3.1. Surface Morphology and Hydrophilicity of the Thin Films
The thicknesses of the obtained films were in the range 10–20 µm and the surface pH was approximately 6.0 in all cases. Ch had low strength and became wrinkled at the edge after being punched (
Figure 2a). The ChG films (
Figure 2b,c) were white and ChG05B1 and ChG05B5 (
Figure 2d,e) were yellowish as a result of the addition of BZ. ChG05B5 (
Figure 2e) was translucent at some areas. FE-SEM images confirmed the interconnected pores structure on the thin film surfaces (
Figure 3). The Ch surfaces had a chitosan nanofiber aggregate structure with densely packed nanosized-fine fibers (
Figure 3a). The addition of GPTMS led to the formation of larger pores with hundreds nanometer-scale diameter (
Figure 3b,d). The size of the nanofiber bundles was approximately 100 nm and they contained 2–5 fibers. The porous structures were maintained when BZ was added (
Figure 3e). However, the excess BZ covered the thin film surfaces (ChG05B5) and filled the pores (
Figure 3f). The translucent appearance of ChG05B5 (
Figure 2e) is attributed to the deposition of BZ on the thin film surfaces.
The contact angles (
Table 2) decreased in the order: Ch (91.9 ± 9.8°) > ChG01 (80.8 ± 4.9°) > ChG05 (76.4 ± 2.0°) > ChG10 (71.1 ± 3.6°), indicating that the addition of GPTMS improved the wettability of the films. The addition of BZ reduced the contact angle with water. BZ was expected to be easily released under wet conditions. The specific BET surface areas of the thin films were in the range 11.0 m
2 g
−1 to 47.8 m
2 g
−1 (
Table 3). The water vapor adsorption and desorption isotherms of the films (
Figure 4) at 298 K exhibited type II shape, according to the standard classification of the International Union of Pure and Applied Chemistry (IUPAC) [
22]. The water vapor adsorption and desorption curves did not coincide with the tendency of wettability and all films showed hysteresis behavior at the high relative pressure. This type II shape suggests that the thin films had pores that were greater than 50 nm in diameter and that the pores filled with the multilayers of adsorbed water at high relative pressure. The desorption curve was above the adsorption curve and the hysteresis loop did not closed (low pressure hysteresis) [
23]. This indicates that the desorption of water was more difficult than adsorption owing to the capillary forces. The amount of water adsorption was lower for ChG01 and ChG05 than for Ch and ChG10.
3.2. Microstructure and Mechanical Properties of the Thin Films
Dropping water onto the thin films caused Ch to immediately shrink, while the ChG series showed no change in appearance, even when the amount of GPTMS added was small (
Figure 5 and
Video S1 and
Video S2). The addition of BZ led to a slight recovery of the instability toward water. The amount of free amino groups in the films was estimated using the ninhydrin method (
Figure 6). The number of free amino groups was not proportional to the amount of GPTMS. The results suggest that the percentage of free amino groups was 30% in ChG01 (chitosan/GPTMS = 0.1 molar ratio), 20% in ChG05 (chitosan/GPTMS = 0.5 molar ratio) and 10% in ChG10 (chitosan/GPTMS = 1.0 molar ratio). The presence of positively charged amino groups and negatively –Si–O
− groups favors electrostatic interactions [
14,
15,
16,
17]. Therefore, in ChG01 and ChG05 chitosan and GPTMS interacted through ring-opening reactions and electrostatic interactions. In case of ChG01, 10% is from the ring-opening and 60% is from the ionic interaction. In ChG01, polycondensation between Si–OH groups is less likely than for the other films because of the small amount of GPTMS and the remaining Si–OH groups can interact with the amino groups. As the amount of GPTMS added increased, the polycondensation of Si–OH groups led to the formation of Si–O–Si networks; therefore, the electrostatic interaction decreased, but the degree of epoxy ring-opening increased.
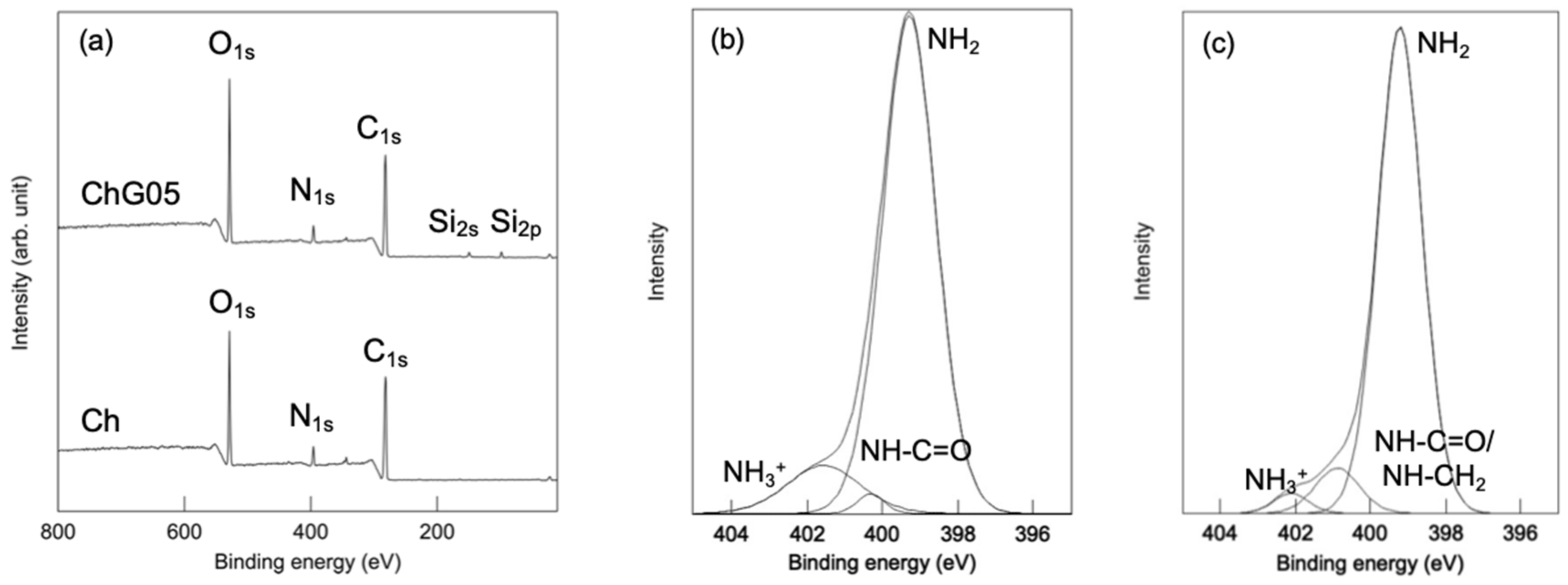
The wide scan XPS spectra of both Ch and ChG05 (
Figure 7,
Table 4) detected O
1s, N
1s and C
1s peaks at around 540.0, 400.0 and 290.0 eV, respectively. Peaks at 150.0 and 99.0 eV assigned to Si
2s and Si
2p [
24], respectively, were only detected for ChG05. The presence of nitrogen in three different chemical states on the thin films surfaces was confirmed from their N
1s XPS spectra (
Figure 7b,c). Ch had a strong N
1s peak at 399.3 eV attributed to the nitrogen atom in the –NH
2 group and two less intense peaks at 400.3 and 401.6 eV, which suggest the presence of nitrogen atoms from the acetyl amine, –NH–C = O and protonated amino group, –NH
3+ [
25]. For ChG05, the relative peak area of the –NH
3+ peak decreased and that of the –NH–C = O peak increased, which indicates that –NH–CH
2 formed as a result of the reaction between protonated amino groups and epoxy groups of GPTMS. The small changes in the N
1s spectra were consistent with the results of the ninhydrin experiment.
Chitosan matrix signals appeared at 105.1 (C1), 82.6 (C4), 75.8 (C3 and C5), 61.6 (C6), 58.1 (C2), 174.4 (N-acetyl C = O signal) and 24.1 (N-acetyl-methyl CH
3 signal) ppm in the
13C CP-MAS NMR spectra (
Figure 8 and
Figure 9) [
26,
27,
28]. The intensity of C2/C1 in the Ch, ChG05 and ChG10 spectra was 1.52, 1.29 and 1.28. The lower C2/C1 for ChG05 and ChG10 compared with Ch suggested that the amino groups of chitosan reacted with GPTMS. The Cg and Ch peaks corresponding to non ring-opened epoxy groups were detected for ChG10. These peaks are attributed to GPTMS that did not react with the amino groups of chitosan, which correspond to the residual amino groups determined using the ninhydrin methods. The spectra also confirmed the addition of BZ to the films (
Figure 9a). The main peaks of the thin films did not change and the carbons of the methyl groups at 13.0 ppm, the CH
2 carbons adjacent to the cyclic structures at 16.5 ppm and the tertiary carbons with methyl and ethyl groups at 40.3 ppm, were detected and corresponded to BZ [
29]. These observations indicate that BZ did not react with the thin film matrix.
The
29Si CP-MAS NMR spectra (
Figure 10) demonstrate the change of the methoxysilane group of GPTMS due to hybridization. The profiles were deconvolved into three component peaks due to T
1, T
2 and T
3 siloxane units, from which the chemical shift, full-width at half-maximum and relative peak area (%) of each T
n unit were calculated as in a previous study [
14,
15,
16] (
Table 5). T
1, T
2 and T
3 denote R–Si(–OSi)(OCH
3, OH)
2, R–Si(–OSi)
2(OCH
3, OH) and R–Si(–OSi)
3, respectively (R is the organic skeleton derived from GPTMS). The number of Si–O–Si bridging bonds per Si atom, N
bo/Si, derived from condensation of the –Si–OH or –Si–OCH
3 groups at the silane end of a GPTMS molecule, is given by Equation (4), according to simple valence theory:
The Nbo/Si in each sample was approximately 2.0 regardless of the amount of GPTMS and BZ. The addition of BZ did not change the structure surrounding S, which indicates that BZ interacted with the thin film matrix by intermolecular forces without forming a chemical bond.
The mechanical properties of the films were examined using a puncture strength test (
Figure 11). ChG10 was too brittle for the puncture strength test to be carried out. The maximum breaking load was the same with and without GPTMS; however, Ch had greater deformation than ChG01 and ChG05.
Chitosan nanofibers are a few μm length and the distance between amino groups of chitosan is about 1 nm [
30]. The length of GPTMS molecule is about 3 nm [
31] and should be branched on the chitosan nanofiber surface by open-ring reaction. GPTMS are also hydrolyzed in acetic acid (pH 3.0–4.0) to form –Si–OH groups because the hydrolysis of methoxy groups preferentially occur, rather than polycondensation of –Si–OH groups at pH lower 5.5 [
32]. Based on the structure analysis, the interconnected porous structure formation is thought to follow the following steps (
Figure 12): (1) The chitosan nanofibers are dispersed in aqueous solution; (2) GPTMS hydrolyzed by acetic acid to form –Si–OH groups and react with the chitosan nanofibers through opening ring reaction of epoxide; (3) the 2–5 nanofibers aggregated and formed the bundles by polycondensation of GPTMS; (4) The –Si–OH groups are also interacted with the remained amino groups; (5) the bundles form a wall and separate from the water phase on the cellulose nitrate filter; (6) finally, the water is evaporated and the interconnected pores are formed with the bundles forming the walls. The polycondensation of GPTMS are also promoted under dry. The –Si–OH groups and –Si–O–Si– networks improved the surface wettability and reduced the flexibility. The water adsorption depended on the remaining amino groups and the amount of –Si–OH groups and –Si–O–Si– networks from GPTMS.
3.3. Release of BZ and In Vitro Biological Properties
The amount of BZ released from the films after 1 day was small, even in PBS. The release on the agar medium was expected to be slower. As the amount of GPTMS increased, the release of BZ was suppressed (
Table 6). In addition, as the amount of BZ loaded into the films increased, the amount released also increased. The bacitracin molecules interacted with the thin film matrix through the –Si–OH groups derived from GPTMS and the release was suppressed as the amount of GPTMS increased. However, the amount of BZ that could be loaded into the matrix was limited and it easily diffused out at high concentrations of BZ. Ch and ChG05 did not show antibacterial properties against
S. aureus (
Figure 13 and
Table 6). This indicates that the amino groups on these films were not protonated under biological conditions. The antibacterial effect depended on the amout of BZ released. ChG05B5 showed a larger inhibition zone (*
p < 0.05), 21.3 ± 0.1 mm than ChG05B1, 14.1 ± 0.6 mm. The addition of GPTMS slightly suppressed (**
p < 0.01) the inhibition of
S. aureus, but the difference was not marked. The Zn
2+ of BZ electrostatically attached to the negatively charged bacterial surface, where BZ was able to prevent cell wall and enzyme synthesis [
21]. Amino groups in the thin films were able to form complexes with Zn
2+, which retarded the release of BZ.
The effect of BZ on L929 fibroblasts was demonstrated by measuring the cell viability when cultured with an insert (
Figure 14). The films did not touch the cell surface and we were able to examine the effect of the released BZ without shutoff between the cells and medium. Ch, ChG05, ChG05B05 and ChG05B1 did not show toxicity towards L929 and samples exhibited the same cell viability as the Control (medium only). Only cells expose to ChG05B5 showed approximately 25% lower viability than those exposed to the other films and the Control. We concluded the release of BZ between ChB1 and ChG10B1 and ChG05B1 and ChG05B5 in
Figure 15. ChB1 does not have the electrostatic interaction with BZ and released easily. On the other hand, ChG10B1 surface has –Si–OH groups and interact with amino groups of BZ to suppress the release. The higher released of BZ from ChGB1 showed higher antibacterial effects on
S. aureus than ChG10B1. Between ChG05B1 and ChG05B5, the amount of the interacted BZ on the chitosan fiber surfaces are same and the release depends on the starting amounts to occur the excess release. This excess release inhibited the viability of fibroblast. Zn
2+ ions easily absorbed on the cell surfaces and then bacitracin covered them to interfere with supply of nutrients to the cells.


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}