
Dually Crosslinked Polymer Networks Incorporating Dynamic Covalent Bonds

Abstract
:1. Introduction
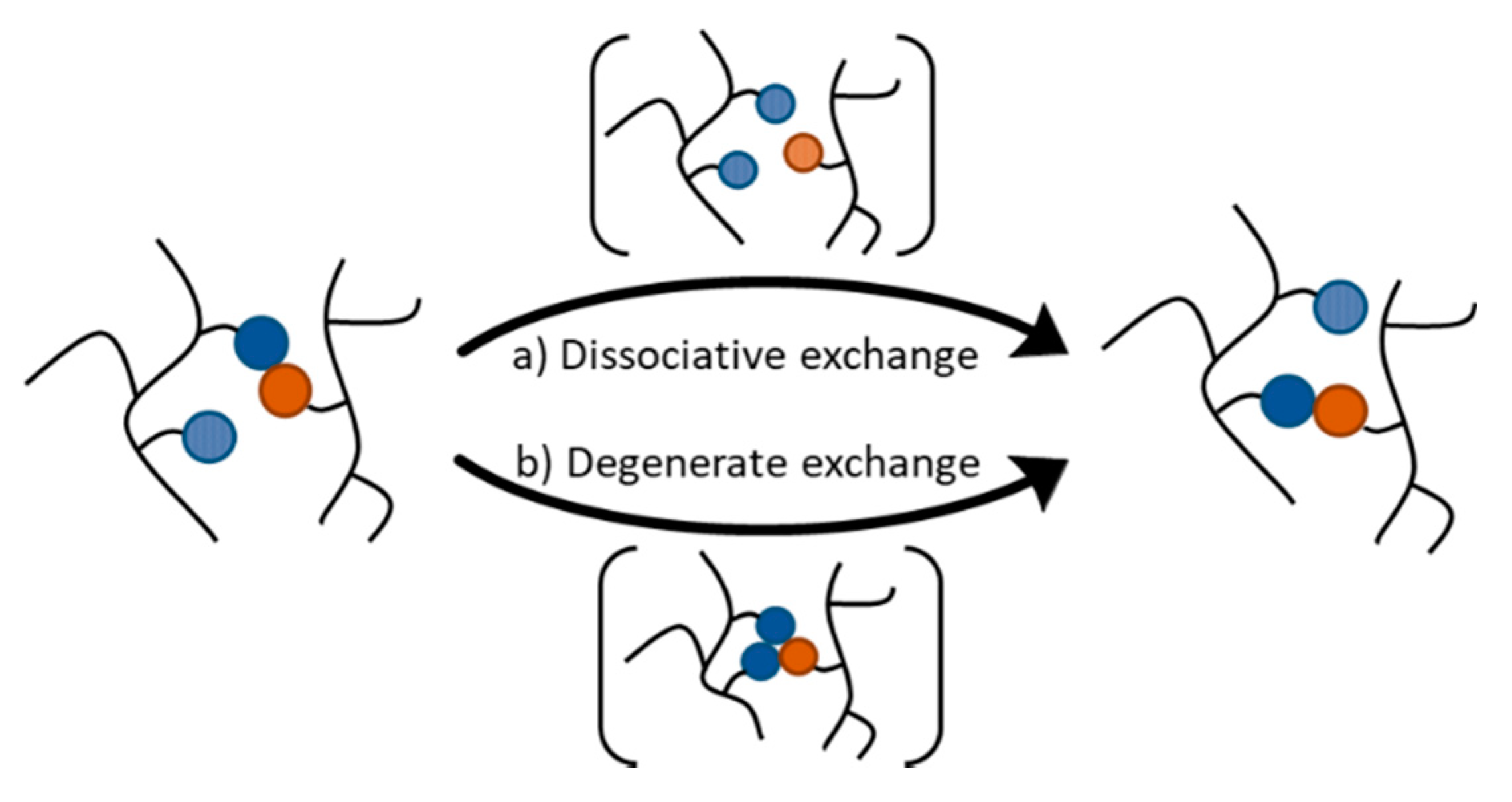
2. Dynamic Covalent Bonds in Polymer Networks
3. Origin of Dual Dynamic Networks: Supramolecular Networks
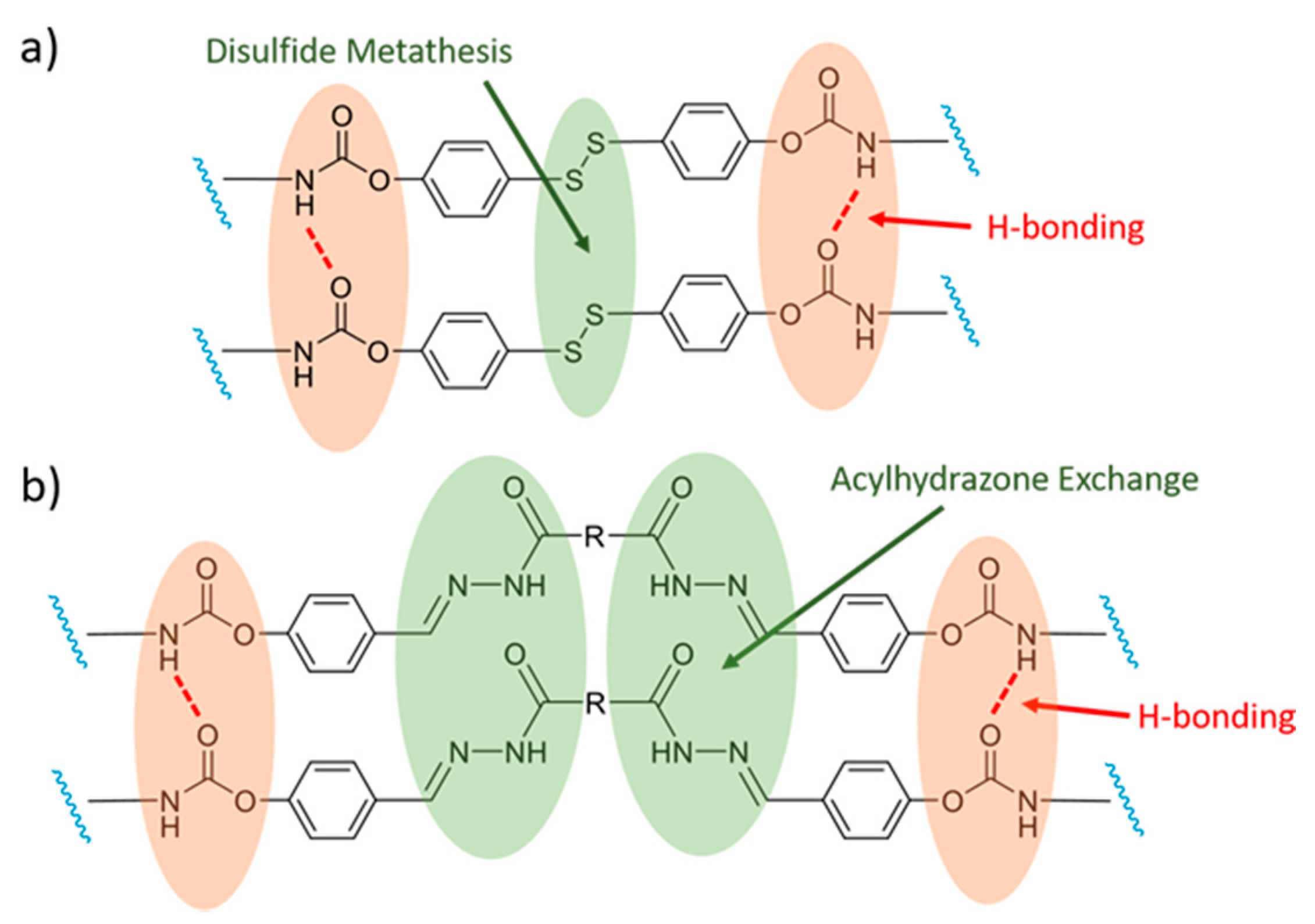
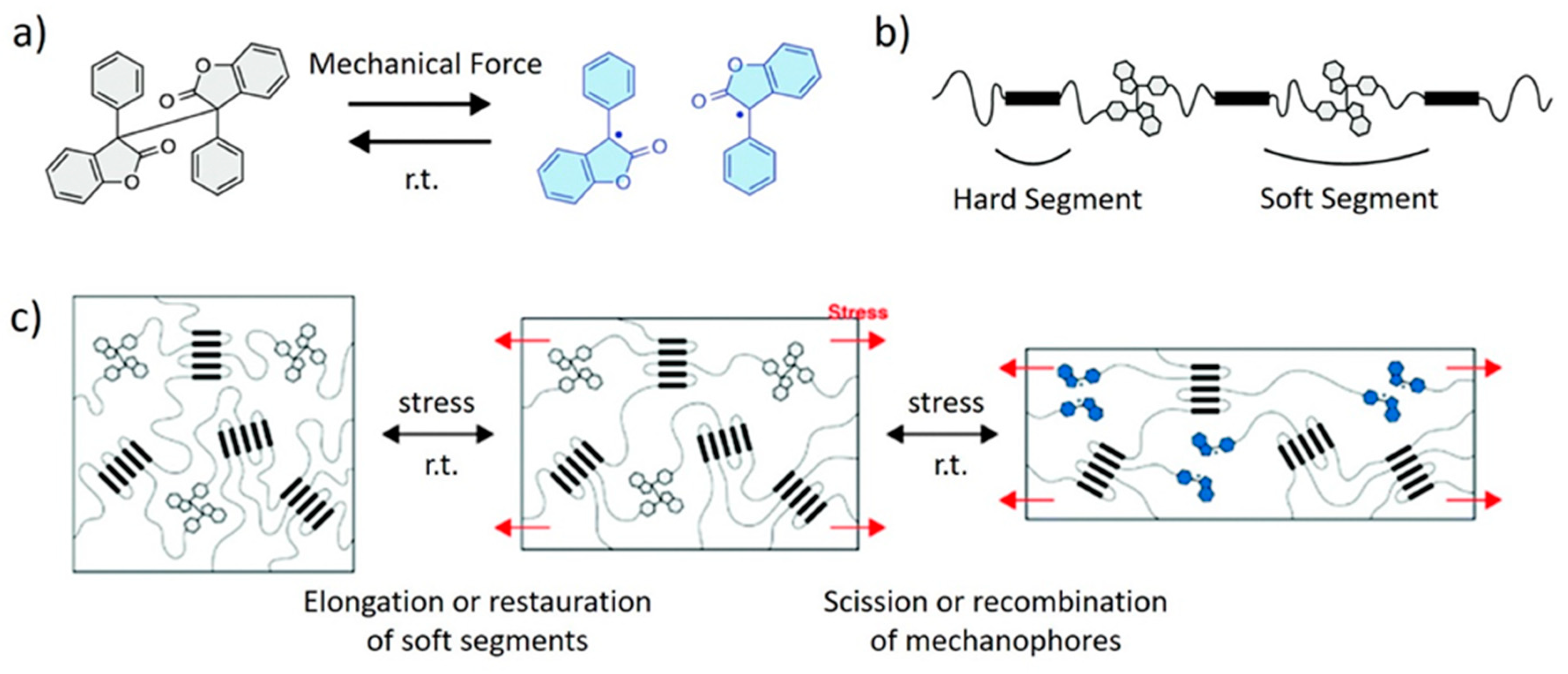
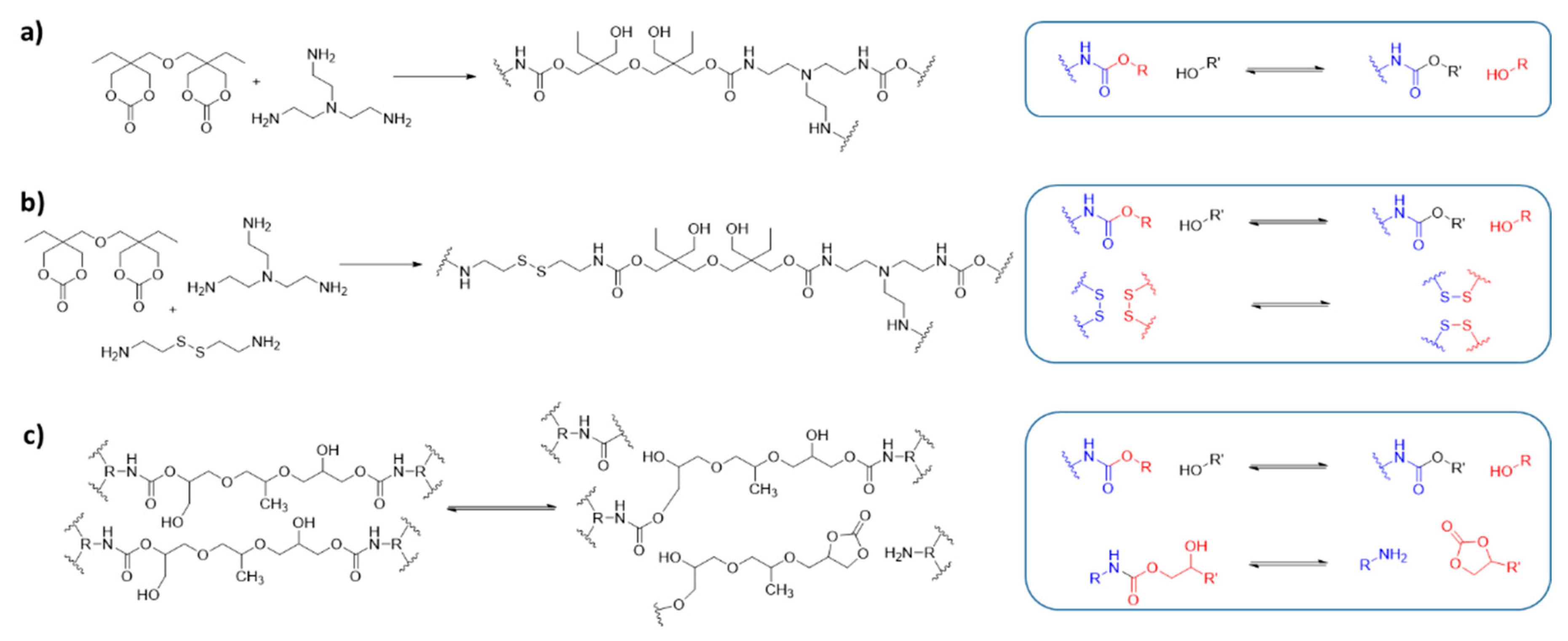
4. Polyurethane Elastomers
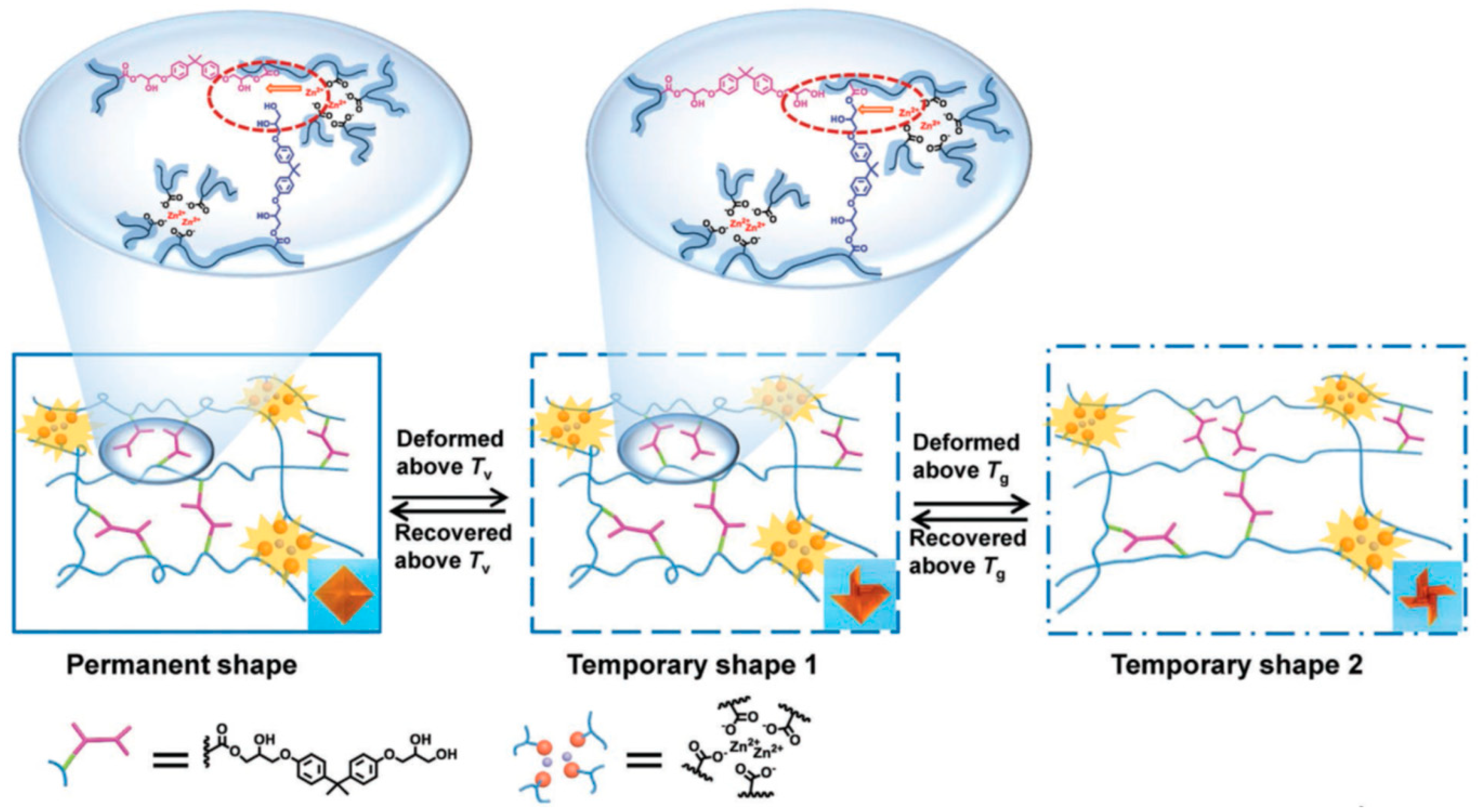
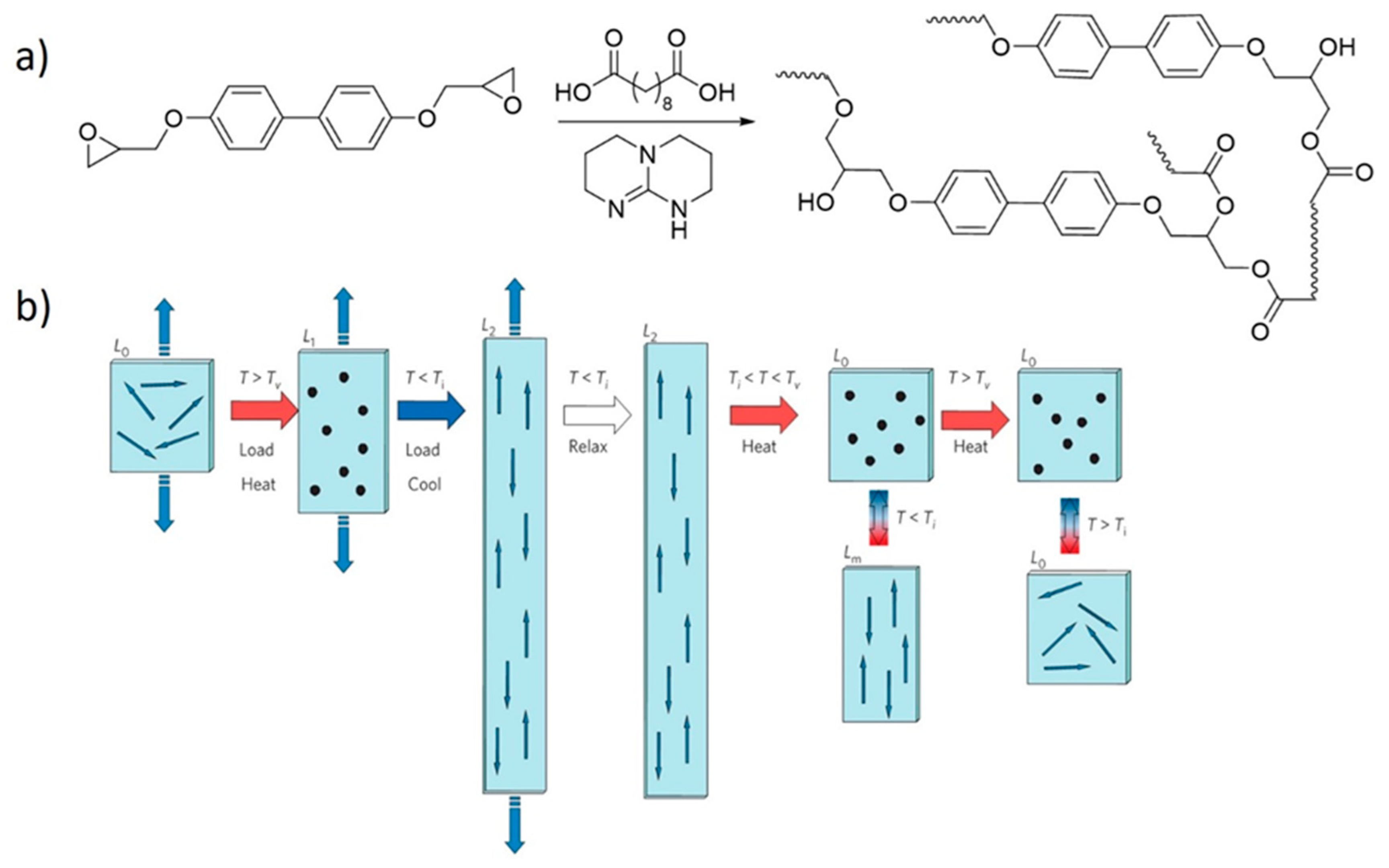
5. Shape Memory Polymers
6. Dual Dynamics in Vitrimers and Related Covalent Adaptable Networks
6.1. Vitrimers and CANs with Two Covalent Dynamic Crosslinking Strategies
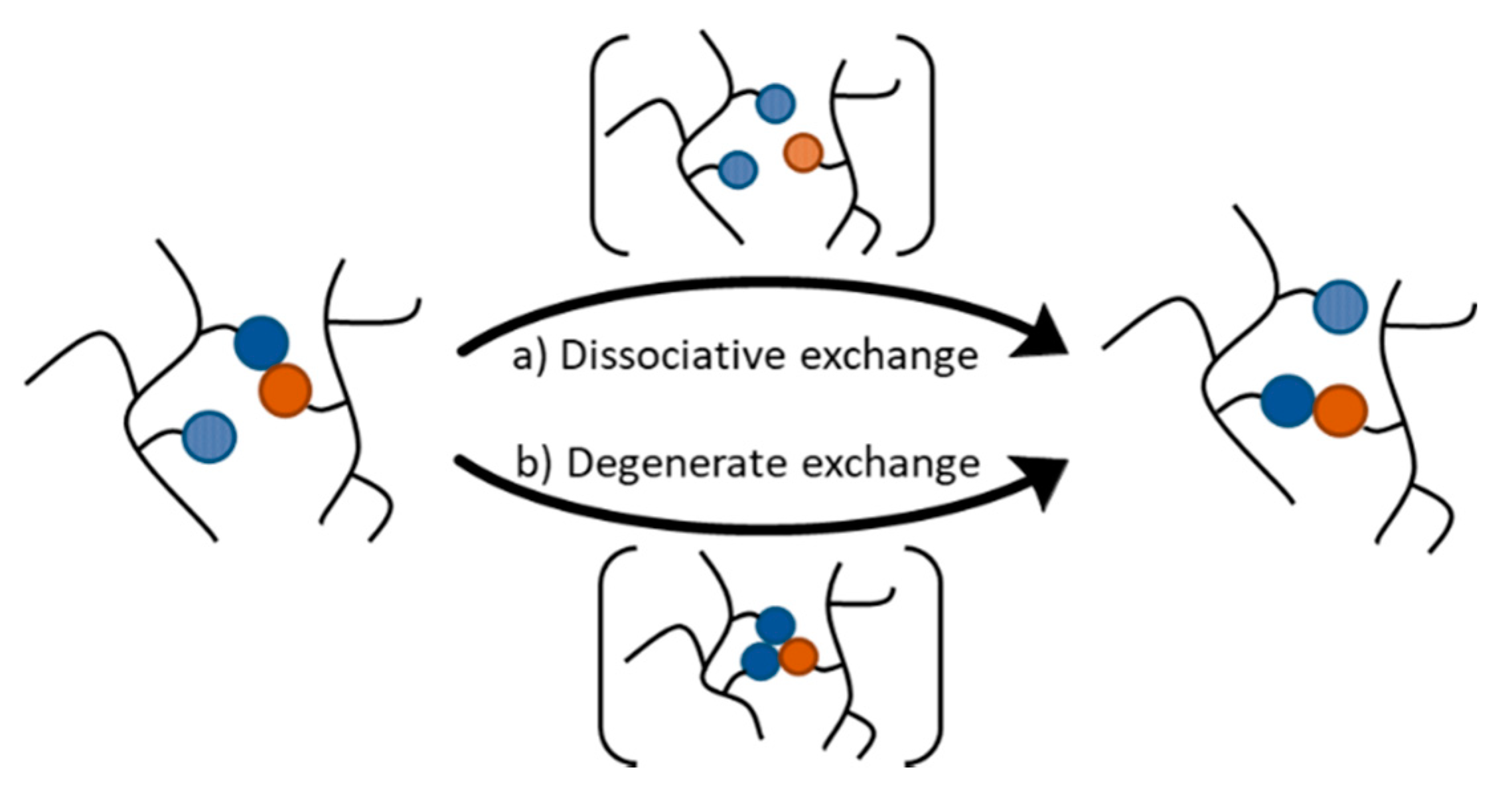
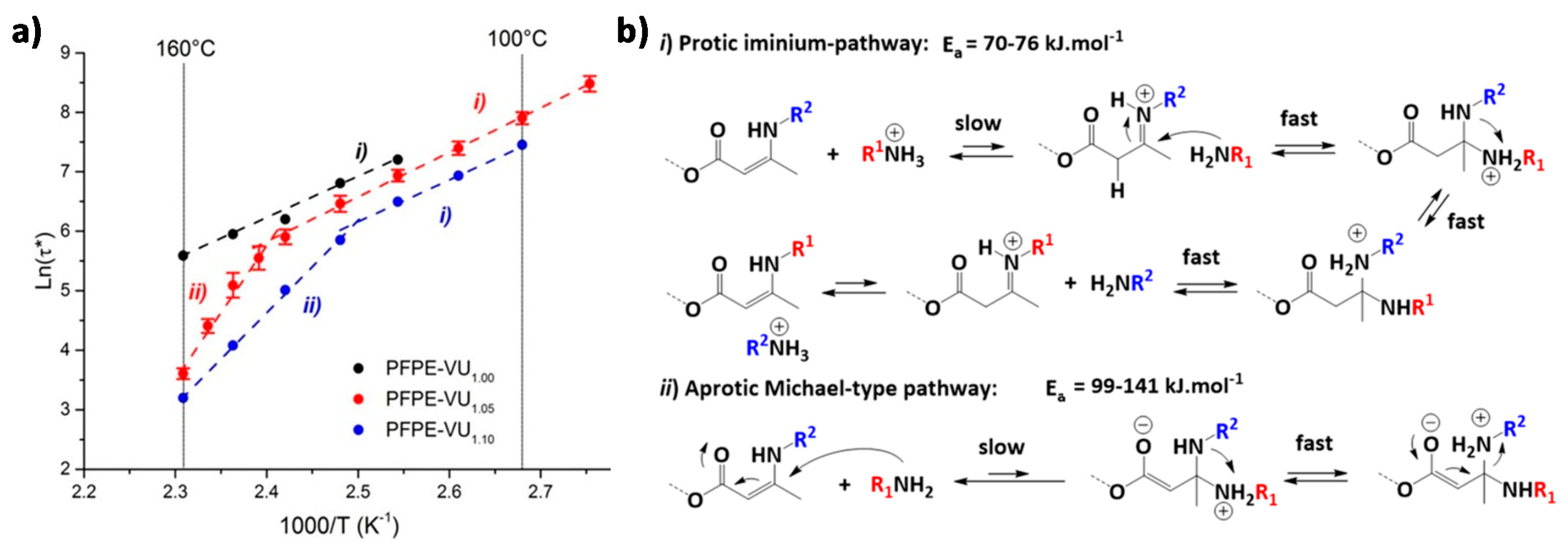
6.2. Vitrimers and CANs with One DCB That Follows Two Different Exchange Mechanisms
6.3. Reinforcement by Supramolecular Interactions
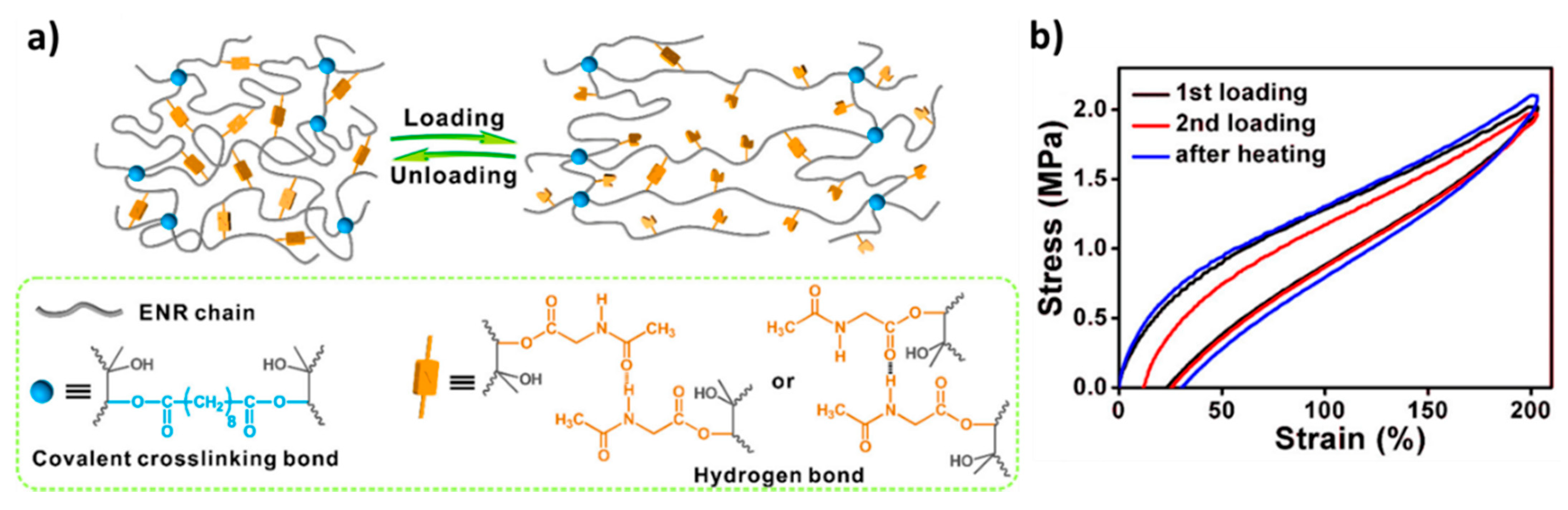
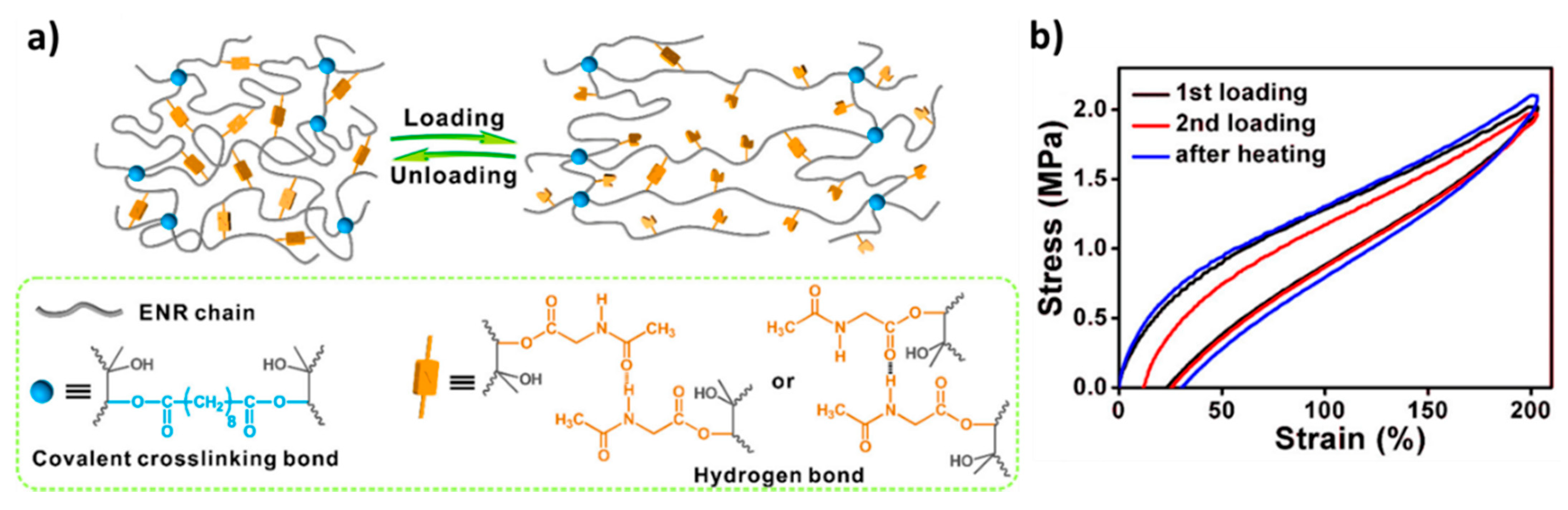
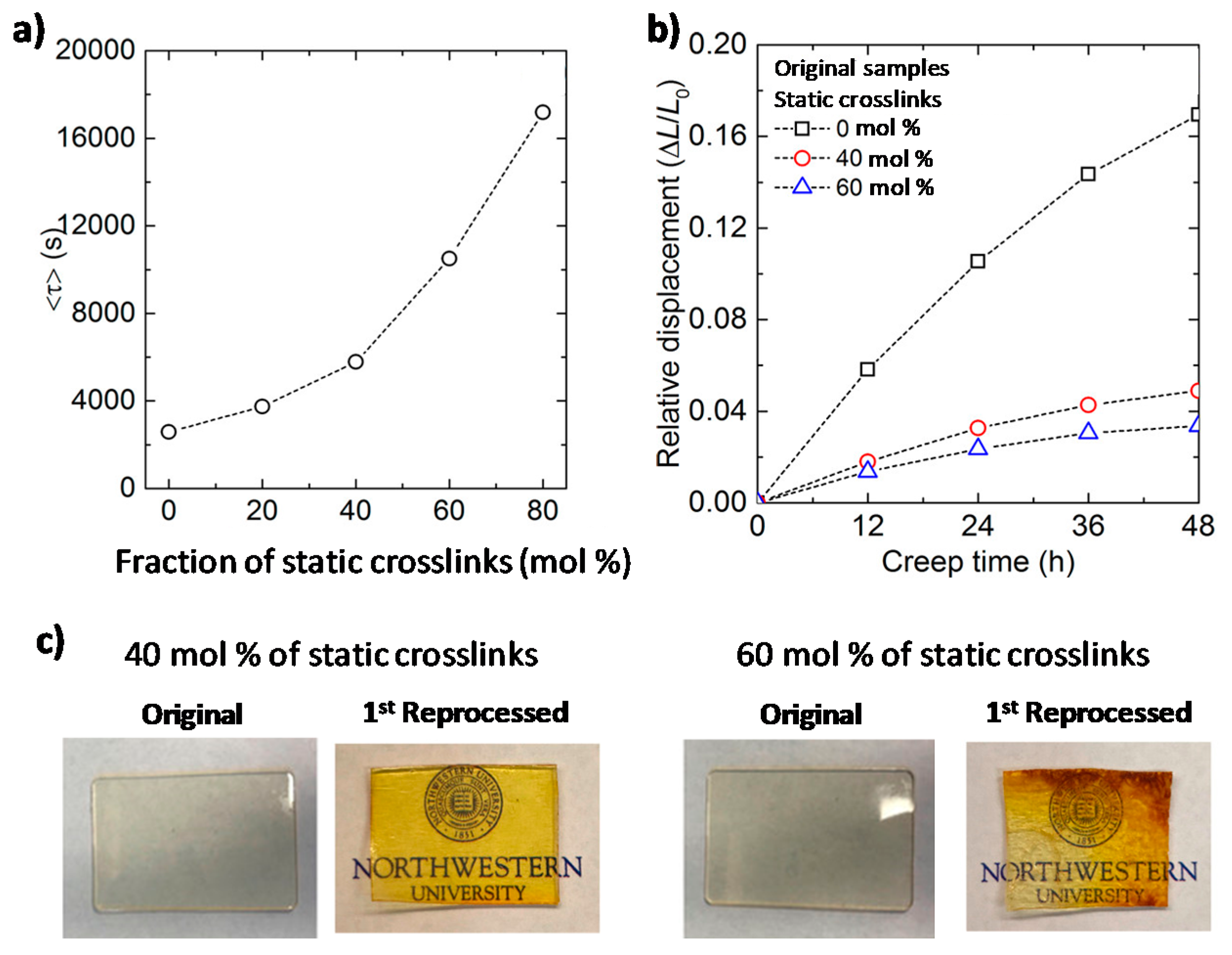
6.4. Reinforcement by Static Covalent Bonds
7. Hydrogels
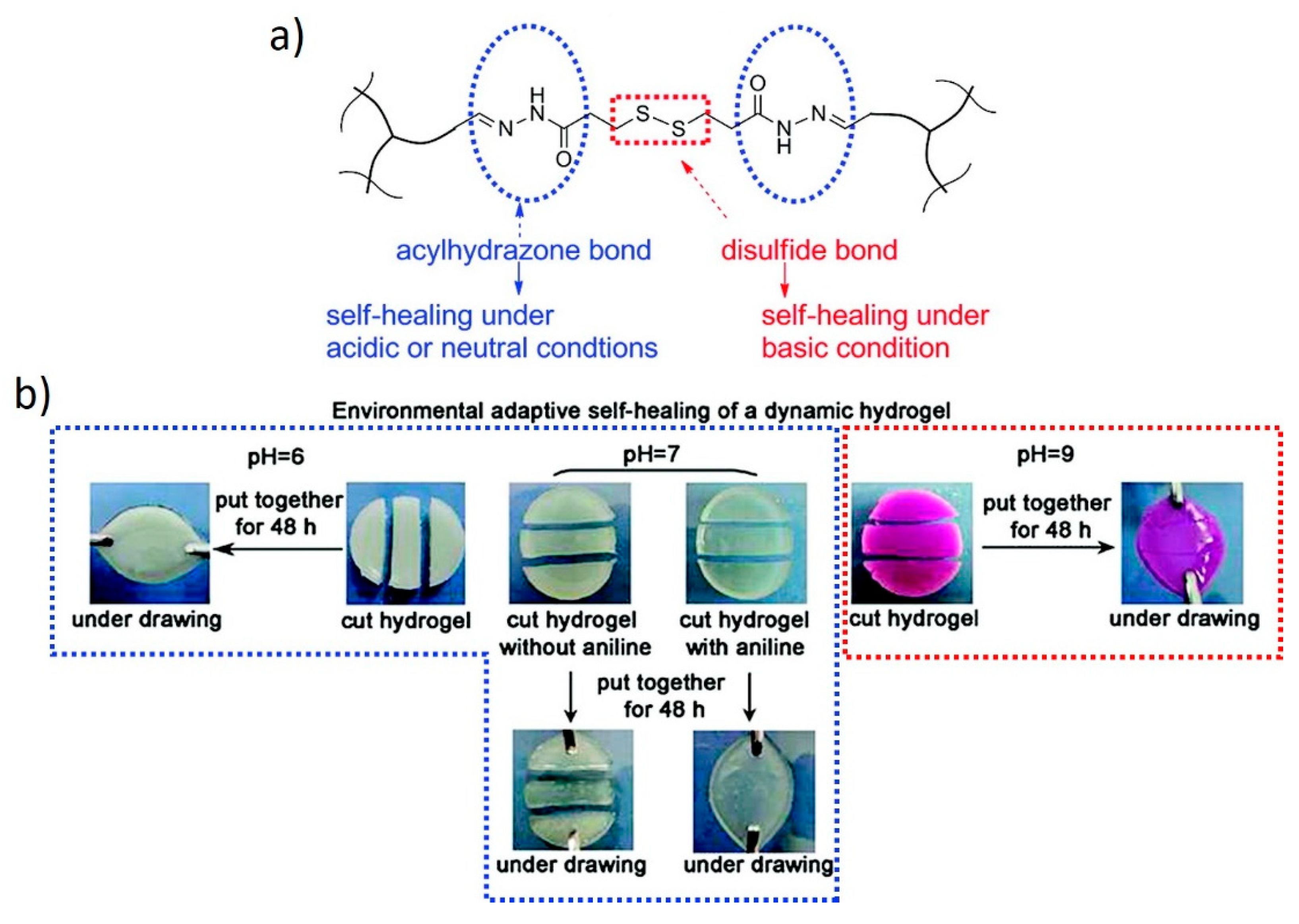
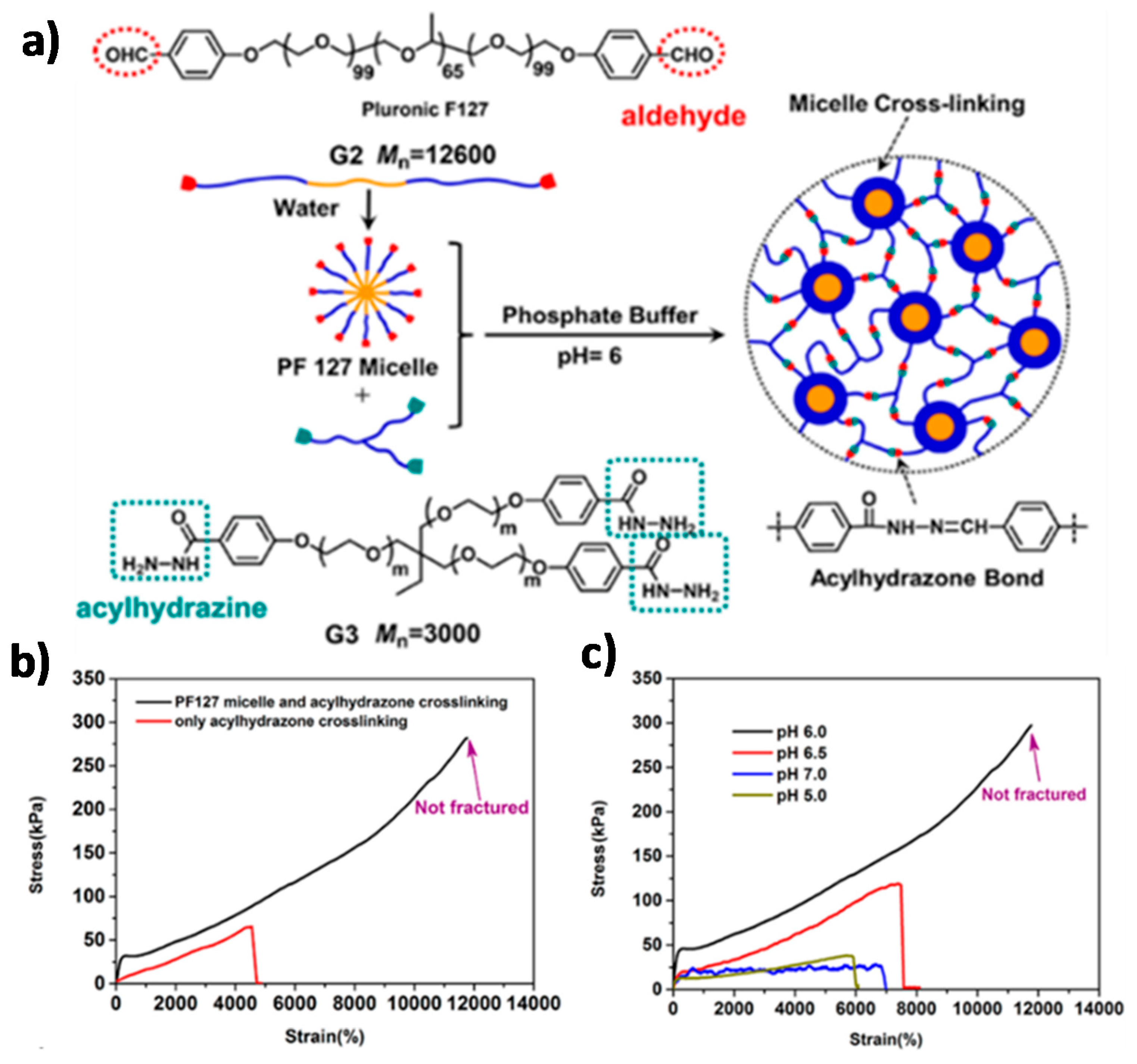
7.1. Hydrogels Combining Dynamic Covalent Bonds and Supramolecular Interactions
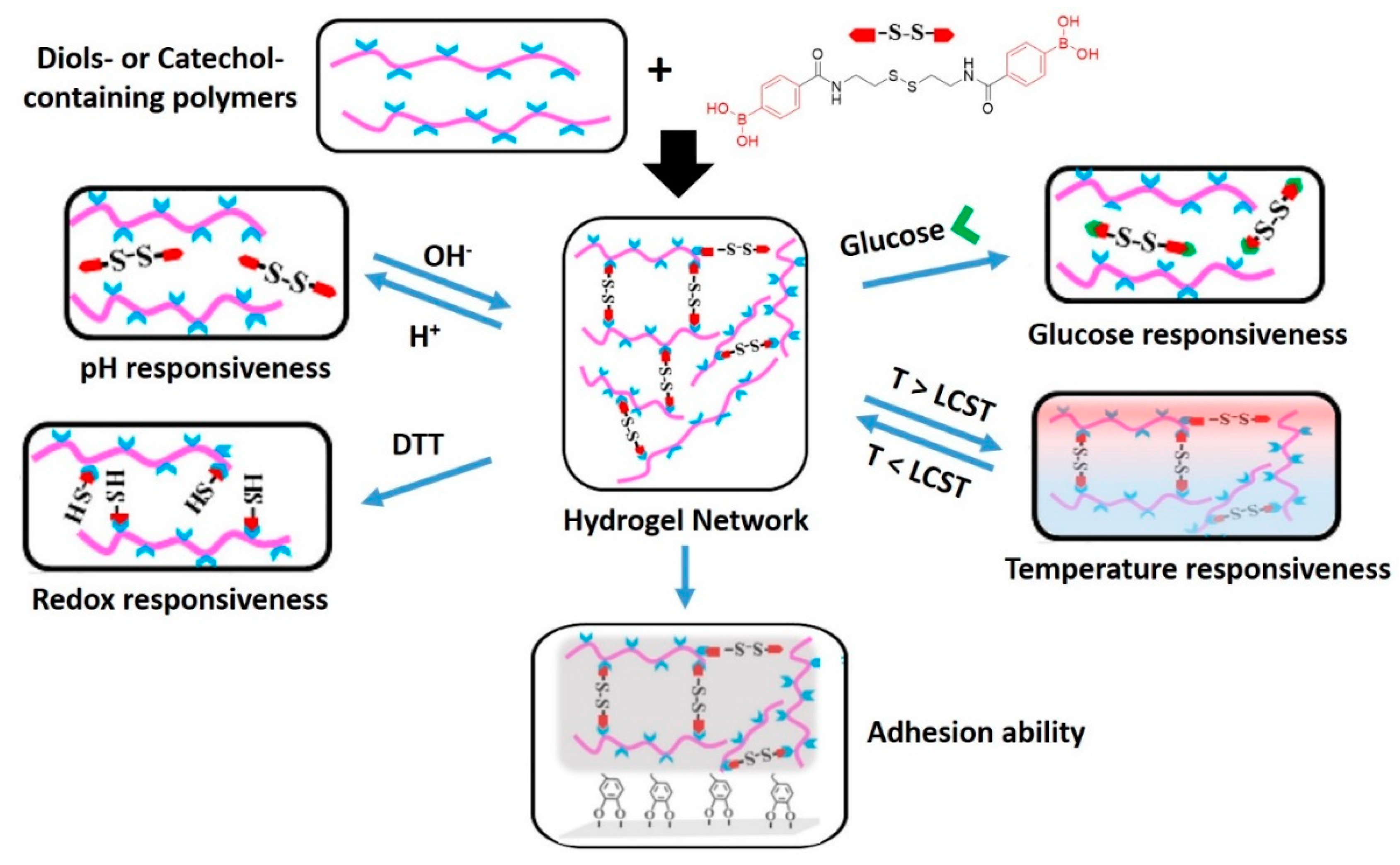
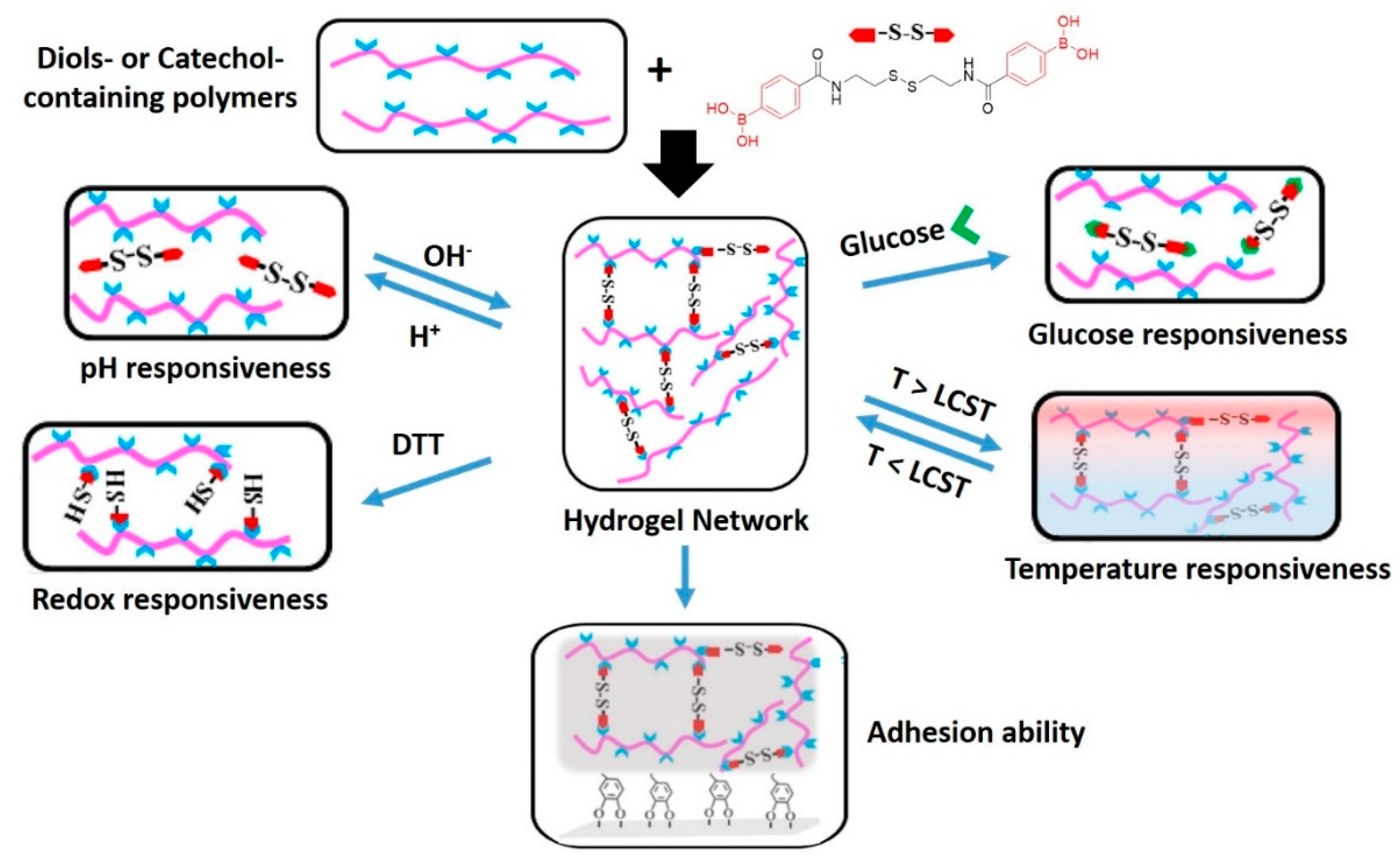
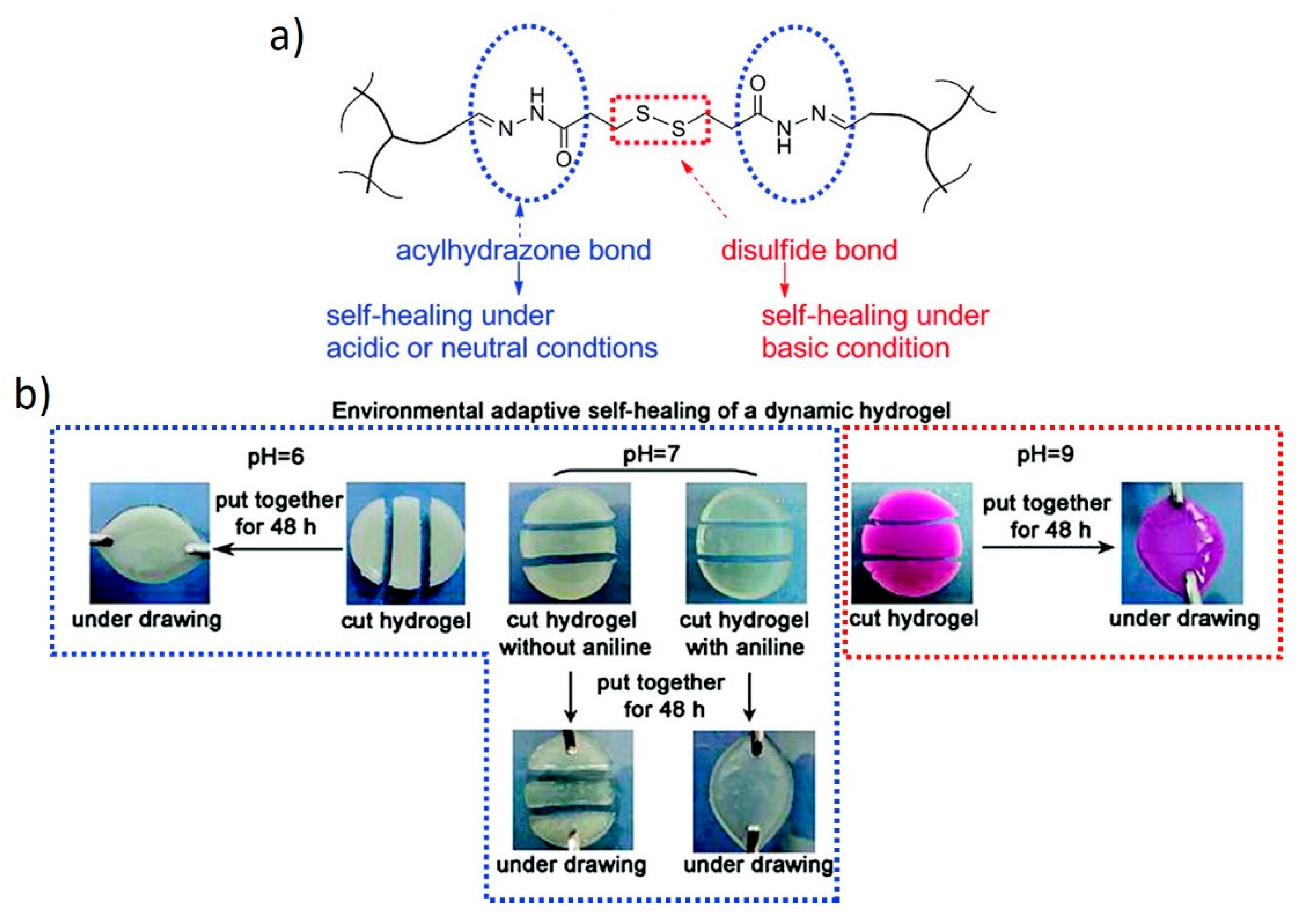
7.2. Hydrogels with Dynamic Covalent Crosslinks of Different Natures
8. Networks Combining Two Different Structures
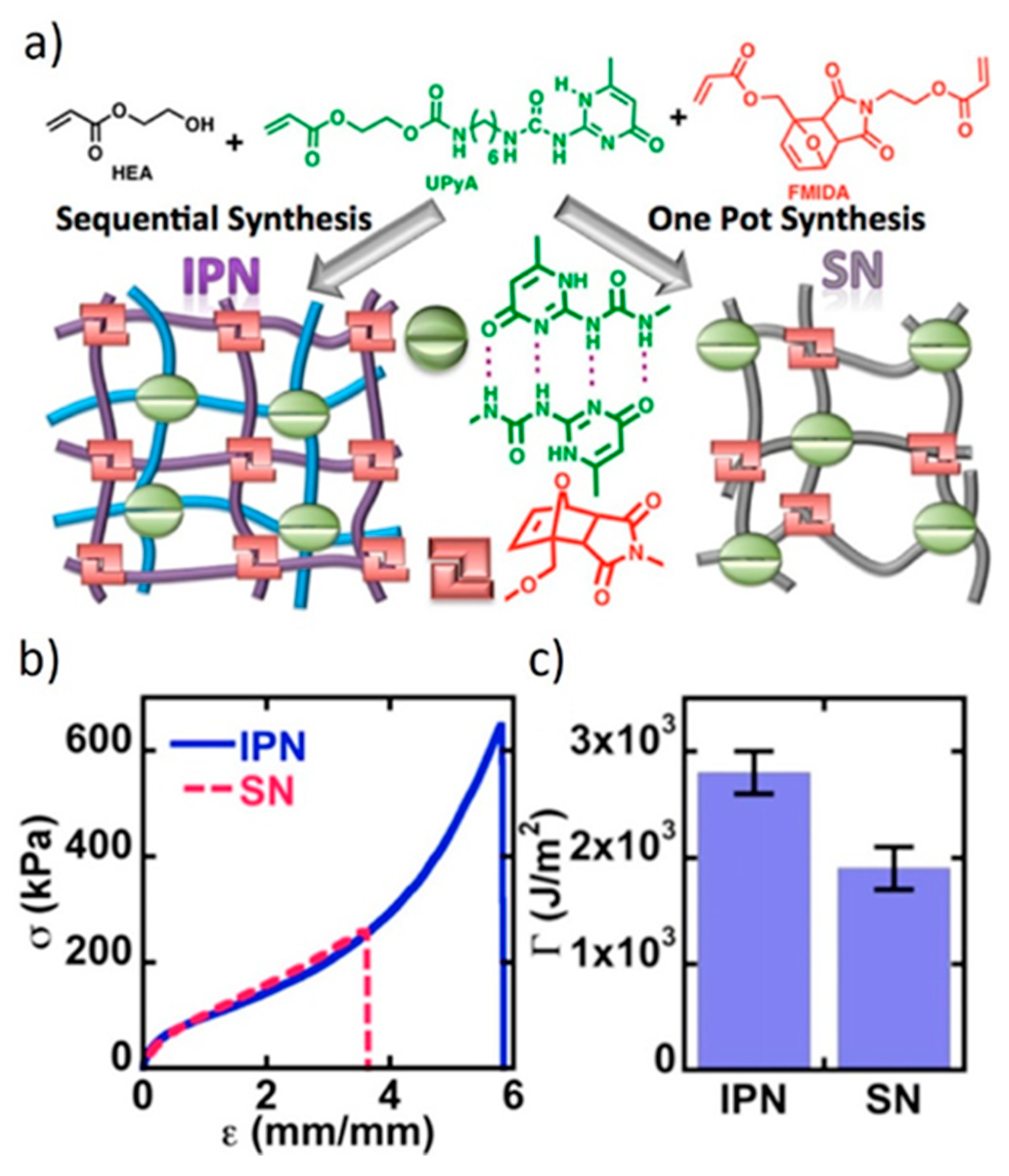
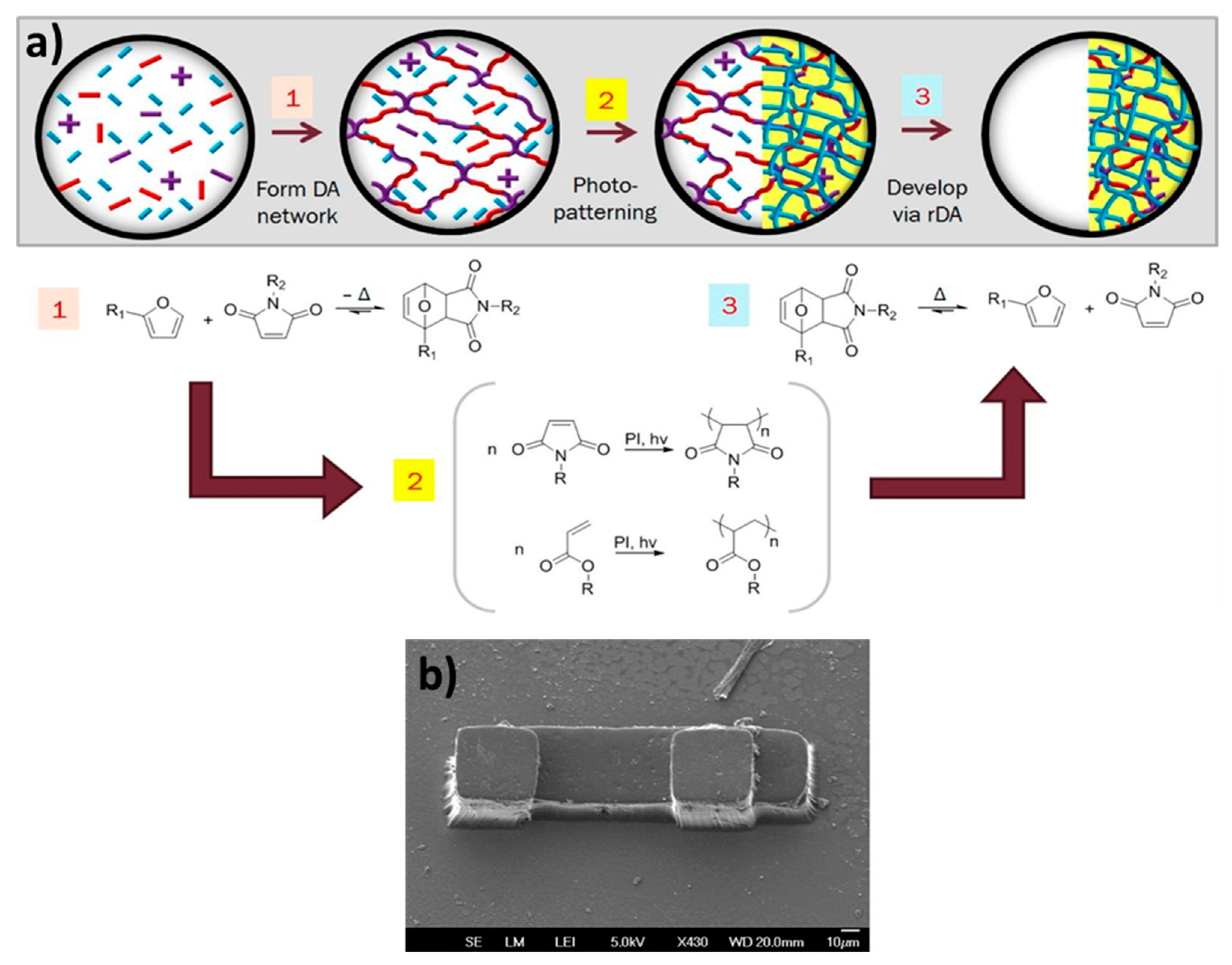
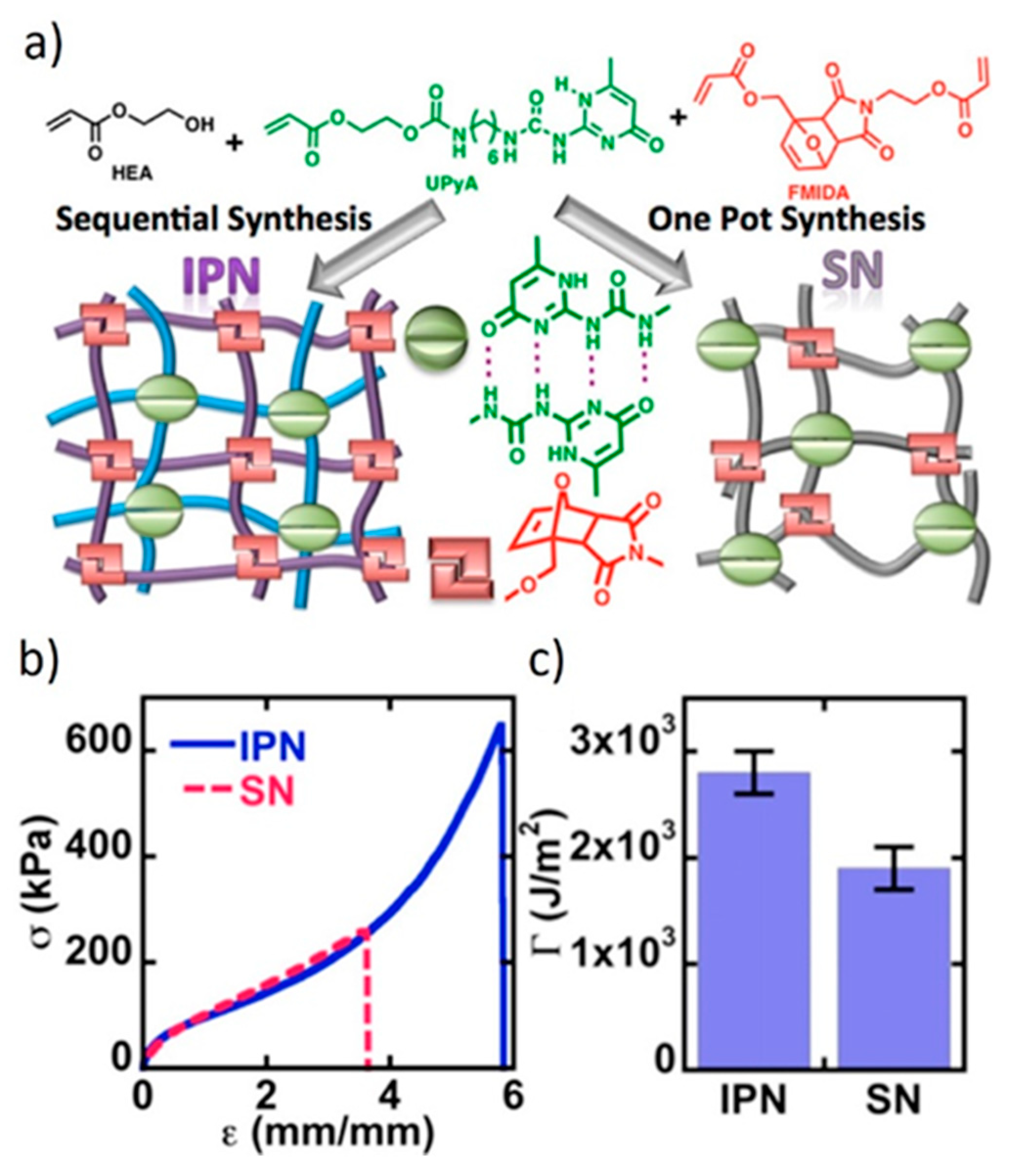
8.1. Interpenetrated Networks
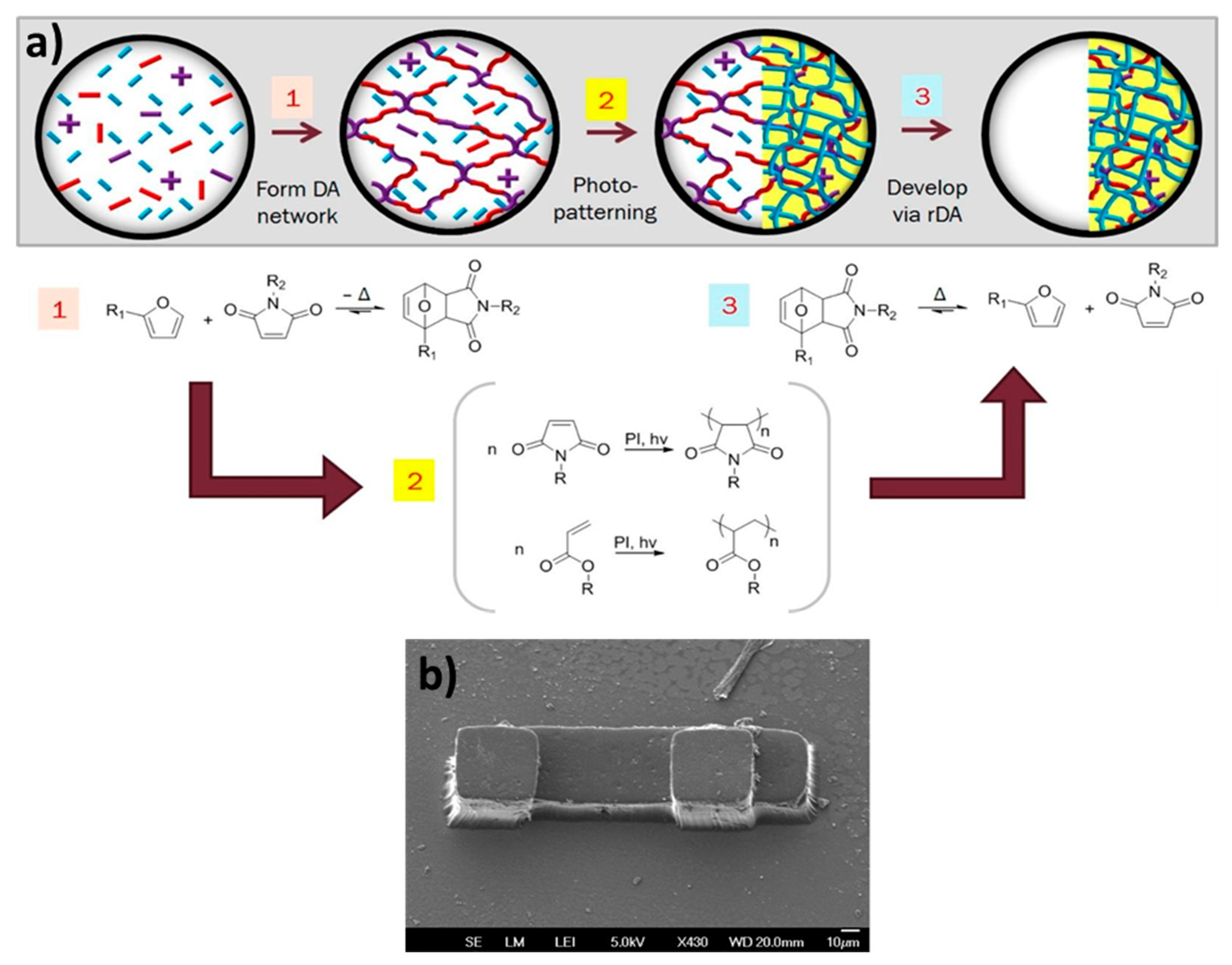
8.2. Combined Networks
9. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Biron, M. Thermoplastics and Thermoplastic Composites: Technical Information for Plastics Users, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2018; p. 944. [Google Scholar]
- Fouquey, C.; Lehn, J.M.; Levelut, A.M. Molecular recognition directed self-assembly of supramolecular liquid crystalline polymers from complementary chiral components. Adv. Mater. 1990, 2, 254–257. [Google Scholar] [CrossRef]
- Biedermann, F.; Schneider, H.-J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies, 1st ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Lehn, J.-M. From supramolecular chemistry towards constitutional dynamic chemistry and adaptive chemistry. Chem. Soc. Rev. 2007, 36, 151–160. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular polymer chemistry-scope and perspectives. Polym. Int. 2002, 51, 825–839. [Google Scholar] [CrossRef]
- Noro, A.; Hayashi, M.; Matsushita, Y. Design and properties of supramolecular polymer gels. Soft Matter 2012, 8, 6416–6429. [Google Scholar] [CrossRef]
- Yan, X.; Wang, F.; Zheng, B.; Huang, F. Stimuli-responsive supramolecular polymeric materials. Chem. Soc. Rev. 2012, 41, 6042–6065. [Google Scholar] [CrossRef]
- Aida, T.; Meijer, E.W.; Stupp, S.I. Functional Supramolecular Polymers. Science 2012, 335, 813–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiffert, S. Supramolecular Polymer Networks and Gels; Springer: Berlin/Heidelberg, Germany, 2015; Volume 268, p. 288. [Google Scholar]
- Voorhaar, L.; Hoogenboom, R. Supramolecular polymer networks: Hydrogels and bulk materials. Chem. Soc. Rev. 2016, 45, 4013–4031. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.-M. Supramolecular chemistry: Where from? Where to? Chem. Soc. Rev. 2017, 46, 2378–2379. [Google Scholar] [CrossRef] [PubMed]
- Kloxin, C.J.; Scott, T.F.; Adzima, B.J.; Bowman, C.N. Covalent Adaptable Networks (CANs): A Unique Paradigm in Crosslinked Polymers. Macromolecules 2010, 43, 2643–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, C.N.; Kloxin, C.J. Covalent Adaptable Networks: Reversible Bond Structures Incorporated in Polymer Networks. Angew. Chem. Int. Ed. 2012, 51, 4272–4274. [Google Scholar] [CrossRef] [PubMed]
- Kloxin, C.J.; Bowman, C.N. Covalent adaptable networks: Smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 2013, 42, 7161–7173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nut, S.R.; Sheran, K.; Wudl, F. A thermally re-mendable cross-linked polymeric material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Winne, J.M.; Leibler, L.; Du Prez, F.E. Dynamic covalent chemistry in polymer networks: A mechanistic perspective. Polym. Chem. 2019, 10, 6091–6108. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Capelot, M.; Unterlass, M.M.; Tournilhac, F.; Leibler, L. Catalytic Control of the Vitrimer Glass Transition. ACS Macro Lett. 2012, 1, 789–792. [Google Scholar] [CrossRef]
- Van Zee, N.J.; Nicolay, R. Vitrimers: Permanently crosslinked polymers with dynamic network topology. Prog. Polym. Sci. 2020, 104, 101233. [Google Scholar] [CrossRef]
- Brutman, J.P.; Delgado, P.A.; Hillmyer, M.A. Polylactide Vitrimers. ACS Macro Lett. 2014, 3, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Breuillac, A.; Kassalias, A.; Nicolay, R. Polybutadiene Vitrimers Based on Dioxaborolane Chemistry and Dual Networks with Static and Dynamic Cross-links. Macromolecules 2019, 52, 7102–7113. [Google Scholar] [CrossRef]
- Obadia, M.M.; Jourdain, A.; Cassagnau, P.; Montarnal, D.; Drockenmuller, E. Tuning the Viscosity Profile of Ionic Vitrimers Incorporating 1,2,3-Triazolium Cross-Links. Adv. Funct. Mater. 2017, 27, 1703258. [Google Scholar] [CrossRef]
- Delahaye, M.; Winne, J.M.; Du Prez, F.E. Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification As a Versatile Dynamic Cross-Linking Chemistry. J. Am. Chem. Soc. 2019, 141, 15277–15287. [Google Scholar] [CrossRef] [PubMed]
- Jourdain, A.; Asbai, R.; Anaya, O.; Chehimi, M.M.; Drockenmuller, E.; Montarnal, D. Rheological Properties of Covalent Adaptable Networks with 1,2,3-Triazolium Cross-Links: The Missing Link between Vitrimers and Dissociative Networks. Macromolecules 2020, 53, 1884–1900. [Google Scholar] [CrossRef] [Green Version]
- Chakma, P.; Morley, C.N.; Sparks, J.L.; Konkolewicz, D. Exploring How Vitrimer-like Properties Can Be Achieved from Dissociative Exchange in Anilinium Salts. Macromolecules 2020, 53, 1233–1244. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajj, R.; Duval, A.; Dhers, S.; Averous, L. Network Design to Control Polyimine Vitrimer Properties: Physical Versus Chemical Approach. Macromolecules 2020, 53, 3796–3805. [Google Scholar] [CrossRef]
- Nicolay, R.; Kamada, J.; Van Wassen, A.; Matyjaszewski, K. Responsive Gels Based on a Dynamic Covalent Trithiocarbonate Cross-Linker. Macromolecules 2010, 43, 4355–4361. [Google Scholar] [CrossRef]
- Sun, H.; Kabb, C.P.; Sims, M.B.; Sumerlin, B.S. Architecture-transformable polymers: Reshaping the future of stimuli-responsive polymers. Prog. Polym. Sci. 2019, 89, 61–75. [Google Scholar] [CrossRef]
- Jiang, Z.; Bhaskaran, A.; Aitken, H.M.; Shackleford, I.C.G.; Connal, L.A. Using Synergistic Multiple Dynamic Bonds to Construct Polymers with Engineered Properties. Macromol. Rapid Commun. 2019, 40, 1900038. [Google Scholar] [CrossRef] [Green Version]
- Chakma, P.; Konkolewicz, D. Dynamic Covalent Bonds in Polymeric Materials. Angew. Chem. Int. Ed. 2019, 58, 9682–9695. [Google Scholar] [CrossRef]
- Scheutz, G.M.; Lessard, J.J.; Sims, M.B.; Sumerlin, B.S. Adaptable Crosslinks in Polymeric Materials: Resolving the Intersection of Thermoplastics and Thermosets. J. Am. Chem. Soc. 2019, 141, 16181–16196. [Google Scholar] [CrossRef]
- Guerre, M.; Taplan, C.; Winne, J.M.; Du Prez, F.E. Vitrimers: Directing chemical reactivity to control material properties. Chem. Sci. 2020, 11, 4855–4870. [Google Scholar] [CrossRef] [Green Version]
- Obadia, M.M.; Mudraboyina, B.P.; Serghei, A.; Montarnal, D.; Drockenmuller, E. Reprocessing and Recycling of Highly Cross-Linked Ion-Conducting Networks through Transalkylation Exchanges of C-N Bonds. J. Am. Chem. Soc. 2015, 137, 6078–6083. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, R.; Wu, Q.; Chen, T.; Sun, P. Bio-Inspired High-Performance and Recyclable Cross-Linked Polymers. Adv. Mater. 2013, 25, 4912–4917. [Google Scholar] [CrossRef]
- Inoue, K.; Yamashiro, M.; Iji, M. Recyclable shape-memory polymer: Poly(lactic acid) crosslinked by a thermoreversible Diels-Alder reaction. J. Appl. Polym. Sci. 2009, 112, 876–885. [Google Scholar] [CrossRef]
- Yu, F.; Cao, X.; Du, J.; Wang, G.; Chen, X. Multifunctional hydrogel with good structure integrity, self-healing, and tissue-adhesive property formed by combining diels-alder click reaction and acylhydrazone bond. ACS Appl. Mater. Interfaces 2015, 7, 24023–24031. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.M.; Lensmeyer, E.E.; Zhang, B.; Chakma, P.; Flum, J.A.; Via, J.J.; Sparks, J.L.; Konkolewicz, D. Effect of Polymer Network Architecture, Enhancing Soft Materials Using Orthogonal Dynamic Bonds in an Interpenetrating Network. ACS Macro Lett. 2017, 6, 495–499. [Google Scholar] [CrossRef]
- Berg, G.J.; Gong, T.; Fenoli, C.R.; Bowman, C.N. A Dual-Cure, Solid-State Photoresist Combining a Thermoreversible Diels-Alder Network and a Chain Growth Acrylate Network. Macromolecules 2014, 47, 3473–3482. [Google Scholar] [CrossRef]
- Rekondo, A.; Martin, R.; Ruiz de Luzuriaga, A.; Cabanero, G.; Grande, H.J.; Odriozola, I. Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater. Horiz. 2014, 1, 237–240. [Google Scholar] [CrossRef]
- Aoki, D.; Teramoto, Y.; Nishio, Y. SH-Containing Cellulose Acetate Derivatives: Preparation and Characterization as a Shape Memory-Recovery Material. Biomacromolecules 2007, 8, 3749–3757. [Google Scholar] [CrossRef]
- Ruiz de Luzuriaga, A.; Martin, R.; Markaide, N.; Rekondo, A.; Cabanero, G.; Rodriguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horiz. 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Deng, G.; Li, F.; Yu, H.; Liu, F.; Liu, C.; Sun, W.; Jiang, H.; Chen, Y. Dynamic Hydrogels with an Environmental Adaptive Self-Healing Ability and Dual Responsive Sol-Gel Transitions. ACS Macro Lett. 2012, 1, 275–279. [Google Scholar] [CrossRef]
- Peng, W.L.; You, Y.; Xie, P.; Rong, M.Z.; Zhang, M.Q. Adaptable interlocking macromolecular networks with homogeneous architecture made from immiscible single networks. Macromolecules 2020, 53, 584–593. [Google Scholar] [CrossRef]
- Kolomiets, E.; Lehn, J.-M. Double dynamers: Molecular and supramolecular double dynamic polymers. Chem. Commun. 2005, 1519–1521. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, H.; Yang, H.; Hao, X.; Tang, Q.; Zhang, X. An Injectable Interpenetrating Polymer Network Hydrogel with Tunable Mechanical Properties and Self-Healing Abilities. Macromol. Chem. Phys. 2017, 218, 1700348. [Google Scholar] [CrossRef]
- Collins, J.; Nadgorny, M.; Xiao, Z.; Connal, L.A. Doubly Dynamic Self-Healing Materials Based on Oxime Click Chemistry and Boronic Acids. Macromol. Rapid Commun. 2017, 38, 1600760. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, Z.; Jia, P.; Zhang, M.; Bo, C.; Feng, G.; Hu, L.; Zhou, Y. Tunable “soft and stiff”, self-healing, recyclable, thermadapt shape memory biomass polymers based on multiple hydrogen bonds and dynamic imine bonds. J. Mater. Chem. A 2019, 7, 13400–13410. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wang, H.; Huang, X.; Huang, G.; Wu, J. Weldable, malleable and programmable epoxy vitrimers with high mechanical properties and water insensitivity. Chem. Eng. J. 2019, 368, 61–70. [Google Scholar] [CrossRef]
- Li, Y.; Yang, L.; Zeng, Y.; Wu, Y.; Wei, Y.; Tao, L. Self-Healing Hydrogel with a Double Dynamic Network Comprising Imine and Borate Ester Linkages. Chem. Mater. 2019, 31, 5576–5583. [Google Scholar] [CrossRef]
- Zheng, N.; Fang, Z.; Zou, W.; Zhao, Q.; Xie, T. Thermoset Shape-Memory Polyurethane with Intrinsic Plasticity Enabled by Transcarbamoylation. Angew. Chem., Int. Ed. 2016, 55, 11421–11425. [Google Scholar] [CrossRef]
- Chen, X.; Li, L.; Jin, K.; Torkelson, J.M. Reprocessable polyhydroxyurethane networks exhibiting full property recovery and concurrent associative and dissociative dynamic chemistry via transcarbamoylation and reversible cyclic carbonate aminolysis. Polym. Chem. 2017, 8, 6349–6355. [Google Scholar] [CrossRef]
- Pei, Z.; Yang, Y.; Chen, Q.; Terentjev, E.M.; Wei, Y.; Ji, Y. Mouldable liquid-crystalline elastomer actuators with exchangeable covalent bonds. Nat. Mater. 2014, 13, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Saed, M.O.; Gablier, A.; Terentejv, E.M. Liquid Crystalline Vitrimers with Full or Partial Boronic-Ester Bond Exchange. Adv. Funct. Mater. 2020, 30, 1906458. [Google Scholar] [CrossRef]
- Cash, J.J.; Kubo, T.; Dobbins, D.J.; Sumerlin, B.S. Maximizing the symbiosis of static and dynamic bonds in self-healing boronic ester networks. Polym. Chem. 2018, 9, 2011–2020. [Google Scholar] [CrossRef]
- Imato, K.; Kanehara, T.; Nojima, S.; Ohishi, T.; Higaki, Y.; Takahara, A.; Otsuka, H. Repeatable mechanochemical activation of dynamic covalent bonds in thermoplastic elastomers. Chem. Commun. 2016, 52, 10482–10485. [Google Scholar] [CrossRef] [Green Version]
- Lendlein, A.; Jiang, H.; Juenger, O.; Langer, R. Light-induced shape-memory polymers. Nature 2005, 434, 879–882. [Google Scholar] [CrossRef]
- McBride, M.K.; Martinez, A.M.; Cox, L.; Alim, M.; Childress, K.; Beiswinger, M.; Podgorski, M.; Worrell, B.T.; Killgore, J.; Bowman, C.N. A readily programmable, fully reversible shape-switching material. Sci. Adv. 2018, 4, eaat4634. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.; Buhler, E.; Lehn, J.-M. Double dynamic self-healing polymers: Supramolecular and covalent dynamic polymers based on the bis-iminocarbohydrazide motif. Polym. Int. 2014, 63, 1400–1405. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Mechanically Robust, Self-Healable, and Highly Stretchable “Living” Crosslinked Polyurethane Based on a Reversible C-C Bond. Adv. Funct. Mater. 2018, 28, 1706050. [Google Scholar] [CrossRef]
- Zhang, B.; Ke, J.; Vakil, J.R.; Cummings, S.C.; Digby, Z.A.; Sparks, J.L.; Ye, Z.; Zanjani, M.B.; Konkolewicz, D. Dual-dynamic interpenetrated networks tuned through macromolecular architecture. Polym. Chem. 2019, 10, 6290–6304. [Google Scholar] [CrossRef]
- Zhang, B.; Digby, Z.A.; Flum, J.A.; Foster, E.M.; Sparks, J.L.; Konkolewicz, D. Self-healing, malleable and creep limiting materials using both supramolecular and reversible covalent linkages. Polym. Chem. 2015, 6, 7368–7372. [Google Scholar] [CrossRef]
- Jian, X.; Hu, Y.; Zhou, W.; Xiao, L. Self-healing polyurethane based on disulfide bond and hydrogen bond. Polym. Adv. Technol. 2018, 29, 463–469. [Google Scholar] [CrossRef]
- Liu, J.; Ma, X.; Tong, Y.; Lang, M. Self-healing polyurethane based on ditelluride bonds. Appl. Surf. Sci. 2018, 455, 318–325. [Google Scholar] [CrossRef]
- Xu, W.M.; Rong, M.Z.; Zhang, M.Q. Sunlight driven self-healing, reshaping and recycling of a robust, transparent and yellowing-resistant polymer. J. Mater. Chem. A 2016, 4, 10683–10690. [Google Scholar] [CrossRef]
- Neal, J.A.; Mozhdehi, D.; Guan, Z. Enhancing Mechanical Performance of a Covalent Self-Healing Material by Sacrificial Noncovalent Bonds. J. Am. Chem. Soc. 2015, 137, 4846–4850. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, Z.; Wu, S.; Guo, B. Integrating Sacrificial Bonds into Dynamic Covalent Networks toward Mechanically Robust and Malleable Elastomers. ACS Macro Lett. 2019, 8, 193–199. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, Z.; Wang, D.; Wu, S.; Guo, B. Biomimetic design of elastomeric vitrimers with unparalleled mechanical properties, improved creep resistance and retained malleability by metal-ligand coordination. J. Mater. Chem. A 2019, 7, 26867–26876. [Google Scholar] [CrossRef]
- Wang, S.; Ma, S.; Li, Q.; Xu, X.; Wang, B.; Huang, K.; Liu, Y.; Zhu, J. Facile Preparation of Polyimine Vitrimers with Enhanced Creep Resistance and Thermal and Mechanical Properties via Metal Coordination. Macromolecules 2020, 53, 2919–2931. [Google Scholar] [CrossRef]
- Sanchez-Moran, H.; Ahmadi, A.; Vogler, B.; Roh, K.-H. Oxime Cross-Linked Alginate Hydrogels with Tunable Stress Relaxation. Biomacromolecules 2019, 20, 4419–4429. [Google Scholar] [CrossRef]
- Niu, X.; Wang, F.; Xing, K.; Zhang, R.; Wang, X.; Li, X.; Chen, T.; Sun, P.; Shi, A.-C. Dual Cross-linked Vinyl Vitrimer with Efficient Self-Catalysis Achieving Triple-Shape-Memory Properties. Macromol. Rapid Commun. 2019, 40, 1900313. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Rios, O.; Keum, J.K.; Kessler, M.R. Liquid crystalline epoxy networks with exchangeable disulfide bonds. Soft Matter 2017, 13, 5021–5027. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Rios, O.; Keum, J.K.; Kessler, M.R. Photo-responsive liquid crystalline epoxy networks with exchangeable disulfide bonds. RSC Adv. 2017, 7, 37248–37254. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tian, H.; He, Q.; Cai, S. Reprogrammable, Reprocessible, and Self-Healable Liquid Crystal Elastomer with Exchangeable Disulfide Bonds. ACS Appl. Mater. Interfaces 2017, 9, 33119–33128. [Google Scholar] [CrossRef]
- Wen, Z.; McBride, M.K.; Zhang, X.; Han, X.; Martinez, A.M.; Shao, R.; Zhu, C.; Visvanathan, R.; Clark, N.A.; Wang, Y.; et al. Reconfigurable LC Elastomers: Using a Thermally Programmable Monodomain To Access Two-Way Free-Standing Multiple Shape Memory Polymers. Macromolecules 2018, 51, 5812–5819. [Google Scholar] [CrossRef]
- Yang, Y.; Pei, Z.; Li, Z.; Wei, Y.; Ji, Y. Making and Remaking Dynamic 3D Structures by Shining Light on Flat Liquid Crystalline Vitrimer Films without a Mold. J. Am. Chem. Soc. 2016, 138, 2118–2121. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Terentjev, E.M.; Zhang, Y.; Chen, Q.; Zhao, Y.; Wei, Y.; Ji, Y. Reprocessable Thermoset Soft Actuators. Angew. Chem., Int. Ed. 2019, 58, 17474–17479. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Yang, Y.; Chen, Q.; Wei, Y.; Ji, Y. Regional Shape Control of Strategically Assembled Multishape Memory Vitrimers. Adv. Mater. 2016, 28, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rios, O.; Keum, J.K.; Chen, J.; Kessler, M.R. Photoresponsive Liquid Crystalline Epoxy Networks with Shape Memory Behavior and Dynamic Ester Bonds. ACS Appl. Mater. Interfaces 2016, 8, 15750–15757. [Google Scholar] [CrossRef]
- Lu, X.; Guo, S.; Tong, X.; Xia, H.; Zhao, Y. Tunable Photocontrolled Motions Using Stored Strain Energy in Malleable Azobenzene Liquid Crystalline Polymer Actuators. Adv. Mater. 2017, 29, 1606467. [Google Scholar] [CrossRef]
- Chen, Q.; Li, Y.; Yang, Y.; Xu, Y.; Qian, X.; Wei, Y.; Ji, Y. Durable liquid-crystalline vitrimer actuators. Chem. Sci. 2019, 10, 3025–3030. [Google Scholar] [CrossRef] [Green Version]
- Lawton, M.I.; Tillman, K.R.; Mohammed, H.S.; Kuang, W.; Shipp, D.A.; Mather, P.T. Anhydride-Based Reconfigurable Shape Memory Elastomers. ACS Macro Lett. 2016, 5, 203–207. [Google Scholar] [CrossRef]
- Defize, T.; Riva, R.; Raquez, J.-M.; Dubois, P.; Jerome, C.; Alexandre, M. Thermoreversibly Crosslinked Poly(ε-caprolactone) as Recyclable Shape-Memory Polymer Network. Macromol. Rapid Commun. 2011, 32, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Niu, Y.; Huang, C.; Xiao, L.; Chen, Z.; Yang, K.; Wang, Y. Self-healable and recyclable triple-shape PPDO-PTMEG co-network constructed through thermoreversible Diels-Alder reaction. Polym. Chem. 2012, 3, 1390–1393. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, Q.; Yang, L.; Zou, W.; Xi, X.; Xie, T. Exploring Dynamic Equilibrium of Diels-Alder Reaction for Solid State Plasticity in Remoldable Shape Memory Polymer Network. ACS Macro Lett. 2016, 5, 805–808. [Google Scholar] [CrossRef]
- Defize, T.; Riva, R.; Thomassin, J.-M.; Alexandre, M.; Herck, N.V.; Prez, F.D.; Jerome, C. Reversible TAD Chemistry as a Convenient Tool for the Design of (Re)processable PCL-Based Shape-Memory Materials. Macromol. Rapid Commun. 2017, 38, 1600517. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Liu, G.; Dong, X.; Wang, D. Triple-shape memory epoxy based on Diels-Alder adduct molecular switch. Polymer 2016, 84, 1–9. [Google Scholar] [CrossRef]
- Michal, B.T.; Jaye, C.A.; Spencer, E.J.; Rowan, S.J. Inherently Photohealable and Thermal Shape-Memory Polydisulfide Networks. ACS Macro Lett. 2013, 2, 694–699. [Google Scholar] [CrossRef]
- Zhang, S.; Pan, L.; Xia, L.; Sun, Y.; Liu, X. Dynamic polysulfide shape memory networks derived from elemental sulfur and their dual thermo-/photo-induced solid-state plasticity. React. Funct. Polym. 2017, 121, 8–14. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, D. A Novel Self-Healing Polyurethane Based on Disulfide Bonds. Macromol. Chem. Phys. 2016, 217, 1191–1196. [Google Scholar] [CrossRef]
- Fang, Z.; Zheng, N.; Zhao, Q.; Xie, T. Healable, Reconfigurable, Reprocessable Thermoset Shape Memory Polymer with Highly Tunable Topological Rearrangement Kinetics. ACS Appl. Mater. Interfaces 2017, 9, 22077–22082. [Google Scholar] [CrossRef]
- Zheng, N.; Hou, J.; Xu, Y.; Fang, Z.; Zou, W.; Zhao, Q.; Xie, T. Catalyst-Free Thermoset Polyurethane with Permanent Shape Reconfigurability and Highly Tunable Triple-Shape Memory Performance. ACS Macro Lett. 2017, 6, 326–330. [Google Scholar] [CrossRef]
- Zhao, Q.; Zou, W.; Luo, Y.; Xie, T. Shape memory polymer network with thermally distinct elasticity and plasticity. Sci. Adv. 2016, 2, e1501297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Hao, C.; Wang, L.; Li, Y.; Liu, W.; Xin, J.; Zhang, J. Eugenol-Derived Biobased Epoxy: Shape Memory, Repairing, and Recyclability. Macromolecules 2017, 50, 8588–8597. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, W.; Zhu, S. Reversible Shape Memory Polymer from Semicrystalline Poly(ethylene-co-vinyl acetate) with Dynamic Covalent Polymer Networks. Macromolecules 2018, 51, 8956–8963. [Google Scholar] [CrossRef]
- Röttger, M.; Domenech, T.; van der Weegen, R.; Breuillac, A.; Nicolaÿ, R.; Leibler, L. High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis. Science 2017, 356, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, R.G.; Tournilhac, F.; Leibler, L. Phase Separation and Self-Assembly in Vitrimers: Hierarchical Morphology of Molten and Semicrystalline Polyethylene/Dioxaborolane Maleimide Systems. Macromolecules 2018, 52, 432–443. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Deng, G.; Zhou, L.; Li, Z.; Chen, Y. Ultrastretchable, Self-Healable Hydrogels Based on Dynamic Covalent Bonding and Triblock Copolymer Micellization. ACS Macro Lett. 2017, 6, 881–886. [Google Scholar] [CrossRef]
- Altuna, F.I.; Casado, U.; dell’Erba, I.E.; Luna, L.; Hoppe, C.E.; Williams, R.J.J. Epoxy vitrimers incorporating physical crosslinks produced by self-association of alkyl chains. Polym. Chem. 2020, 11, 1337–1347. [Google Scholar] [CrossRef]
- Statz, R.J. History of Polyolefins. In History of Polyolefins; Seymour, R.B., Cheng, T., Eds.; Springer: Berlin/Heidelberg, Germany, 1986; pp. 172–192. [Google Scholar] [CrossRef]
- Legge, N.R. Thermoplastic elastomers—Three decades of progress. Rubber Chem. Technol. 1989, 62, 529–547. [Google Scholar] [CrossRef]
- Spontak, R.J.; Patel, N.P. Thermoplastic elastomers: Fundamentals and applications. Curr. Opin. Colloid Interface Sci. 2000, 5, 334–341. [Google Scholar] [CrossRef]
- Amin, S.; Amin, M. Thermoplastic elastomeric (TPE) materials and their use in outdoor electrical insulation. Rev. Adv. Mater. Sci. 2011, 29, 15–30. [Google Scholar]
- Fazli, A.; Rodrigue, D. Waste rubber recycling: A review on the evolution and properties of thermoplastic elastomers. Materials 2020, 13, 782. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, H.; Kimura, I.; Saito, K.; Ono, H. Infrared studies on segmented polyurethane-urea elastomers. J. Macromol. Sci. Phys. 1974, B10, 591–618. [Google Scholar] [CrossRef]
- Petrovic, Z.S.; Ferguson, J. Polyurethane elastomers. Prog. Polym. Sci. 1991, 16, 695–836. [Google Scholar] [CrossRef]
- Wang, X.; Wei, Y.; Chen, D.; Bai, Y. Synthesis and properties of room-temperature self-healing polyurethane elastomers. J. Macromol. Sci. Part. A Pure Appl. Chem. 2017, 54, 956–966. [Google Scholar] [CrossRef]
- Otsuka, H.; Nagano, S.; Kobashi, Y.; Maeda, T.; Takahara, A. A dynamic covalent polymer driven by disulfide metathesis under photoirradiation. Chem. Commun. 2010, 46, 1150–1152. [Google Scholar] [CrossRef] [PubMed]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Xu, M.; Cheng, B.; Sheng, Y.; Zhou, J.; Wang, M.; Jiang, X.; Lu, X. High-Performance Cross-Linked Self-Healing Material Based on Multiple Dynamic Bonds. ACS Appl. Polym. Mater. 2020, 2, 2228–2237. [Google Scholar] [CrossRef]
- Lendlein, A.; Kelch, S. Shape-memory polymers. Angew. Chem. Int. Ed. 2002, 41, 2034–2057. [Google Scholar] [CrossRef]
- Liu, C.; Qin, H.; Mather, P.T. Review of progress in shape-memory polymers. J. Mater. Chem. 2007, 17, 1543–1558. [Google Scholar] [CrossRef]
- Behl, M.; Razzaq, M.Y.; Lendlein, A. Multifunctional Shape-Memory Polymers. Adv. Mater. 2010, 22, 3388–3410. [Google Scholar] [CrossRef]
- Leng, J.; Lan, X.; Liu, Y.; Du, S. Shape-memory polymers and their composites: Stimulus methods and applications. Prog. Mater. Sci. 2011, 56, 1077–1135. [Google Scholar] [CrossRef]
- Hu, J.; Zhu, Y.; Huang, H.; Lu, J. Recent advances in shape-memory polymers: Structure, mechanism, functionality, modeling and applications. Prog. Polym. Sci. 2012, 37, 1720–1763. [Google Scholar] [CrossRef]
- Hager, M.D.; Bode, S.; Weber, C.; Schubert, U.S. Shape memory polymers: Past, present and future developments. Prog. Polym. Sci. 2015, 49–50, 3–33. [Google Scholar] [CrossRef]
- Davis, F.J. Liquid-crystalline elastomers. J. Mater. Chem. 1993, 3, 551–562. [Google Scholar] [CrossRef]
- Finkelmann, H.; Nishikawa, E.; Pereira, G.G.; Warner, M. A New Opto-Mechanical Effect in Solids. Phys. Rev. Lett. 2001, 87, 015501. [Google Scholar] [CrossRef]
- Mayer, S.; Zentel, R. Liquid crystalline polymers and elastomers. Curr. Opin. Solid State Mater. Sci. 2003, 6, 545–551. [Google Scholar] [CrossRef]
- Warner, M.; Terentjev, E.M. Liquid Crystal Elastomers; Oxford University Press: Oxford, UK, 2003; 424p. [Google Scholar]
- Lewis, C.L.; Dell, E.M. A review of shape memory polymers bearing reversible binding groups. J. Polym. Sci. Part. B Polym. Phys. 2016, 54, 1340–1364. [Google Scholar] [CrossRef]
- Wu, Y.; Wei, Y.; Ji, Y. Polymer actuators based on covalent adaptable networks. Polym. Chem. 2020, 11, 5297–5320. [Google Scholar] [CrossRef]
- Hoekstra, D.C.; Schenning, A.P.H.J.; Debije, M.G. Epoxide and oxetane based liquid crystals for advanced functional materials. Soft Matter 2020, 16, 5106–5119. [Google Scholar] [CrossRef]
- Jin, B.; Song, H.; Jiang, R.; Song, J.; Zhao, Q.; Xie, T. Programming a crystalline shape memory polymer network with thermo- and photo-reversible bonds toward a single-component soft robot. Sci. Adv. 2018, 4, eaao3865. [Google Scholar] [CrossRef] [Green Version]
- Wool, R.P.; O’Connor, K.M. A theory of crack healing in polymers. J. Appl. Phys. 1981, 52, 5953–5963. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Torkelson, J.M. Reprocessable Polymer Networks via Thiourethane Dynamic Chemistry: Recovery of Cross-link Density after Recycling and Proof-of-Principle Solvolysis Leading to Monomer Recovery. Macromolecules 2019, 52, 8207–8216. [Google Scholar] [CrossRef]
- Chen, M.; Zhou, L.; Wu, Y.; Zhao, X.; Zhang, Y. Rapid Stress Relaxation and Moderate Temperature of Malleability Enabled by the Synergy of Disulfide Metathesis and Carboxylate Transesterification in Epoxy Vitrimers. ACS Macro Lett. 2019, 8, 255–260. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Cramer, C.J.; Hillmyer, M.A.; Dichtel, W.R. Mechanically Activated, Catalyst-Free Polyhydroxyurethane Vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Hillmyer, M.A.; Dichtel, W.R. Structural effects on the reprocessability and stress relaxation of cross-linked polyhydroxyurethanes. J. Appl. Polym. Sci. 2017, 134, 44984. [Google Scholar] [CrossRef] [Green Version]
- Fortman, D.J.; Snyder, R.L.; Sheppard, D.T.; Dichtel, W.R. Rapidly Reprocessable Cross-Linked Polyhydroxyurethanes Based on Disulfide Exchange. ACS Macro Lett. 2018, 7, 1226–1231. [Google Scholar] [CrossRef]
- Wen, Z.; Han, X.; Fairbanks, B.D.; Yang, K.; Bowman, C.N. Development of thiourethanes as robust, reprocessable networks. Polymer 2020, 202, 122715. [Google Scholar] [CrossRef]
- Roy, C.D.; Brown, H.C. Stability of boronic esters - Structural effects on the relative rates of transesterification of 2-(phenyl)-1,3,2-dioxaborolane. J. Organomet. Chem. 2007, 692, 784–790. [Google Scholar] [CrossRef]
- Guerre, M.; Taplan, C.; Nicolay, R.; Winne, J.M.; Du Prez, F.E. Fluorinated Vitrimer Elastomers with a Dual Temperature Response. J. Am. Chem. Soc. 2018, 140, 13272–13284. [Google Scholar] [CrossRef]
- Denissen, W.; Rivero, G.; Nicolay, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25, 2451–2457. [Google Scholar] [CrossRef]
- Denissen, W.; Droesbeke, M.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Chemical control of the viscoelastic properties of vinylogous urethane vitrimers. Nat. Commun. 2017, 8, 14857–14863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tang, Z.; Liu, Y.; Wu, S.; Guo, B. Mechanically Robust, Self-Healable, and Reprocessable Elastomers Enabled by Dynamic Dual Cross-Links. Macromolecules 2019, 52, 3805–3812. [Google Scholar] [CrossRef]
- Ji, F.; Liu, X.; Lin, C.; Zhou, Y.; Dong, L.; Xu, S.; Sheng, D.; Yang, Y. Reprocessable and Recyclable Crosslinked Polyethylene with Triple Shape Memory Effect. Macromol. Mater. Eng. 2018, 304, 1800528. [Google Scholar] [CrossRef]
- Tanaka, R.; Tonoko, N.; Kihara, S.-i.; Nakayama, Y.; Shiono, T. Reversible star assembly of polyolefins using interconversion between boroxine and boronic acid. Polym. Chem. 2018, 9, 3774–3779. [Google Scholar] [CrossRef]
- Caffy, F.; Nicolaÿ, R. Transformation of polyethylene into a vitrimer by nitroxide radical coupling of a bis-dioxaborolane. Polym. Chem. 2019, 10, 3107–3115. [Google Scholar] [CrossRef]
- Tellers, J.; Pinalli, R.; Soliman, M.; Vachon, J.; Dalcanale, E. Reprocessable vinylogous urethane cross-linked polyethylene via reactive extrusion. Polym. Chem. 2019, 10, 5534–5542. [Google Scholar] [CrossRef]
- Kar, G.P.; Saed, M.O.; Terentjev, E.M. Scalable upcycling of thermoplastic polyolefins into vitrimers through transesterification. J. Mater. Chem. A 2020, 8, 24137–24147. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Tournilhac, F.; Cloître, M.; Leibler, L. Linear Viscoelasticity and Flow of Self-Assembled Vitrimers: The Case of a Polyethylene/Dioxaborolane System. Macromolecules 2020, 53, 1852–1866. [Google Scholar] [CrossRef]
- Yang, F.; Pan, L.; Ma, Z.; Lou, Y.; Li, Y.; Li, Y. Highly elastic, strong, and reprocessable cross-linked polyolefin elastomers enabled by boronic ester bonds. Polym. Chem. 2020, 11, 3285–3295. [Google Scholar] [CrossRef]
- Zych, A.; Pinalli, R.; Soliman, M.; Vachon, J.; Dalcanale, E. Polyethylene vitrimers via silyl ether exchange reaction. Polymer 2020, 199, 122567. [Google Scholar] [CrossRef]
- Lessard, J.J.; Scheutz, G.M.; Sung, S.H.; Lantz, K.A.; Epps, T.H., 3rd; Sumerlin, B.S. Block Copolymer Vitrimers. J. Am. Chem Soc. 2020, 142, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, D.; Liu, W.; Li, P.; Liu, J.; Liu, C.; Zhang, J.; Zhao, N.; Xu, J. Recyclable polybutadiene elastomer based on dynamic imine bond. J. Polym. Sci. Part. A Polym. Chem. 2017, 55, 2011–2018. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Jin, K.; Torkelson, J.M. Vitrimers Designed Both To Strongly Suppress Creep and To Recover Original Cross-Link Density after Reprocessing: Quantitative Theory and Experiments. Macromolecules 2018, 51, 5537–5546. [Google Scholar] [CrossRef]
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-X.; Guan, Z. Olefin Metathesis for Effective Polymer Healing via Dynamic Exchange of Strong Carbon-Carbon Double Bonds. J. Am. Chem. Soc. 2012, 134, 14226–14231. [Google Scholar] [CrossRef] [PubMed]
- Legrand, A.; Soulie-Ziakovic, C. Silica-Epoxy Vitrimer Nanocomposites. Macromolecules 2016, 49, 5893–5902. [Google Scholar] [CrossRef]
- Chabert, E.; Vial, J.; Cauchois, J.-P.; Mihaluta, M.; Tournilhac, F. Multiple welding of long fiber epoxy vitrimer composites. Soft Matter 2016, 12, 4838–4845. [Google Scholar] [CrossRef]
- Yoon, J.A.; Kamada, J.; Koynov, K.; Mohin, J.; Nicolaÿ, R.; Zhang, Y.; Balazs, A.C.; Kowalewski, T.; Matyjaszewski, K. Self-Healing Polymer Films Based on Thiol–Disulfide Exchange Reactions and Self-Healing Kinetics Measured Using Atomic Force Microscopy. Macromolecules 2012, 45, 142–149. [Google Scholar] [CrossRef]
- Kolmakov, G.V.; Matyjaszewski, K.; Balazs, A.C. Harnessing Labile Bonds between Nanogel Particles to Create Self-Healing Materials. ACS Nano 2009, 3, 885–892. [Google Scholar] [CrossRef]
- Imbernon, L.; Oikonomou, E.K.; Norvez, S.; Leibler, L. Chemically crosslinked yet reprocessable epoxidized natural rubber via thermo-activated disulfide rearrangements. Polym. Chem. 2015, 6, 4271–4278. [Google Scholar] [CrossRef]
- Spiesschaert, Y.; Guerre, M.; De Baere, I.; van Paepegem, W.; Winne, J.M.; Du Prez, F.E. Dynamic curing agents for amine-hardened epoxy vitrimers with short (re)processing times. Macromolecules 2020, 53, 2485–2495. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Ullah, F.; Othman, M.B.H.; Javed, F.; Ahmad, Z.; Akil, H.M. Classification, processing and application of hydrogels: A review. Mater. Sci. Eng. C 2015, 57, 414–433. [Google Scholar] [CrossRef] [PubMed]
- Jayawarna, V.; Ali, M.; Jowitt, T.A.; Miller, A.F.; Saiani, A.; Gough, J.E.; Ulijn, R.V. Nanostructured hydrogels for three-dimensional cell culture through self-assembly of fluorenylmethoxycarbonyldipeptides. Adv. Mater. 2006, 18, 611–614. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Parente, M.E.; Ochoa Andrade, A.; Ares, G.; Russo, F.; Jimenez-Kairuz, A. Bioadhesive hydrogels for cosmetic applications. Int. J. Cosmet. Sci. 2015, 37, 511–518. [Google Scholar] [CrossRef]
- Bashir, R.; Hilt, J.Z.; Elibol, O.; Gupta, A.; Peppas, N.A. Micromechanical cantilever as an ultrasensitive pH microsensor. Appl. Phys. Lett. 2002, 81, 3091–3093. [Google Scholar] [CrossRef]
- Lin, C.; Gitsov, I. Preparation and Characterization of Novel Amphiphilic Hydrogels with Covalently Attached Drugs and Fluorescent Markers. Macromolecules 2010, 43, 10017–10030. [Google Scholar] [CrossRef]
- Vermonden, T.; Censi, R.; Hennink, W.E. Hydrogels for Protein Delivery. Chem. Rev. 2012, 112, 2853–2888. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Hydrogels for Tissue Engineering. Chem. Rev. 2001, 101, 1869–1879. [Google Scholar] [CrossRef]
- Nguyen, K.T.; West, J.L. Photopolymerizable hydrogels for tissue engineering applications. Biomaterials 2002, 23, 4307–4314. [Google Scholar] [CrossRef]
- Zhu, J.; Marchant, R.E. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devices 2011, 8, 607–626. [Google Scholar] [CrossRef] [PubMed]
- Mohammadinejad, R.; Maleki, H.; Larraneta, E.; Fajardo, A.R.; Nik, A.B.; Shavandi, A.; Sheikhi, A.; Ghorbanpour, M.; Farokhi, M.; Govindh, P.; et al. Status and future scope of plant-based green hydrogels in biomedical engineering. Appl. Mater. Today 2019, 16, 213–246. [Google Scholar] [CrossRef] [Green Version]
- Mitura, S.; Sionkowska, A.; Jaiswal, A. Biopolymers for hydrogels in cosmetics: Review. J. Mater. Sci. Mater. Med. 2020, 31, 50. [Google Scholar] [CrossRef]
- Klein, M.; Poverenov, E. Natural biopolymer-based hydrogels for use in food and agriculture. J. Sci. Food Agric. 2020, 100, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Weiss, R.A. Mechanical behavior of hybrid hydrogels composed of a physical and a chemical network. Polymer 2013, 54, 2174–2182. [Google Scholar] [CrossRef]
- Li, J.; Suo, Z.; Vlassak, J.J. Stiff, strong, and tough hydrogels with good chemical stability. J. Mater. Chem. B 2014, 2, 6708–6713. [Google Scholar] [CrossRef]
- Li, J.; Illeperuma, W.R.K.; Suo, Z.; Vlassak, J.J. Hybrid Hydrogels with Extremely High Stiffness and Toughness. ACS Macro Lett. 2014, 3, 520–523. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, G.; Li, J.; Zeng, S.; Zhang, X.; Zheng, Z.; Ding, X.; Chen, W.; Wang, Q.; Zhang, W. Preparation and self-healing behaviors of poly(acrylic acid)/cerium ions double network hydrogels. Macromol. Res. 2015, 23, 1098–1102. [Google Scholar] [CrossRef]
- Hu, X.; Vatankhah-Varnoosfaderani, M.; Zhou, J.; Li, Q.; Sheiko, S.S. Weak Hydrogen Bonding Enables Hard, Strong, Tough, and Elastic Hydrogels. Adv. Mater. 2015, 27, 6899–6905. [Google Scholar] [CrossRef]
- Hackelbusch, S.; Rossow, T.; Becker, H.; Seiffert, S. Multiresponsive Polymer Hydrogels by Orthogonal Supramolecular Chain Cross-Linking. Macromolecules 2014, 47, 4028–4036. [Google Scholar] [CrossRef]
- Chen, Q.; Yan, X.; Zhu, L.; Chen, H.; Jiang, B.; Wei, D.; Huang, L.; Yang, J.; Liu, B.; Zheng, J. Improvement of Mechanical Strength and Fatigue Resistance of Double Network Hydrogels by Ionic Coordination Interactions. Chem. Mater. 2016, 28, 5710–5720. [Google Scholar] [CrossRef]
- Jia, H.; Huang, Z.; Fei, Z.; Dyson, P.J.; Zheng, Z.; Wang, X. Unconventional Tough Double-Network Hydrogels with Rapid Mechanical Recovery, Self-Healing, and Self-Gluing Properties. ACS Appl. Mater. Interfaces 2016, 8, 31339–31347. [Google Scholar] [CrossRef] [PubMed]
- Brassinne, J.; Gohy, J.-F.; Fustin, C.-A. Orthogonal Control of the Dynamics of Supramolecular Gels from Heterotelechelic Associating Polymers. ACS Macro Lett. 2016, 5, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Niu, R.; Tang, C.; Xia, J.; Ji, F.; Dong, D.; Zhang, H.; Zhang, S.; Li, J.; Yao, F. A Dual-crosslinked strategy to construct physical hydrogels with high strength, toughness, good mechanical recoverability, and shape-memory ability. Macromol. Mater. Eng. 2018, 303, 1700396. [Google Scholar] [CrossRef]
- Guo, Z.; Gu, H.; He, Y.; Zhang, Y.; Xu, W.; Zhang, J.; Liu, Y.; Xiong, L.; Chen, A.; Feng, Y. Dual dynamic bonds enable biocompatible and tough hydrogels with fast self-recoverable, self-healable and injectable properties. Chem. Eng. J. 2020, 388, 124282. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, F.; Li, Z.; Lin, S.; Chen, L.; Liu, L.; Chen, Y. Hydrogel Cross-Linked with Dynamic Covalent Bonding and Micellization for Promoting Burn Wound Healing. ACS Appl. Mater. Interfaces 2018, 10, 25194–25202. [Google Scholar] [CrossRef]
- Guo, Z.; Ma, W.; Gu, H.; Feng, Y.; He, Z.; Chen, Q.; Mao, X.; Zhang, J.; Zheng, L. pH-Switchable and self-healable hydrogels based on ketone type acylhydrazone dynamic covalent bonds. Soft Matter 2017, 13, 7371–7380. [Google Scholar] [CrossRef]
- Guo, R.; Su, Q.; Zhang, J.; Dong, A.; Lin, C.; Zhang, J. Facile Access to Multisensitive and Self-Healing Hydrogels with Reversible and Dynamic Boronic Ester and Disulfide Linkages. Biomacromolecules 2017, 18, 1356–1364. [Google Scholar] [CrossRef]
- Chen, J.; Su, Q.; Guo, R.; Zhang, J.; Dong, A.; Lin, C.; Zhang, J. A Multitasking Hydrogel Based on Double Dynamic Network with Quadruple-Stimuli Sensitiveness, Autonomic Self-Healing Property, and Biomimetic Adhesion Ability. Macromol. Chem. Phys. 2017, 218, 1700166. [Google Scholar] [CrossRef]
- Aleman, J.; Chadwick, A.V.; He, J.; Hess, M.; Horie, K.; Jones, R.G.; Kratochvil, P.; Meisel, I.; Mita, I.; Moad, G.; et al. Definitions of terms relating to the structure and processing of sols, gels, networks, and inorganic-organic hybrid materials. Pure Appl. Chem. 2007, 79, 1801–1829. [Google Scholar] [CrossRef]
- Dragan, E.S. Design and applications of interpenetrating polymer network hydrogels. A review. Chem. Eng. J. 2014, 243, 572–590. [Google Scholar] [CrossRef]
- Ducrot, E.; Chen, Y.; Bulters, M.; Sijbesma, R.P.; Creton, C. Toughening Elastomers with Sacrificial Bonds and Watching Them Break. Science 2014, 344, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Asmarandei, I.; Fundueanu, G.; Cristea, M.; Harabagiu, V.; Constantin, M. Thermo- and pH-sensitive interpenetrating poly(N-isopropylacrylamide)/carboxymethyl pullulan network for drug delivery. J. Polym. Res. 2013, 20, 1–13. [Google Scholar] [CrossRef]
- Yang, J.; Ma, M.; Zhang, X.; Xu, F. Elucidating Dynamics of Precoordinated Ionic Bridges as Sacrificial Bonds in Interpenetrating Network Hydrogels. Macromolecules 2016, 49, 4340–4348. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, F.; Li, M.; Wang, E. pH switching on-off semi-IPN hydrogel based on crosslinked poly(acrylamide-co-acrylic acid) and linear polyallyamine. Polymer 2005, 46, 7695–7700. [Google Scholar] [CrossRef]
- Wu, W.; Wang, D.-S. A fast pH-responsive interpenetrating polymer network hydrogel: Synthesis and controlled drug delivery. React. Funct. Polym. 2010, 70, 684–691. [Google Scholar] [CrossRef]
- Xing, Z.; Wang, C.; Yan, J.; Zhang, L.; Li, L.; Zha, L. Dual stimuli responsive hollow nanogels with IPN structure for temperature controlling drug loading and pH triggering drug release. Soft Matter 2011, 7, 7992–7997. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, J.; Han, J.; Zhu, Y.; Huang, H.; Li, J.; Tang, B. Two-way shape memory polymer with “switch-spring” composition by interpenetrating polymer network. J. Mater. Chem. A 2014, 2, 18816–18822. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Gong, J.P. Why are double network hydrogels so tough? Soft Matter 2010, 6, 2583–2590. [Google Scholar] [CrossRef]
- Haque, M.A.; Kurokawa, T.; Gong, J.P. Super tough double network hydrogels and their application as biomaterials. Polymer 2012, 53, 1805–1822. [Google Scholar] [CrossRef]
- Gong, J.P. Materials both tough and soft. Science 2014, 344, 161–162. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dynamic Bonds | Triggers | Polymeric Systems | Imparted Functions |
|---|---|---|---|
| Diels-Alder cycloadduct | T | PURs [36], SMPs [37], hydrogels [38], IPNs [39], combined networks [40] | reshapeability, enhanced mechanical properties, self-healing ability, facilitated synthesis, structural stability, shape memory |
| Disulfide | redox, light, T, pH | PURs [41], SMPs [42], vitrimers [43], hydrogels [44], IPNs [45] | reshapeability, self-healing ability, enhanced mechanical properties, responsiveness, facilitated synthesis, shape memory |
| Acylhydrazone | pH | supramolecular polymers [46], hydrogels [44], IPNs [47] | reshapeability, self-healing ability, responsiveness, facilitated synthesis, injectability |
| Oxime | T, pH, addition of molecules | hydrogels [48] | structural stability, self-healing ability, enhanced mechanical properties |
| Imine | T, pH, addition of molecules | SMPs [49], vitrimers [50], combined networks [51] | reshapeability, self-healing ability, enhanced mechanical properties, facilitated synthesis |
| Urethane/Urea | T, catalysts | SMPs [52], vitrimers [53] | reshapeability, enhanced mechanical properties, facilitated synthesis |
| Ester | T, catalysts | SMPs [54], vitrimers [54] | reshapeability, facilitated synthesis, shape memory |
| Boronic ester | T, pH, addition of molecules | SMPs [55], vitrimers [56], hydrogels [48], IPNs [47], combined networks [51] | reshapeability, self-healing ability, enhanced mechanical properties, responsiveness, facilitated synthesis |
| C–C (scission) | mechanical force, T | PURs [57] | self-healing ability, responsiveness, enhanced mechanical properties |
| Cycloadduct of cinnamic acid derivatives | light | SMPs [58] | shape memory |
| Allyl sulfide and trithiocarbonate | T, radicals, catalysts | SMPs [59], vitrimers [29] | reshapeability, self-healing ability, facilitated synthesis |
| Physical Interactions | Combined DCBs | Polymeric Systems | Imparted Functions |
|---|---|---|---|
| Hydrogen bonds | acylhydrazone [46,60], C–C [57,61], DA cyclo-adduct [36,39,62,63], disulfide [41,64,65,66], imine [49], olefin [67], ester [68] | supramolecular polymers, PURs, SMPs, vitrimers, hydrogels, IPNs | self-healing ability, enhanced mechanical properties |
| Metal-ligand coordination | boronic ester [69], imine [70], oxime [71], ester [72] | SMPs, vitrimers, hydrogels | structural stability, enhanced mechanical properties, creep, solvent, and acid resistance |
| Liquid crystals | allyl sulfide [59], boronic ester [55], disulfide [73,74,75], carbamate [76], ester [54,77,78,79,80,81,82] | SMPs | shape memory |
| Microphase separation | anhydride [83], DA cycloadduct [37,84,85,86,87,88], imine [49], disulfide [89,90,91], urethane/urea [52,92,93], ester [72,77,94,95,96], boronic esters [97,98] | SMPs, vitrimers | shape memory, self-healing ability |
| Self-assemblies | acylhydrazone [99], ester [100] | vitrimers, hydrogels | self-healing ability, enhanced mechanical properties |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammer, L.; Van Zee, N.J.; Nicolaÿ, R. Dually Crosslinked Polymer Networks Incorporating Dynamic Covalent Bonds. Polymers 2021, 13, 396. https://doi.org/10.3390/polym13030396
Hammer L, Van Zee NJ, Nicolaÿ R. Dually Crosslinked Polymer Networks Incorporating Dynamic Covalent Bonds. Polymers. 2021; 13(3):396. https://doi.org/10.3390/polym13030396
Chicago/Turabian StyleHammer, Larissa, Nathan J. Van Zee, and Renaud Nicolaÿ. 2021. "Dually Crosslinked Polymer Networks Incorporating Dynamic Covalent Bonds" Polymers 13, no. 3: 396. https://doi.org/10.3390/polym13030396
APA StyleHammer, L., Van Zee, N. J., & Nicolaÿ, R. (2021). Dually Crosslinked Polymer Networks Incorporating Dynamic Covalent Bonds. Polymers, 13(3), 396. https://doi.org/10.3390/polym13030396