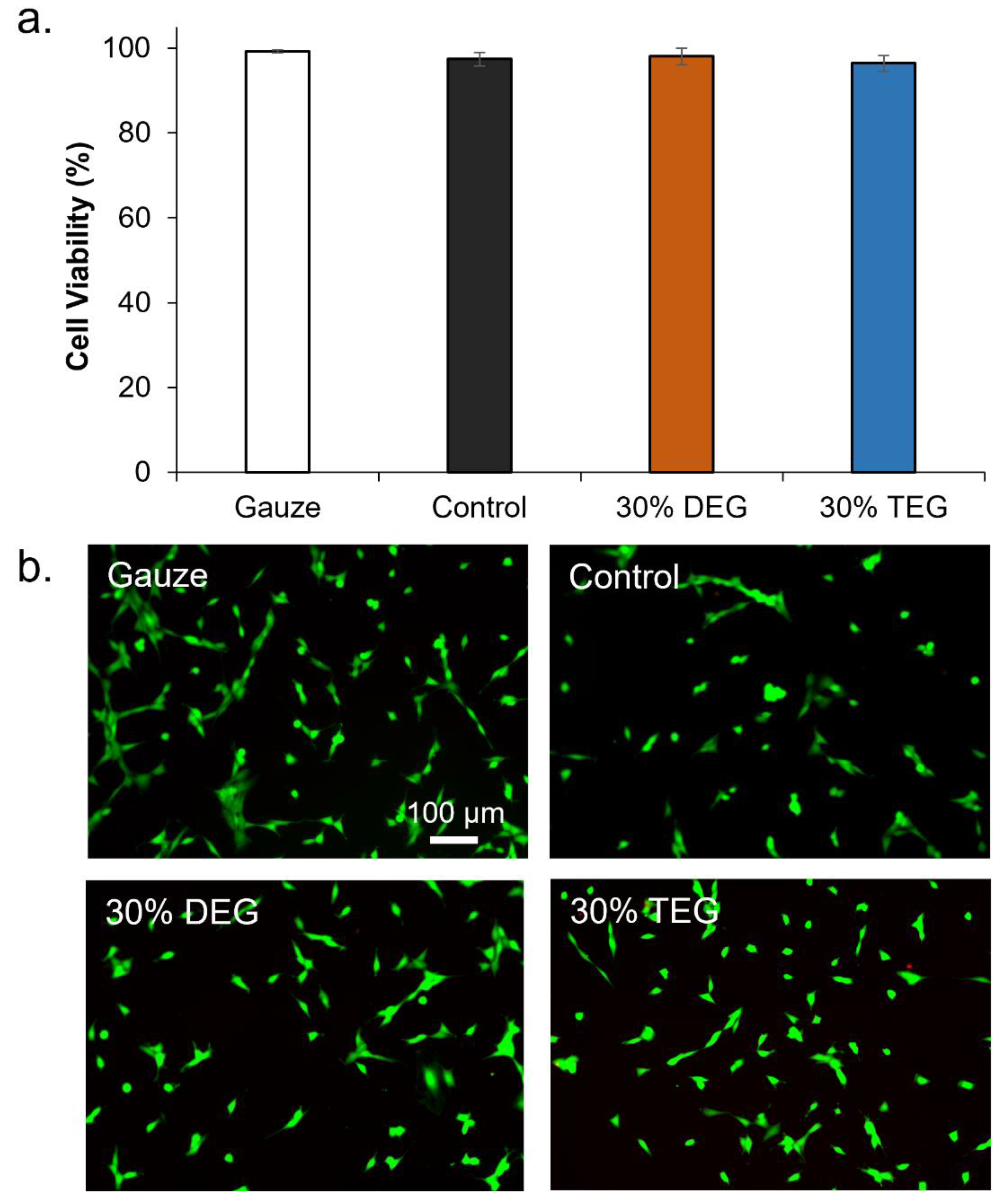
Cytocompatibility: Sample cytocompatibility was tested using 3T3 Swiss mouse fibroblasts (ATCC-CCL92; ATCC, Manassas, VA, USA). Cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM, high glucose GlutaMAX), supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S, Gibco) at 37 °C/5% CO
2. Cells from passage 11 were used after three days of culture. Cells were seeded onto a 24-well tissue culture polystyrene plate at 10,000 cells/well and incubated for 24 h at 37 °C/5% CO
2 for 24 h. Samples were cleaned using water, 20% Contrad solution, and isopropyl alcohol, and then soaked in 1× PBS overnight prior to testing to leach out any alcohol. Samples (n = 3) were placed in each well along with positive controls (media-only with cells, n = 3), and negative controls (media-only with no cells). Samples were incubated with cells, and viability was assessed after 24 h using a Live/Dead assay (ThermoFisher Scientific, Waltham, MA, USA). Briefly, cells were stained with green fluorescent calcein-AM (live cells) and red-fluorescent ethidium homodimer-1 (dead cells) for 15 min at room temperature while protecting from light. Cells were imaged using an inverted microscope (Leica, DMI6000) at 10× magnification to determine the number of live (green) and dead (red) cells. Three images were captured for each sample. Cell viability of each sample (x) was measured as:
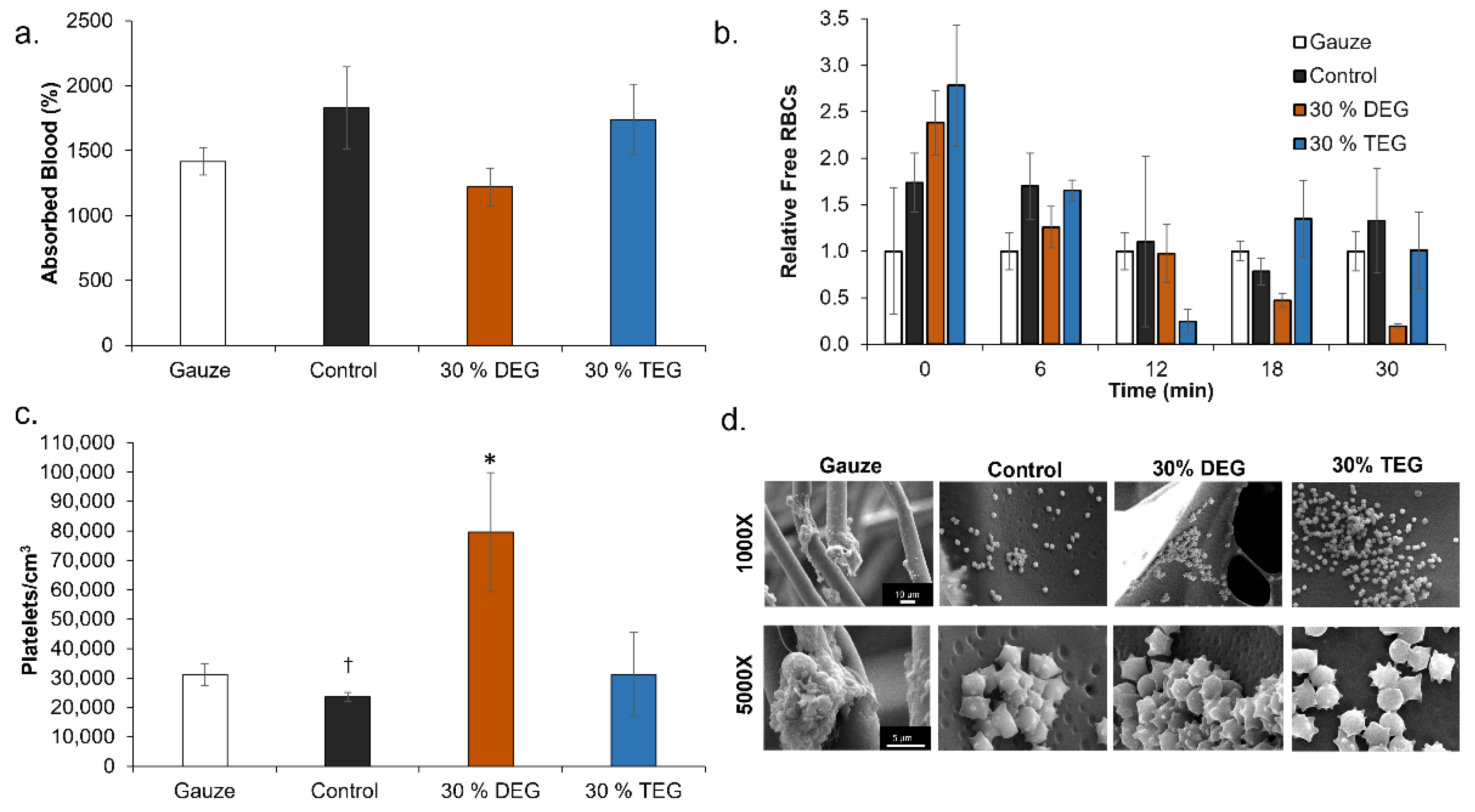
Blood Interactions: Porcine blood (Lampire Biological Laboratories, Pipersville, PA, USA) anticoagulated with Na-Citrate upon collection was stored at 4 °C for up to 3 weeks from the bleed date, according to supplier guidelines. Control, 30% DEG, and 30% TEG foams were washed and dried prior to characterization in all studies. QuikClot Combat Gauze was included as a clinical control. Blood absorption was analyzed by weighing dry samples (n = 3; ~50 mg) and incubating them in blood at 37 °C. Samples were weighed at 24 h, and blood absorption was calculated as:
where
Wb is the mass of the sample in blood and
Wd is the dry mass.
Platelet attachment was measured via LDH cytotoxicity assay kit (Cayman Chemical, Ann Arbor, MI, USA). Platelet-rich plasma (PRP) was collected by centrifuging whole blood at 3000 rpm for 15 min to obtain a standard curve. PRP was diluted with PBS to obtain multiple concentrations (100, 50, 25, 12.5, and 6%) to generate a standard. Hemocytometer counts were acquired at each PRP concentration (n = 4) to quantify the standard values. SMP foams (n = 4) were cut to equal surface area and placed in individual wells in a 24-well plate. Gauze was used as a clinical control. One milliliter of blood was added to each well and the soaked samples were incubated at 37 °C for 30 min. PBS was used to wash out any unattached platelets. Samples were then added to wells on a separate plate containing 1 mL PBS and 100 µL of 10% Triton X-100 to lyse unattached platelets. Supernatant (100 µL) from each well was added to wells on a separate 96 well plate along with 100 µL of LDH reaction solution. The mixture was incubated at 37 °C for 30 min on an orbital shaker. Microplate reader was then used to obtain absorbance values from each sample at 490 nm.
Samples that were washed with PBS to remove unattached platelets were then imaged via SEM to observe activity states and platelet activation. Prior to imaging, samples were soaked in 2% glutaraldehyde solution (Electron Microscopy Sciences, Hartfield, PA, USA) to fix them and later dehydrated. To achieve complete dehydration, samples were soaked in a series of ethanol concentrations: (1) 30 min in 50% ethanol, (2) 30 min in 70% ethanol, (3) 30 min in 95% ethanol and finally (4) 30 min in 95% ethanol. Post dehydration samples were dried overnight in a vacuum oven at 50 °C and −30 inches Hg. SEM analysis was performed using JEOL NeoScope JSM-5600 (JEOL USA, Peabody, MA, USA) operated at 10 kV. Images were captured at regions of interest at 1000× and 5000× magnification. These images were later analyzed via ImageJ assess platelet aggregations and activation (morphology change).
The time required for coagulation was measured for each sample (n = 4) by placing them in 1.5 mL microcentrifuge tubes and exposing them to blood. One tube was maintained as a negative control with no sample. Samples were weighed and cut to have the same surface area throughout. Blood was brought to room temperature and the Na-citrate anticoagulant was reversed by adding 1 M CaCl2 solution to obtain a net 0.01 M CaCl2 solution. Then, 50 µL of this blood was added to each sample tube. The clotting process was stopped at each time point (every 6 min over 30 min) by adding 1 mL DI water to each tube to lyse the free red blood cells. These tubes were centrifuged at 2300 rpm for 15 min, inverted, and images were captured using a digital camera (AKASO V50 Pro Native, AKASO, Frederick, MD, USA). The relative amount of hemoglobin released at each time point was determined by adding 200 µL of the lysate from each tube to a 96-well plate and measuring the absorbance at 540 nm using a BioTek Synergy 2 Multimode Microplate Reader (Winooski, VT, USA).
Statistical Analysis: Measurements are presented as mean ± standard deviations. Student’s t-tests were performed to determine differences between ether foams and controls. Statistical significance was taken as p < 0.05.
4. Discussion
Overall, it was observed that adding ether-containing monomers, DEG and TEG, resulted in increased pore interconnectivity and reduction in T
g compared to control foams. We hypothesize that the increased hydrophilicity of DEG and TEG enabled increased interactions between the monomers in the pre-polymer and the chemical blowing agent (water) and/or the surfactant, which resulted in pore opening in the 30% DEG and TEG foams. This phenomenon could be advantageous in applications that require increased interconnectivity without relying on physical or mechanical modifications like mechanical reticulation, [
17] plasma treatment, [
27], and/or the addition of physical blowing agents.
TEG-containing foams had slightly lower Tg’s compared to corollary DEG-containing foams and increasing TEG and DEG content induced further decreases in Tg. Reduction in foam Tg is attributed to increased hydrophilicity and flexibility of DEG and TEG, which corresponds with the contact angle measurements. Additionally, replacing the tri-functional TEA with difunctional DEG or TEG theoretically reduces foam crosslink density, which would result in lower Tg. However, all foams had dry Tg well above room temperature, which would enable their stable storage in the secondary shape. Exposure to water at 37 °C results in a reduction in Tg due to plasticization by water molecules penetrating the inner structure of the foams. This reduced Tg aids in rapid volume recovery once implanted in the body and exposed to water in body temperature blood.
The factors that determine the volume recovery of foams are wet Tg, pore size, and hydrophilicity. Higher hydrophilicity (lower contact angle) allows for easier water absorption that corresponds to faster plasticization of foams, which is also indicated by a lower wet Tg. Larger pore size can also increase water penetration speed into foams, further accelerating volume recovery. Compared to control foams, DEG and TEG foams have a higher hydrophilicity (lower contact angle) due to the addition of hydrophilic ether linkages. Among the ether-containing foams, 30% DEG and 30% TEG foams have increased hydrophilicity compared to 15% DEG and 15% TEG foams and have a correspondingly faster volume recovery expected. This increased volume recovery may be valuable in rapidly filling wounds during implantation.
The increased elongation at break and decreased stiffness of the ether-containing foams were expected due to the overall decreases in crosslink density and increased chain flexibility of ether linkages. The penetration of water molecules into the polymer network and interruption of hydrogen bonds allows the polymer chains to move more freely, as indicated by the overall decreased modulus and increased elongation at break of all samples in the wet conditions compared to foams tested in dry conditions. The dry measurements are important for considering material handling prior to implantation, and all materials are mechanically robust and easy to handle in the dry state. The wet measurements provide information about the material properties after implantation, which is important for matching native tissue properties. Again, all materials are mechanically within the range of soft tissues, and the differences between the ether foams and controls are reduced in the wet state [
28]. In future work, the ether foams could be modified with stiffer diisocyanate species to increase the modulus if needed [
16].
When incubated in an accelerated hydrolytic degradation medium containing 0.1 M NaOH, all samples remained stable, with no significant mass loss due to the lack of hydrolytically labile linkages. This result agrees with previous work on this material system that consistently shows high hydrolytic stability. [
7,
29] In oxidative degradation medium containing 3% H
2O
2, control foams physically broke apart after ~42 days, while the other formulations maintained their geometry for longer times throughout the degradation time frame. These physical changes were accompanied by an increase in the mass-loss rate in control foams. The breaking apart of control foams may be attributed to their relatively high brittleness, evidenced by the lowest elongation at break (0.17 ± 0.04 mm/mm), compared to the other formulations. During the degradation study, foams are repeatedly subjected to minor mechanical forces during the weekly washing and drying steps. Since the control foams are more brittle, they may break apart more easily and be more susceptible to bulk erosion, despite maintaining T
g values throughout degradation. The ether-containing foams were more flexible and less susceptible to these stresses, as demonstrated by their increased overall physical integrity throughout degradation, which translates to slower and more consistent degradation rates. The ether linkages appear to remain intact during the degradation process, as evidenced in FTIR spectra. This stability of the ether linkages could potentially contribute to their increased stability and more consistent degradation profiles.
While the overall changes in degradation rates were not hugely different, the introduction of ether linkages increased the oxidative stability by ~40% (an increase from 72 days for control to 100+ days for ether foams). Additionally, the observed surface erosion and maintenance of pore structure over longer time frames may be beneficial for graded load transfer during new tissue formation as the SMP foams degrade. Finally, the ability to tune degradation independently of thermal and shape memory properties enables easy transition to ether-containing foams to increase degradation rates without altering storage or implantation considerations.
While future studies will require more in-depth analysis of biocompatibility after implantation and cytocompatibility of degradation byproducts, the addition of TEG and DEG does not affect the cytocompatibility of SMP foams. In terms of blood absorption, the increased absorption by TEG foams compared to DEG foams is attributed to increased hydrophilicity, which increases fluid uptake. The increased absorption by control foams may be attributed to their closed pore structure, which increases blood retention compared to open pore ether foams.
Various surface characteristics, like surface charge, relative hydrophilicity, and surface roughness, can impact protein absorption and subsequent blood and/or cell interactions with biomaterials. Thus, blood interactions must be considered when making chemical changes in any biomaterial system, particularly for embolic applications. All samples had complete clotting within 12 min as seen by a reduction in free RBCs. The highest clotting at later time points (>18 min) was observed on 30% DEG foams, which corresponds with the higher platelet attachment observed on these foams, both in the quantified LDH assay and the qualitative SEM imaging. The 30% TEG foams had similar platelet attachment values to clinical gauze control, and control foams showed the lowest number of attached platelets. When visualized using SEM, gauze promoted thrombus formation within the testing time frame, while all SMP foams had aggregated and activated platelets with similar trends observed in relative platelet numbers on each surface. This result shows that incorporating ether linkages into the SMP foams enhanced platelet attachment and activation, which may translate to increased efficacy of these materials in embolic applications and provides a new tool for increasing clotting in SMP foams. These results are analyzed in a static model. Going further, we will focus on analyzing clotting capabilities using a dynamic in vitro hemorrhage model where blood is allowed to flow through the foams [
30].
These smart biomaterials with increased biostability and excellent biocompatibility have a wide application in multiple tissue engineering applications. One such application involves the use as a temporary embolic device in minimally invasive medical applications that may require removal after a certain time point. The removal process can be avoided using these biomaterials.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}