
Strategies for the Development of pH-Responsive Synthetic Polypeptides and Polymer-Peptide Hybrids: Recent Advancements

 , and
, and 
Abstract
:1. Introduction
2. Overview of pH-Responsive Polypeptide and Polymer-Peptide Hybrid Preparation
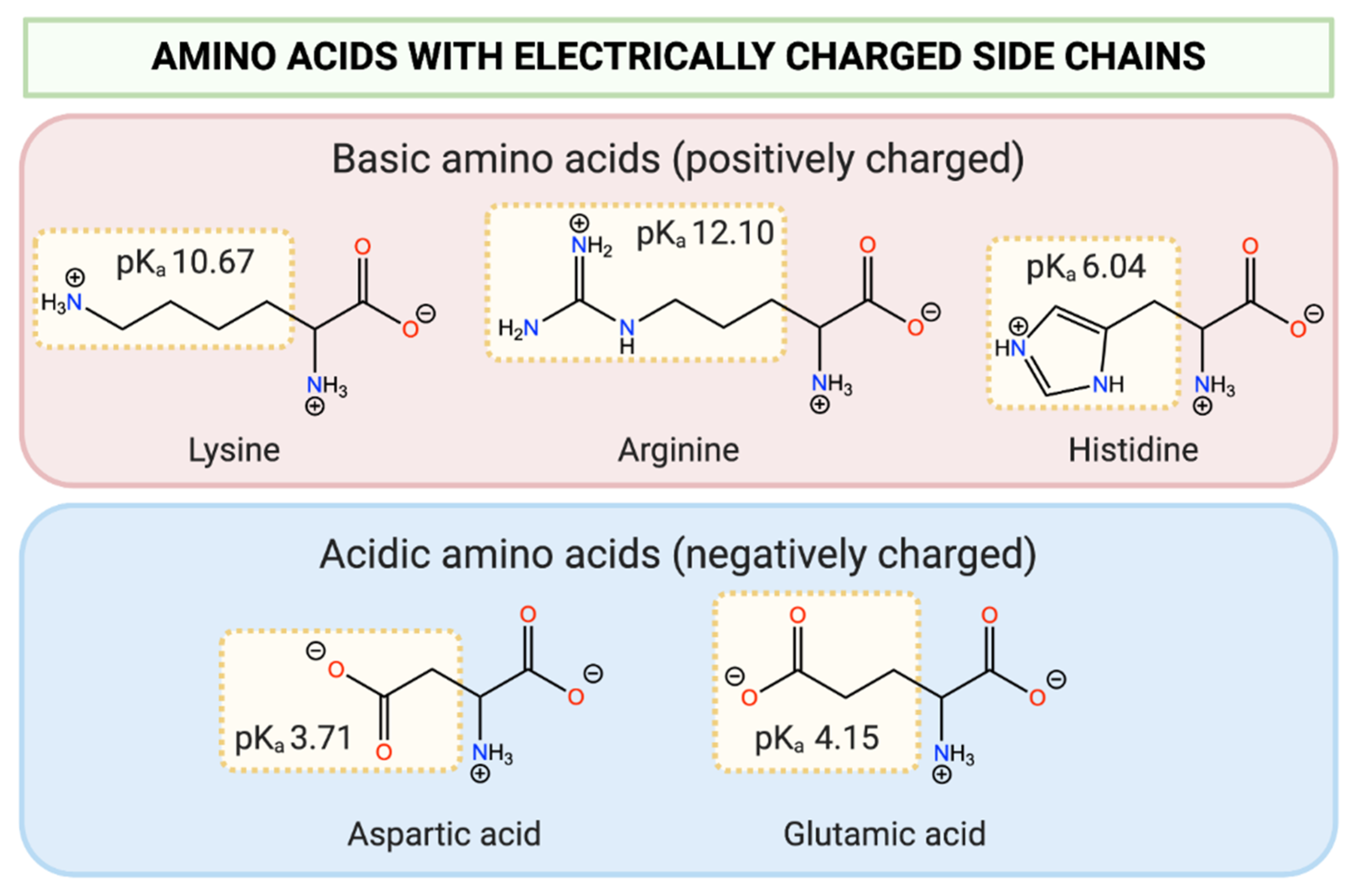
2.1. Ionisation-Induced pH-Responsivity
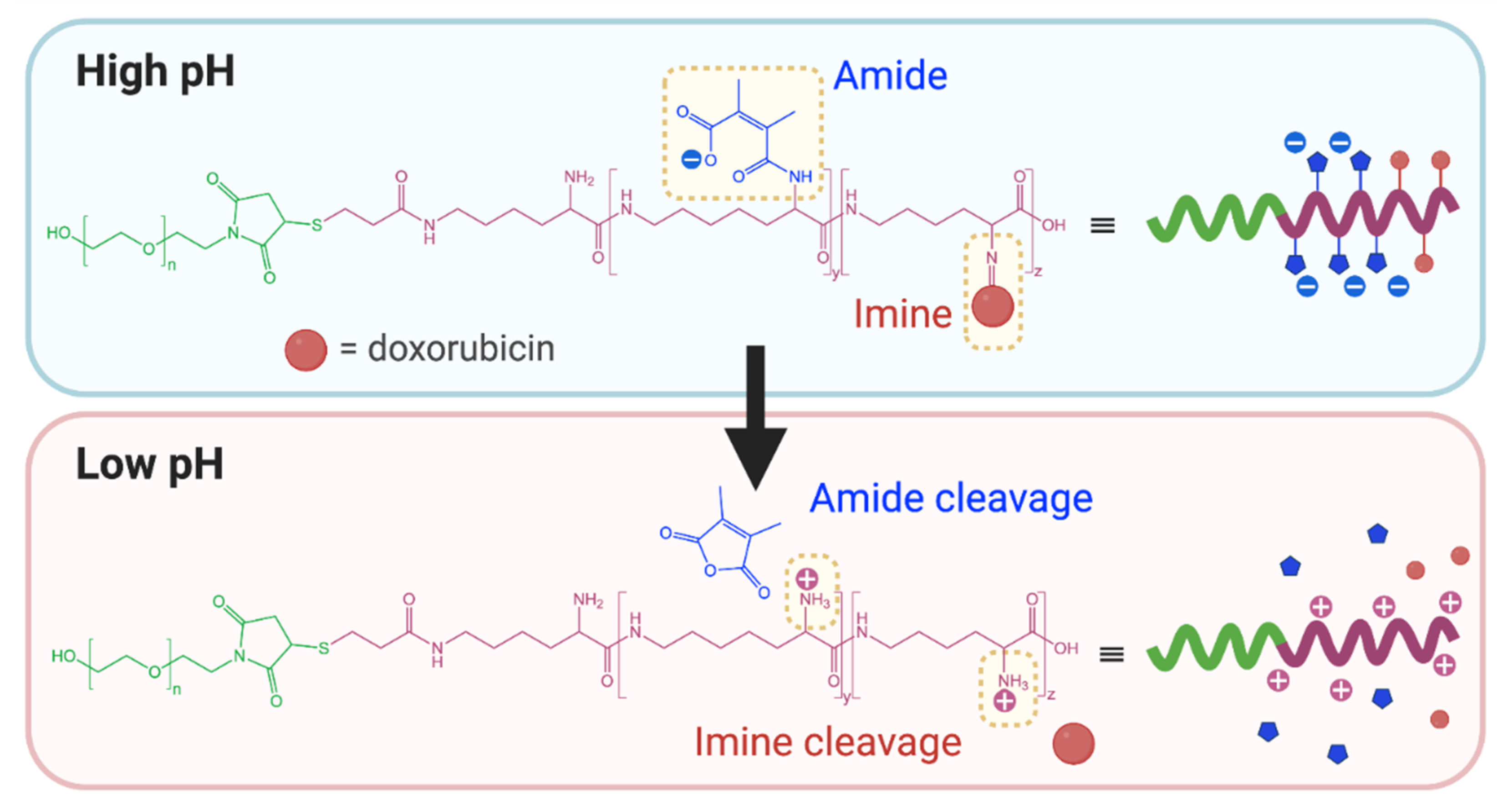
2.2. pH-Responsivity Triggered by Functional Group Cleavage
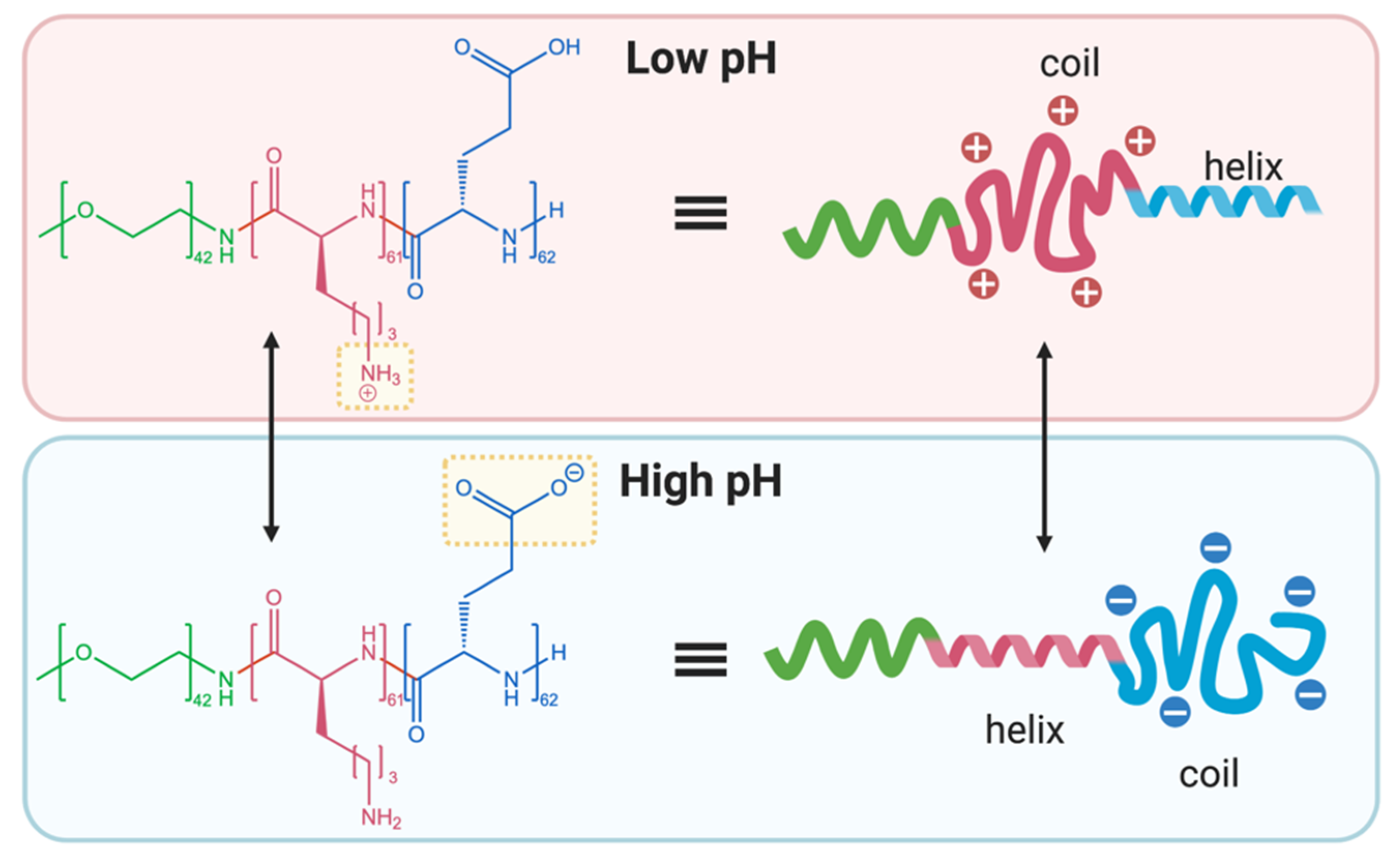
2.3. pH-Responsivity Triggered by Changes to Polypeptide Secondary Structure
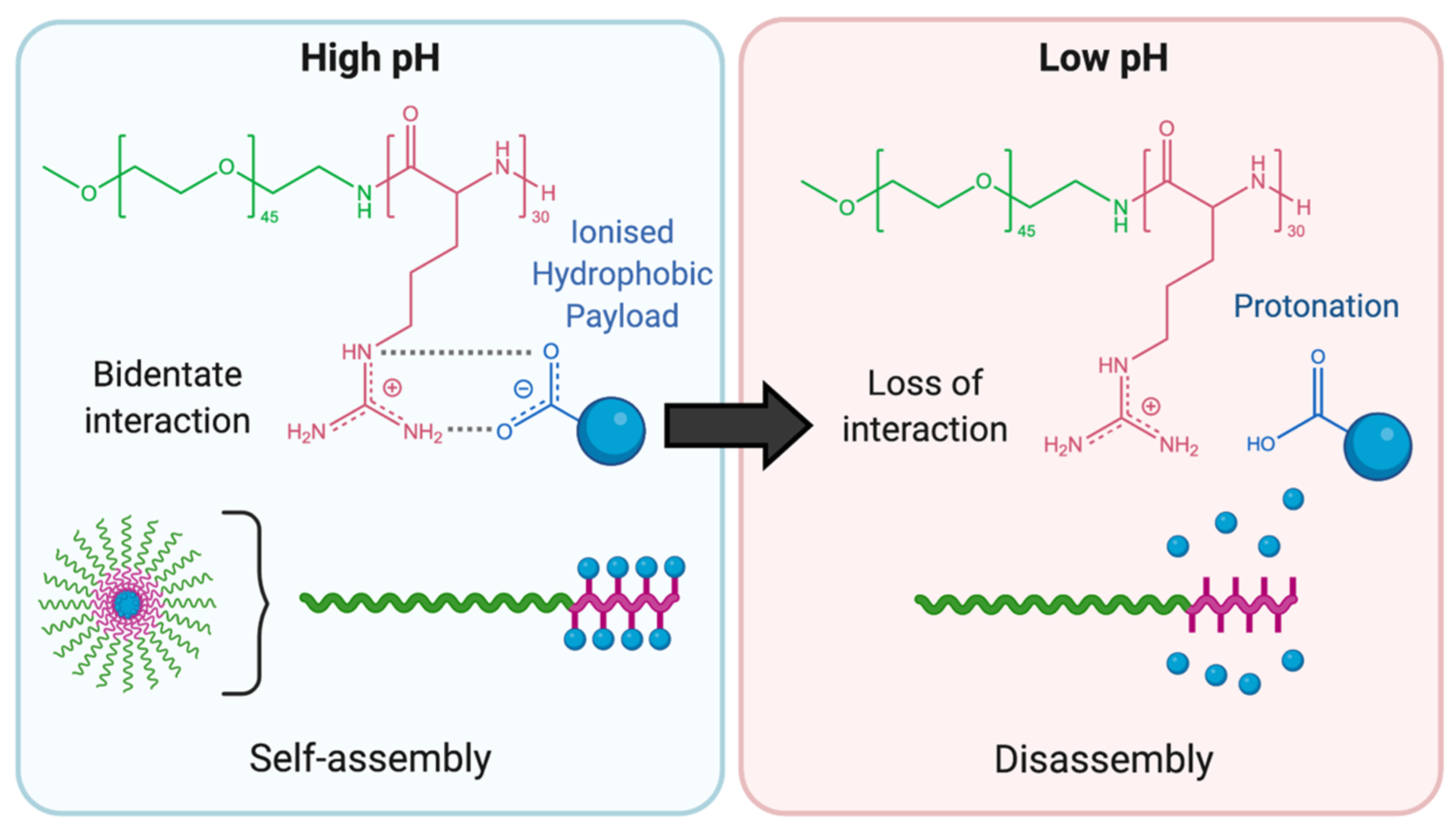
2.4. pH-Responsivity Triggered by Supramolecular Interactions
3. Conclusions and Future Perspectives
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dupre, J.; Ross, S.A.; Watson, D.; Brown, J.C. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 1973, 37, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.L.; Shelton, R.L.; Athar, S. The osmoregulation of vasopressin. Kidney Int. 1976, 10, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.J.; Sulistio, A.; Ladewig, K.; Wong, E.H.H.; Blencowe, A.; Qiao, G.G. Peptide-based star polymers as potential siRNA carriers. Aust. J. Chem. 2014, 67, 592–597. [Google Scholar] [CrossRef]
- Lam, S.J.; Wong, E.H.H.; O’Brien-Simpson, N.M.; Pantarat, N.; Blencowe, A.; Reynolds, E.C.; Qiao, G.G. Bionano interaction study on antimicrobial star-shaped peptide polymer nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 33446–33456. [Google Scholar] [CrossRef] [PubMed]
- Sulistio, A.; Gurr, P.A.; Blencowe, A.; Qiao, G.G. Peptide-based star polymers: The rising star in functional polymers. Aust. J. Chem. 2012, 65, 978–984. [Google Scholar] [CrossRef]
- Johnson, R.P.; Uthaman, S.; John, J.V.; Lee, H.R.; Lee, S.J.; Park, H.; Park, I.-K.; Suh, H.; Kim, I. Poly(PEGA)-b-poly(l-lysine)-b-poly(l-histidine) hybrid vesicles for tumoral pH-triggered intracellular delivery of doxorubicin hydrochloride. ACS Appl. Mater. Interfaces 2015, 7, 21770–21779. [Google Scholar] [CrossRef]
- Kataoka, K.; Matsumoto, T.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Fukushima, S.; Okamoto, K.; Kwon, G.S. Doxorubicin-loaded poly(ethylene glycol)–poly(β-benzyl-l-aspartate) copolymer micelles: Their pharmaceutical characteristics and biological significance. J. Control. Release 2000, 64, 143–153. [Google Scholar] [CrossRef]
- Zhang, P.; Gao, Z.; Cui, J.; Hao, J. Dual-stimuli-responsive polypeptide nanoparticles for photothermal and photodynamic therapy. ACS Appl. Bio Mater. 2020, 3, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Deming, T.J. Synthetic polypeptides for biomedical applications. Prog. Polym. Sci. 2007, 32, 858–875. [Google Scholar] [CrossRef]
- Song, Z.; Tan, Z.; Cheng, J. Recent advances and future perspectives of synthetic polypeptides from N-carboxyanhydrides. Macromolecules 2019, 52, 8521–8539. [Google Scholar] [CrossRef]
- Lim, Z.W.; Varma, V.B.; Ramanujan, R.V.; Miserez, A. Magnetically responsive peptide coacervates for dual hyperthermia and chemotherapy treatments of liver cancer. Acta Biomater. 2020, 110, 221–230. [Google Scholar] [CrossRef]
- Lin, W.; Ma, G.; Yuan, Z.; Qian, H.; Xu, L.; Sidransky, E.; Chen, S. Development of zwitterionic polypeptide nanoformulation with high doxorubicin loading content for targeted drug delivery. Langmuir 2019, 35, 1273–1283. [Google Scholar] [CrossRef]
- Deming, T.J. Polypeptide and Polypeptide Hybrid Copolymer Synthesis via NCA Polymerization. In Peptide Hybrid Polymers; Klok, H.-A., Schlaad, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1–18. [Google Scholar] [CrossRef]
- Schlaad, H.; Antonietti, M. Block copolymers with amino acid sequences: Molecular chimeras of polypeptides and synthetic polymers. Eur. Phys. J. E 2003, 10, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Rasines Mazo, A.; Allison-Logan, S.; Karimi, F.; Chan, N.J.-A.; Qiu, W.; Duan, W.; O’Brien-Simpson, N.M.; Qiao, G.G. Ring opening polymerization of α-amino acids: Advances in synthesis, architecture and applications of polypeptides and their hybrids. Chem. Soc. Rev. 2020, 49, 4737–4834. [Google Scholar] [CrossRef]
- Deng, C.; Wu, J.; Cheng, R.; Meng, F.; Klok, H.-A.; Zhong, Z. Functional polypeptide and hybrid materials: Precision synthesis via α-amino acid N-carboxyanhydride polymerization and emerging biomedical applications. Prog. Polym. Sci. 2014, 39, 330–364. [Google Scholar] [CrossRef]
- Jing, P.; Rudra, J.S.; Herr, A.B.; Collier, J.H. Self-assembling peptide-polymer hydrogels designed from the coiled coil region of fibrin. Biomacromolecules 2008, 9, 2438–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühnle, R.I.; Gebauer, D.; Börner, H.G. Calcium ions as bioinspired triggers to reversibly control the coil-to-helix transition in peptide-polymer conjugates. Soft Matter 2011, 7, 9616–9619. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Fu, H.; Wang, R.; Pacheco, L.A.; Wang, X.; Lin, Y.; Cheng, J. Secondary structures in synthetic polypeptides from N-carboxyanhydrides: Design, modulation, association, and material applications. Chem. Soc. Rev. 2018, 47, 7401–7425. [Google Scholar] [CrossRef]
- Yang, C.; Shi, Z.; Feng, C.; Li, R.; Luo, S.; Li, X.; Ruan, L. An adjustable pH-responsive drug delivery system based on self-assembly polypeptide-modified mesoporous silica. Macromol. Biosci. 2020, 20, 2000034. [Google Scholar] [CrossRef]
- Zhang, W.; Garg, S.; Eldi, P.; Zhou, F.H.-h.; Johnson, I.R.D.; Brooks, D.A.; Lam, F.; Rychkov, G.; Hayball, J.; Albrecht, H. Targeting prostate cancer cells with genetically engineered polypeptide-based micelles displaying gastrin-releasing peptide. Int. J. Pharm. 2016, 513, 270–279. [Google Scholar] [CrossRef]
- Saiki, I.; Murata, J.; Iida, J.; Sakurai, T.; Nishi, N.; Matsuno, K.; Azuma, I. Antimetastatic effects of synthetic polypeptides containing repeated structures of the cell adhesive Arg-Gly-Asp (RGD) and Tyr-Ile-Gly-Ser-Arg (YIGSR) sequences. Br. J. Cancer 1989, 60, 722–728. [Google Scholar] [CrossRef]
- Dathe, M.; Schümann, M.; Wieprecht, T.; Winkler, A.; Beyermann, M.; Krause, E.; Matsuzaki, K.; Murase, O.; Bienert, M. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry 1996, 35, 12612–12622. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Poon, Z.; Engler, A.C.; Bonner, D.K.; Hammond, P.T. Enhanced stability of polymeric micelles based on postfunctionalized poly(ethylene glycol)-b-poly(γ-propargyl l-glutamate): The substituent effect. Biomacromolecules 2012, 13, 1315–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, J.R.; Deming, T.J. Glycopolypeptides via living polymerization of glycosylated-l-lysine N-carboxyanhydrides. J. Am. Chem. Soc. 2010, 132, 15068–15071. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Schlaad, H. Thiol−ene clickable polypeptides. Macromolecules 2010, 43, 4445–4448. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, D. General route toward side-chain-functionalized α-helical polypeptides. Biomacromolecules 2010, 11, 1585–1592. [Google Scholar] [CrossRef]
- Guo, J.; Huang, Y.; Jing, X.; Chen, X. Synthesis and characterization of functional poly(γ-benzyl-l-glutamate) (PBLG) as a hydrophobic precursor. Polymer 2009, 50, 2847–2855. [Google Scholar] [CrossRef]
- Matsumura, Y. Preclinical and clinical studies of NK012, an SN-38-incorporating polymeric micelles, which is designed based on EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 184–192. [Google Scholar] [CrossRef]
- Matsumura, Y.; Hamaguchi, T.; Ura, T.; Muro, K.; Yamada, Y.; Shimada, Y.; Shirao, K.; Okusaka, T.; Ueno, H.; Ikeda, M.; et al. Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin. Br. J. Cancer 2004, 91, 1775–1781. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387. [Google Scholar]
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Li, Z. Thermoresponsive polypeptides from pegylated poly-l-glutamates. Biomacromolecules 2011, 12, 2859–2863. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhuang, X.; Xiao, C.; Cheng, Y.; Zhao, L.; He, C.; Tang, Z.; Chen, X. Preparation of photo-cross-linked pH-responsive polypeptide nanogels as potential carriers for controlled drug delivery. J. Mater. Chem. 2011, 21, 11383–11391. [Google Scholar] [CrossRef]
- Zhao, S.; Zhu, H.; Chen, Z.; Shuai, S.; Zhang, N.; Liu, Y.; Rao, Z.; Li, Y.; Zhao, C.; Zhou, K.; et al. Preparation and properties of a temperature- and pH- responsive polypeptide hydrogel. Mater. Res. Express 2019, 6, 085711. [Google Scholar] [CrossRef]
- Kim, J.H.; Li, Y.; Kim, M.S.; Kang, S.W.; Jeong, J.H.; Lee, D.S. Synthesis and evaluation of biotin-conjugated pH-responsive polymeric micelles as drug carriers. Int. J. Pharm. 2012, 427, 435–442. [Google Scholar] [CrossRef]
- Li, K.; Li, D.; Zhao, L.; Chang, Y.; Zhang, Y.; Cui, Y.; Zhang, Z. Calcium-mineralized polypeptide nanoparticle for intracellular drug delivery in osteosarcoma chemotherapy. Bioact. Mater. 2020, 5, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Min, K.H.; Kim, J.-H.; Bae, S.M.; Shin, H.; Kim, M.S.; Park, S.; Lee, H.; Park, R.-W.; Kim, I.-S.; Kim, K.; et al. Tumoral acidic pH-responsive MPEG-poly(β-amino ester) polymeric micelles for cancer targeting therapy. J. Control. Release 2010, 144, 259–266. [Google Scholar] [CrossRef]
- He, Q.; Chen, J.; Yan, J.; Cai, S.; Xiong, H.; Liu, Y.; Peng, D.; Mo, M.; Liu, Z. Tumor microenvironment responsive drug delivery systems. Asian J. Pharm. Sci. 2020, 15, 416–448. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbs, M.; McSheehy, P.M.J.; Griffiths, J.R.; Bashford, C.L. Causes and consequences of tumour acidity and implications for treatment. Mol. Med. Today 2000, 6, 15–19. [Google Scholar] [CrossRef]
- Kim, D.; Lee, E.S.; Oh, K.T.; Gao, Z.G.; Bae, Y.H. Doxorubicin-loaded polymeric micelle overcomes multidrug resistance of cancer by double-targeting folate receptor and early endosomal pH. Small 2008, 4, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- Kongkatigumjorn, N.; Cortez-Jugo, C.; Czuba, E.; Wong, A.S.M.; Hodgetts, R.Y.; Johnston, A.P.R.; Such, G.K. Probing endosomal escape using pHlexi nanoparticles. Macromol. Biosci. 2017, 17, 1600248. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef]
- Iversen, T.-G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Jiang, X.; Dausend, J.; Hafner, M.; Musyanovych, A.; Röcker, C.; Landfester, K.; Mailänder, V.; Nienhaus, G.U. Specific effects of surface amines on polystyrene nanoparticles in their interactions with mesenchymal stem cells. Biomacromolecules 2010, 11, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, K.; Huang, G.; Hensley, C.; Huang, X.; Ma, X.; Zhao, T.; Sumer, B.D.; DeBerardinis, R.J.; Gao, J. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat. Mater. 2014, 13, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Ninan, N.; Forget, A.; Shastri, V.P.; Voelcker, N.H.; Blencowe, A. Antibacterial and anti-inflammatory pH-responsive tannic acid-carboxylated agarose composite hydrogels for wound healing. ACS Appl. Mater. Interfaces 2016, 8, 28511–28521. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Si, X.; Ding, X.; Duan, L.; Xiao, C. pH-responsive hydrogels based on the self-assembly of short polypeptides for controlled release of peptide and protein drugs. J. Polym. Res. 2019, 26, 278. [Google Scholar] [CrossRef]
- Park, J.; Choi, Y.; Chang, H.; Um, W.; Ryu, J.H.; Kwon, I.C. Alliance with EPR effect: Combined strategies to improve the EPR effect in the tumor microenvironment. Theranostics 2019, 9, 8073–8090. [Google Scholar] [CrossRef]
- Lide, D.R. Handbook of Chemistry and Physics, 72nd ed.; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Wang, P.; Liu, W.; Liu, S.; Yang, R.; Pu, Y.; Zhang, W.; Wang, X.; Liu, X.; Ren, Y.; Chi, B. pH-responsive nanomicelles of poly(ethylene glycol)-poly(ε-caprolactone)-poly(L-histidine) for targeted drug delivery. J. Biomater. Sci. Polym. Ed. 2020, 31, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Lei, S.; Chang, L.; Wan, D. Smart pH-responsive nanoparticles in a model tumor microenvironment for enhanced cellular uptake. J. Mater. Sci. 2019, 54, 1692–1702. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, J.; Huang, Z.; Chen, Y.; Pan, S.; Hu, H.; Qiao, M.; Chen, D.; Zhao, X. Stimuli-responsive release and efficient siRNA delivery in non-small cell lung cancer by a poly(l-histidine)-based multifunctional nanoplatform. J. Mater. Chem. B 2020, 8, 1616–1628. [Google Scholar] [CrossRef] [PubMed]
- Sattari, S.; Dadkhah Tehrani, A.; Adeli, M. pH-responsive hybrid hydrogels as antibacterial and drug delivery systems. Polymers 2018, 10, 660. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Zhang, P.; Huang, C.; Song, Y.; Garg, S.; Luan, Y. Co-delivery of doxorubicin hydrochloride and verapamil hydrochloride by pH-sensitive polymersomes for the reversal of multidrug resistance. RSC Adv. 2015, 5, 77986–77995. [Google Scholar] [CrossRef]
- Li, Z.; Li, J.; Huang, J.; Zhang, J.; Cheng, D.; Shuai, X. Synthesis and characterization of pH-responsive copolypeptides vesicles for siRNA and chemotherapeutic drug co-delivery. Macromol. Biosci. 2015, 15, 1497–1506. [Google Scholar] [CrossRef]
- Xiao, Y.; Tang, C.; Chen, Y.; Lang, M. Dual stimuli-responsive polypeptide prepared by thiol-ene click reaction of poly(l-cysteine) and N, N-dimethylaminoethyl acrylate. Biopolymers 2019, 110, e23318. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.C.; Bonner, D.K.; Buss, H.G.; Cheung, E.Y.; Hammond, P.T. The synthetic tuning of clickable pH responsive cationic polypeptides and block copolypeptides. Soft Matter 2011, 7, 5627–5637. [Google Scholar] [CrossRef]
- Guo, Z.; Sui, J.; Ma, M.; Hu, J.; Sun, Y.; Yang, L.; Fan, Y.; Zhang, X. pH-Responsive charge switchable PEGylated ε-poly-l-lysine polymeric nanoparticles-assisted combination therapy for improving breast cancer treatment. J. Control. Release 2020, 326, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, J.; Zhang, Y.; Xiao, C.; Zhuang, X.; Chen, X. Polyion complex micelles with gradient pH-sensitivity for adjustable intracellular drug delivery. Polym. Chem. 2015, 6, 397–405. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, Z.; Guo, J.; Hao, J.; Zhang, P.; Cui, J. Polypeptide nanoparticles with pH-sheddable PEGylation for improved drug delivery. Langmuir 2020, 36, 13656–13662. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-S.; Li, Z.-Y.; Zhu, J.-Y.; Han, K.; Zeng, Z.-Y.; Hong, W.; Li, W.-X.; Jia, H.-Z.; Liu, Y.; Zhuo, R.-X.; et al. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small 2015, 11, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Tao, A.; Huang, G.L.; Igarashi, K.; Hong, T.; Liao, S.; Stellacci, F.; Matsumoto, Y.; Yamasoba, T.; Kataoka, K.; Cabral, H. Polymeric micelles loading proteins through concurrent ion complexation and pH-cleavable covalent bonding for in vivo delivery. Macromol. Biosci. 2020, 20, 1900161. [Google Scholar] [CrossRef]
- Liu, N.; Li, B.; Gong, C.; Liu, Y.; Wang, Y.; Wu, G. A pH- and thermo-responsive poly(amino acid)-based drug delivery system. Colloids Surf. B 2015, 136, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Shan, M.; Di, X.; Gong, C.; Zhang, L.; Wang, Y.; Wu, G. A dual pH- and reduction-responsive anticancer drug delivery system based on PEG–SS–poly(amino acid) block copolymer. RSC Adv. 2017, 7, 30242–30249. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Z.; Liu, L.; Jiang, W.; Li, S.; Wang, Y.; Yan, L. NIR imaging-guided combined photodynamic therapy and chemotherapy by a pH-responsive amphiphilic polypeptide prodrug. Biomater. Sci. 2017, 5, 313–321. [Google Scholar] [CrossRef]
- Arroyo-Crespo, J.J.; Armiñán, A.; Charbonnier, D.; Balzano-Nogueira, L.; Huertas-López, F.; Martí, C.; Tarazona, S.; Forteza, J.; Conesa, A.; Vicent, M.J. Tumor microenvironment-targeted poly-L-glutamic acid-based combination conjugate for enhanced triple negative breast cancer treatment. Biomaterials 2018, 186, 8–21. [Google Scholar] [CrossRef]
- Takemoto, H.; Inaba, T.; Nomoto, T.; Matsui, M.; Liu, X.; Toyoda, M.; Honda, Y.; Taniwaki, K.; Yamada, N.; Kim, J.; et al. Polymeric modification of gemcitabine via cyclic acetal linkage for enhanced anticancer potency with negligible side effects. Biomaterials 2020, 235, 119804. [Google Scholar] [CrossRef]
- Zhou, C.; Shi, Z.; Xu, F.; Ling, Y.; Tang, H. Preparation and properties of thermo- and pH-responsive polypeptide bearing OEG and aldehyde pendants. Colloid Polym. Sci. 2020, 298, 1293–1302. [Google Scholar] [CrossRef]
- Lee, S.J.; Min, K.H.; Lee, H.J.; Koo, A.N.; Rim, H.P.; Jeon, B.J.; Jeong, S.Y.; Heo, J.S.; Lee, S.C. Ketal cross-linked poly(ethylene glycol)-poly(amino acid)s copolymer micelles for efficient intracellular delivery of doxorubicin. Biomacromolecules 2011, 12, 1224–1233. [Google Scholar] [CrossRef]
- Rose, G.D.; Wolfenden, R. Hydrogen bonding, hydrophobicity, packing, and protein folding. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 381–415. [Google Scholar] [CrossRef] [PubMed]
- Blout, E.R.; Idelson, M. Polypeptides. VI. Poly-α-L-glutamic acid: Preparation and helix-coil conversions. J. Am. Chem. Soc. 1956, 78, 497–498. [Google Scholar] [CrossRef]
- Parker, R.C.; Applegate, K.; Slutsky, L.J. Ultrasonic study of the helix—coil transition in poly-L-lysine. J. Phys. Chem. 1966, 70, 3018–3019. [Google Scholar] [CrossRef]
- Sun, J.; Černoch, P.; Völkel, A.; Wei, Y.; Ruokolainen, J.; Schlaad, H. Aqueous self-assembly of a protein-mimetic ampholytic block copolypeptide. Macromolecules 2016, 49, 5494–5501. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhang, X.; Liu, X.; Li, J.; Luan, Y. pH-responsive poly(ethylene glycol)-poly(ϵ-caprolactone)-poly(glutamic acid) polymersome as an efficient doxorubicin carrier for cancer therapy. Polym. Int. 2017, 66, 1579–1586. [Google Scholar] [CrossRef]
- Chen, P.; Qiu, M.; Deng, C.; Meng, F.; Zhang, J.; Cheng, R.; Zhong, Z. pH-responsive chimaeric pepsomes based on asymmetric poly(ethylene glycol)-b-poly(l-leucine)-b-poly(l-glutamic acid) triblock copolymer for efficient loading and active intracellular delivery of doxorubicin hydrochloride. Biomacromolecules 2015, 16, 1322–1330. [Google Scholar] [CrossRef]
- Mostoufi, H.; Yousefi, G.; Tamaddon, A.-M.; Firuzi, O. Reversing multi-drug tumor resistance to Paclitaxel by well-defined pH-sensitive amphiphilic polypeptide block copolymers via induction of lysosomal membrane permeabilization. Colloids Surf. B Biointerfaces 2019, 174, 17–27. [Google Scholar] [CrossRef]
- Tinajero-Díaz, E.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. pH-Responsive diblock copolymers made of ω-pentadecalactone and ionically charged α-amino acids. Eur. Polym. J. 2019, 120, 109244. [Google Scholar] [CrossRef]
- Chen, B.-Y.; Huang, Y.-F.; Huang, Y.-C.; Wen, T.-C.; Jan, J.-S. Alkyl chain-grafted poly(l-lysine) vesicles with tunable molecular assembly and membrane permeability. ACS Macro Lett. 2014, 3, 220–223. [Google Scholar] [CrossRef]
- Praveen, K.; Das, S.; Dhaware, V.; Pandey, B.; Mondal, B.; Gupta, S.S. pH-responsive “supra-amphiphilic” nanoparticles based on homoarginine polypeptides. ACS Appl. Bio Mater. 2019, 2, 4162–4172. [Google Scholar] [CrossRef]
- Li, Y.; Lai, Y.; Xu, X.; Zhang, X.; Wu, Y.; Hu, C.; Gu, M.S.Z. Capsid-like supramolecular dendritic systems as pH-responsive nanocarriers for drug penetration and site-specific delivery. Nanomedicine 2016, 12, 355–364. [Google Scholar] [CrossRef]
- Meng, F.; Sun, J.; Li, Z. Stimuli-responsive polypeptide-based supramolecular hydrogels mediated by Ca2+ ion cross-linking. Chin. J. Chem. 2019, 37, 1137–1141. [Google Scholar] [CrossRef]
- Ni, Y.; Sun, J.; Wei, Y.; Fu, X.; Zhu, C.; Li, Z. Two-dimensional supramolecular assemblies from pH-responsive poly(ethyl glycol)-b-poly(l-glutamic acid)-b-poly(N-octylglycine) triblock copolymer. Biomacromolecules 2017, 18, 3367–3374. [Google Scholar] [CrossRef]
- Acharya, A.; Das, I.; Chandhok, D.; Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid Med. Cell Longev. 2010, 3, 23–34. [Google Scholar] [CrossRef]
- Alsuraifi, A.; Curtis, A.; Lamprou, D.A.; Hoskins, C. Stimuli Responsive Polymeric Systems for Cancer Therapy. Pharmaceutics 2018, 10, 136. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dharmayanti, C.; Gillam, T.A.; Klingler-Hoffmann, M.; Albrecht, H.; Blencowe, A. Strategies for the Development of pH-Responsive Synthetic Polypeptides and Polymer-Peptide Hybrids: Recent Advancements. Polymers 2021, 13, 624. https://doi.org/10.3390/polym13040624
Dharmayanti C, Gillam TA, Klingler-Hoffmann M, Albrecht H, Blencowe A. Strategies for the Development of pH-Responsive Synthetic Polypeptides and Polymer-Peptide Hybrids: Recent Advancements. Polymers. 2021; 13(4):624. https://doi.org/10.3390/polym13040624
Chicago/Turabian StyleDharmayanti, Cintya, Todd A. Gillam, Manuela Klingler-Hoffmann, Hugo Albrecht, and Anton Blencowe. 2021. "Strategies for the Development of pH-Responsive Synthetic Polypeptides and Polymer-Peptide Hybrids: Recent Advancements" Polymers 13, no. 4: 624. https://doi.org/10.3390/polym13040624
APA StyleDharmayanti, C., Gillam, T. A., Klingler-Hoffmann, M., Albrecht, H., & Blencowe, A. (2021). Strategies for the Development of pH-Responsive Synthetic Polypeptides and Polymer-Peptide Hybrids: Recent Advancements. Polymers, 13(4), 624. https://doi.org/10.3390/polym13040624