
Use of Solid-State NMR Spectroscopy for the Characterization of Molecular Structure and Dynamics in Solid Polymer and Hybrid Electrolytes

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Solid State NMR Spectroscopy Techniques
2.1. Transference Number Determination
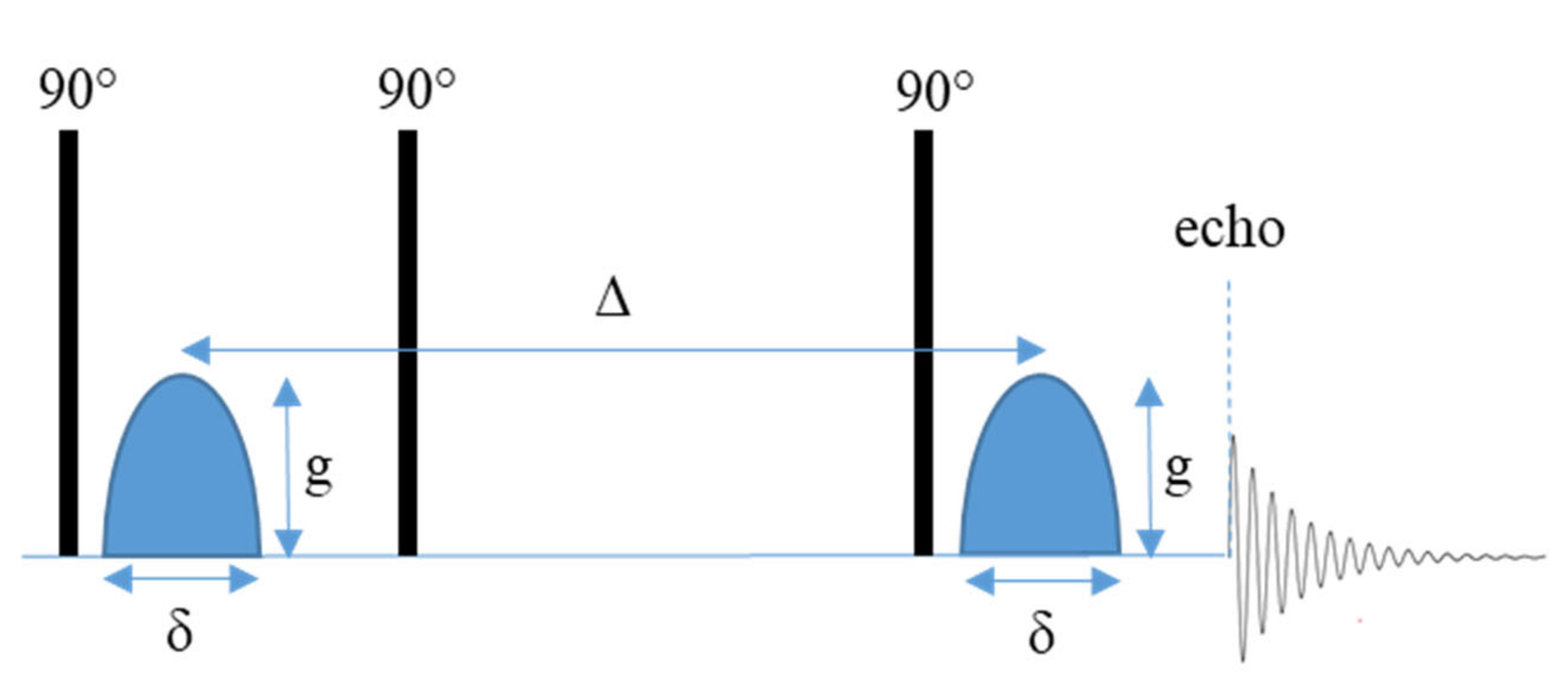
2.1.1. Pulse Field Gradient NMR
2.1.2. Electrophoretic NMR
2.2. Variable Temperature NMR
2.2.1. Spin Lattice Relaxation (T1)
2.2.2. Spin-Spin Relaxation (T2)
2.2.3. Linewidth
2.3. Exchange Spectroscopy
2.4. Dipolar Coupling
2.4.1. Cross Polarization
2.4.2. Rotational Echo Double Resonance (REDOR)
2.5. Isotope Enrichment
3. Future Work
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xia, S.; Wu, X.; Zhang, Z.; Cui, Y.; Liu, W. Practical Challenges and Future Perspectives of All-Solid-State Lithium-Metal Batteries. Chem 2019, 5, 753–785. [Google Scholar] [CrossRef]
- Kerman, K.; Luntz, A.; Viswanathan, V.; Chiang, Y.-M.; Chen, Z. Review—Practical Challenges Hindering the Development of Solid State Li Ion Batteries. J. Electrochem. Soc. 2017, 164, A1731–A1744. [Google Scholar] [CrossRef]
- Yao, P.; Yu, H.; Ding, Z.; Liu, Y.; Lu, J.; Lavorgna, M.; Wu, J.; Liu, X. Review on Polymer-Based Composite Electrolytes for Lithium Batteries. Front. Chem. 2019, 7, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucharsky, E.C.; Schell, K.G.; Hupfer, T.; Hoffmann, M.J.; Rohde, M.; Seifert, H.J. Thermal properties and ionic conductivity of Li1,3Ti1,7Al0,3(PO4)3 solid electrolytes sintered by field-assisted sintering. Ionics 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Hupfer, T.; Bucharsky, E.C.; Schell, K.G.; Senyshyn, A.; Monchak, M.; Hoffmann, M.J.; Ehrenberg, H. Evolution of microstructure and its relation to ionic conductivity in Li1+xAlxTi2−x(PO4)3. Solid State Ionics 2016, 288, 235–239. [Google Scholar] [CrossRef]
- Hanghofer, I.; Gadermaier, B.; Wilkening, A.; Rettenwander, D.; Wilkening, H.M.R.; Wilkening, M. Lithium ion dynamics in LiZr2(PO4)3 and Li1.4Ca0.2Zr1.8(PO4)3. Dalton Trans. 2019, 48, 9376–9387. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Sun, C.; Jin, J.; Li, Y.; Chen, C.; Wen, Z. Realization of the Li+ domain diffusion effect via constructing molecular brushes on the LLZTO surface and its application in all-solid-state lithium batteries. J. Mater. Chem. A 2019, 7, 27304–27312. [Google Scholar] [CrossRef]
- Kato, Y.; Hori, S.; Saito, T.; Suzuki, K.; Hirayama, M.; Mitsui, A.; Yonemura, M.; Iba, H.; Kanno, R. High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 2016, 1, 16030. [Google Scholar] [CrossRef]
- Epp, V.; Ma, Q.; Hammer, E.-M.; Tietz, F.; Wilkening, M. Very fast bulk Li ion diffusivity in crystalline Li1.5Al0.5Ti1.5(PO4)3 as seen using NMR relaxometry. Phys. Chem. Chem. Phys. 2015, 17, 32115–32121. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Manthiram, A. A Long Cycle Life, All-Solid-State Lithium Battery with a Ceramic–Polymer Composite Electrolyte. ACS Appl. Energy Mater. 2020, 3, 2916–2924. [Google Scholar] [CrossRef]
- Tominaga, Y.; Yamazaki, K.; Nanthana, V. Effect of Anions on Lithium Ion Conduction in Poly(ethylene carbonate)-based Polymer Electrolytes. J. Electrochem. Soc. 2015, 162, A3133–A3136. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Li, S.-P.; Fan, L.-Z.; Nan, C.-W.; Goodenough, J.B. PEO/garnet composite electrolytes for solid-state lithium batteries: From “ceramic-in-polymer” to “polymer-in-ceramic. ” Nano Energy 2018, 46, 176–184. [Google Scholar] [CrossRef]
- Bonizzoni, S.; Ferrara, C.; Berbenni, V.; Anselmi-Tamburini, U.; Mustarelli, P.; Tealdi, C. NASICON-type polymer-in-ceramic composite electrolytes for lithium batteries. Phys. Chem. Chem. Phys. 2019, 21, 6142–6149. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhu, P.; Jia, H.; Du, Z.; Zhu, J.; Orenstein, R.; Cheng, H.; Wu, N.; Dirican, M.; Zhang, X. Garnet-rich composite solid electrolytes for dendrite-free, high-rate, solid-state lithium-metal batteries. Energy Storage Mater. 2020, 26, 448–456. [Google Scholar] [CrossRef]
- Liu, W.; Lee, S.W.; Lin, D.; Shi, F.; Wang, S.; Sendek, A.D.; Cui, Y. Enhancing ionic conductivity in composite polymer electrolytes with well-aligned ceramic nanowires. Nat. Energy 2017, 2. [Google Scholar] [CrossRef]
- Boaretto, N.; Meabe, L.; Martinez-Ibañez, M.; Armand, M.; Zhang, H. Review—Polymer Electrolytes for Rechargeable Batteries: From Nanocomposite to Nanohybrid. J. Electrochem. Soc. 2020, 167, 070524. [Google Scholar] [CrossRef]
- Liang, M.; Liu, Y.; Xiao, B.; Yang, S.; Wang, Z.; Han, H. An analytical model for the transverse permeability of gas diffusion layer with electrical double layer effects in proton exchange membrane fuel cells. Int. J. Hydrog. Energy 2018, 43, 17880–17888. [Google Scholar] [CrossRef]
- Liang, M.; Fu, C.; Xiao, B.; Luo, L.; Wang, Z. A fractal study for the effective electrolyte diffusion through charged porous media. Int. J. Heat Mass Transf. 2019, 137, 365–371. [Google Scholar] [CrossRef]
- West, K.; Zachau-Christiansen, B.; Jacobsen, T.; Hiort-Lorenzen, E.; Skaarup, S. Poly(ethylene oxide)-sodium perchlorate electrolytes in solid-state sodium cells. Br. Polym. J. 1988, 20, 243–246. [Google Scholar] [CrossRef]
- Wang, Y.; Song, S.; Xu, C.; Hu, N.; Molenda, J.; Lu, L. Development of solid-state electrolytes for sodium-ion battery–A short review. Nano Mater. Sci. 2019, 1, 91–100. [Google Scholar] [CrossRef]
- Colò, F.; Bella, F.; Nair, J.R.; Destro, M.; Gerbaldi, C. Cellulose-based novel hybrid polymer electrolytes for green and efficient Na-ion batteries. Electrochim. Acta 2015, 174, 185–190. [Google Scholar] [CrossRef]
- Hashmi, S.; Chandra, S. Experimental investigations on a sodium-ion-conducting polymer electrolyte based on poly(ethylene oxide) complexed with NaPF6. Mater. Sci. Eng. B 1995, 34, 18–26. [Google Scholar] [CrossRef]
- Luo, H.; Liang, X.; Wang, L.; Zheng, A.; Liu, C.; Feng, J. Highly mobile segments in crystalline poly(ethylene oxide)8:NaPF6 electrolytes studied by solid-state NMR spectroscopy. J. Chem. Phys. 2014, 140, 074901. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, M.; He, D.; Hu, J.; Wang, X.; Gong, C.; Xie, X.; Xue, Z. Comb-like solid polymer electrolyte based on polyethylene glycol-grafted sulfonated polyether ether ketone. Electrochim. Acta 2017, 255, 396–404. [Google Scholar] [CrossRef]
- Bhide, A.; Hariharan, K. Composite polymer electrolyte based on (PEO)6:NaPO3 dispersed with BaTiO3. Polym. Int. 2008, 57, 523–529. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, K.; Ding, F.; Liu, X. Recent advances in solid polymer electrolytes for lithium batteries. Nano Res. 2017, 10, 4139–4174. [Google Scholar] [CrossRef]
- Xu, K. Electrolytes and Interphases in Li-Ion Batteries and Beyond. Chem. Rev. 2014, 114, 11503–11618. [Google Scholar] [CrossRef]
- Pecher, O.; Carretero-González, J.; Griffith, K.J.; Grey, C.P. Materials’ Methods: NMR in Battery Research. Chem. Mater. 2017, 29, 213–242. [Google Scholar] [CrossRef] [Green Version]
- Keeler, J. Understanding NMR Spectroscopy, 2nd ed.; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Roach, D.J.; Dou, S.; Colby, R.H.; Mueller, K.T. Nuclear magnetic resonance investigation of dynamics in poly(ethylene oxide)-based lithium polyether-ester-sulfonate ionomers. J. Chem. Phys. 2012, 136, 14510. [Google Scholar] [CrossRef]
- Daigle, J.-C.; Arnold, A.A.; Vijh, A.; Zaghib, K. Solid-State NMR Study of New Copolymers as Solid Polymer Electrolytes. Magnetochemistry 2018, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Morales, D.J.; Greenbaum, S. NMR Investigations of Crystalline and Glassy Solid Electrolytes for Lithium Batteries: A Brief Review. Int. J. Mol. Sci. 2020, 21, 3402. [Google Scholar] [CrossRef] [PubMed]
- Zujovic, Z.; Kilmartin, P.A.; Travas-Sejdic, J. The Applications of Solid-State NMR to Conducting Polymers. The Special Case on Polyaniline. Molecules 2020, 25, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbrent, S.; Greenbaum, S. Using nuclear magnetic resonance spectroscopy in polymer electrolyte research. In Polymer Electrolytes; Elsevier: Amsterdam, The Netherlands, 2010; pp. 278–313. [Google Scholar]
- Price, W. Pulsed-Field Gradient Nuclear Magnetic Resonance as a Tool for Studying Translational Diffusion: Part 1; Basic Theory. Concept Magn. Reson 1997, 9, 299–336. [Google Scholar] [CrossRef]
- Sun, B.; Mindemark, J.; Morozov, E.V.; Costa, L.T.; Bergman, M.; Johansson, P.; Fang, Y.; Furó, I.; Brandell, D. Ion transport in polycarbonate based solid polymer electrolytes: Experimental and computational investigations. Phys. Chem. Chem. Phys. 2016, 18, 9504–9513. [Google Scholar] [CrossRef]
- Liu, X.; Ding, G.; Zhou, X.; Li, S.; He, W.; Chai, J.; Pang, C.; Liu, Z.; Cui, G. An interpenetrating network poly (diethylene glycol carbonate)-based polymer electrolyte for solid state lithium batteries. J. Mater. Chem. A 2017, 5, 11124–11130. [Google Scholar] [CrossRef]
- Shah, F.U.; Gnezdilov, O.I.; Gusain, R.; Filippov, A. Transport and Association of Ions in Lithium Battery Electrolytes Based on Glycol Ether Mixed with Halogen-Free Orthoborate Ionic Liquid. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef] [Green Version]
- Cotts, R.; Hoch, M.; Sun, T.; Markert, J. Pulsed field gradient stimulated echo methods for improved NMR diffusion measurements in heterogeneous systems. J. Magn. Reson. 1989, 83, 252–266. [Google Scholar] [CrossRef]
- Tanner, J.E. Use of the Stimulated Echo in NMR Diffusion Studies. J. Chem. Phys. 1970, 52, 2523–2526. [Google Scholar] [CrossRef]
- Engelke, S.; Marbella, L.E.; Trease, N.M.; De Volder, M.; Grey, C.P. Three-dimensional pulsed field gradient NMR measurements of self-diffusion in anisotropic materials for energy storage applications. Phys. Chem. Chem. Phys. 2019, 21, 4538–4546. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; De Menezes, S.M.C.; Goodfellow, R.; Granger, P. NMR nomenclature: Nuclear spin properties and conventions for chemical shifts. IUPAC Recommendations 2001. International Union of Pure and Applied Chemistry. Physical Chemistry Division. Commission on Molecular Structure and Spectroscopy. Magn. Reson. Chem. 2002, 40, 489–505. [Google Scholar] [CrossRef]
- Adebahr, J.; Best, A.S.; Byrne, N.; Jacobsson, P.; Macfarlane, D.R.; Forsyth, M. Ion transport in polymer electrolytes containing nanoparticulate TiO2: The influence of polymer morphology. Phys. Chem. Chem. Phys. 2003, 5, 720–725. [Google Scholar] [CrossRef]
- Hosseinioun, A.; Nürnberg, P.; Schönhoff, M.; Diddens, D.; Paillard, E. Improved lithium ion dynamics in crosslinked PMMA gel polymer electrolyte. RSC Adv. 2019, 9, 27574–27582. [Google Scholar] [CrossRef] [Green Version]
- Foran, G.; Mankovsky, D.; Verdier, N. The Impact of Absorbed Solvent on the Performance of Solid Polymer Electrolytes for Use in Solid-State Lithium Batteries. Science 2020, 23, 10. [Google Scholar]
- Greenbaum, S.G. NMR studies of ion mobility and association in polyether-based polymer electrolytes. Polym. Adv. Technol. 1993, 4, 172–178. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, Y.; Liang, X.; Lei, Y.; Yuan, T.; Lu, H.; Liu, Z.; Cao, Y.; Feng, J. Novel Sodium–Poly(tartaric acid)Borate-Based Single-Ion Conducting Polymer Electrolyte for Sodium–Metal Batteries. ACS Appl. Energy Mater. 2020, 3, 10053–10060. [Google Scholar] [CrossRef]
- Verdier, N.; Lepage, D.; Zidani, R.; Prébé, A.; Aymé-Perrot, D.; Pellerin, C.; Dollé, M.; Rochefort, D. Cross-Linked Polyacrylonitrile-Based Elastomer Used as Gel Polymer Electrolyte in Li-Ion Battery. ACS Appl. Energy Mater. 2019, 3, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Fong, K.D.; Self, J.; Diederichsen, K.M.; Wood, B.M.; McCloskey, B.D.; Persson, K.A. Ion Transport and the True Transference Number in Nonaqueous Polyelectrolyte Solutions for Lithium Ion Batteries. ACS Central Sci. 2019, 5, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Mindemark, J.; Lacey, M.J.; Bowden, T.; Brandell, D. Beyond PEO—Alternative host materials for Li + -conducting solid polymer electrolytes. Prog. Polym. Sci. 2018, 81, 114–143. [Google Scholar] [CrossRef]
- Evans, J.; Vincent, C.A.; Bruce, P.G. Electrochemical measurement of transference numbers in polymer electrolytes. Polymer 1987, 28, 2324–2328. [Google Scholar] [CrossRef]
- Pesko, D.M.; Timachova, K.; Bhattacharya, R.; Smith, M.C.; Villaluenga, I.; Newman, J.; Balsara, N.P. Negative Transference Numbers in Poly(ethylene oxide)-Based Electrolytes. J. Electrochem. Soc. 2017, 164, E3569–E3575. [Google Scholar] [CrossRef] [Green Version]
- Balsara, N.P.; Newman, J.W. Relationship between Steady-State Current in Symmetric Cells and Transference Number of Electrolytes Comprising Univalent and Multivalent Ions. J. Electrochem. Soc. 2015, 162, A2720–A2722. [Google Scholar] [CrossRef] [Green Version]
- Pesko, D. Complete Electrochemical Characterization of Ion Transport in Polumer Electrolytes; University of California: Berkeley, CA, USA, 2018. [Google Scholar]
- Hittorf, W. Ueber die Wanderungen der Ionen während der Elektrolyse. Ann. der Phys. 1853, 165, 177–211. [Google Scholar] [CrossRef]
- Bruce, P.G.; Hardgrave, M.T.; Vincent, C.A. The determination of transference numbers in solid polymer electrolytes using the Hittorf method. Solid State Ion. 1992, 53–56, 1087–1094. [Google Scholar] [CrossRef]
- Kaneko, F.; Wada, S.; Nakayama, M.; Wakiha ra, M.; Kuroki, S. Dynamic Transport in Li-Conductive Polymer Electrolytes Plasticized with Poly(ethylene glycol)-Borate/Aluminate Ester. ChemPhysChem 2009, 10, 1911–1915. [Google Scholar] [CrossRef]
- Chintapalli, M.; Timachova, K.; Olson, K.R.; Mecham, S.J.; Devaux, D.; DeSimone, J.M.; Balsara, N.P. Relationship between Conductivity, Ion Diffusion, and Transference Number in Perfluoropolyether Electrolytes. Macromol 2016, 49, 3508–3515. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, Y.; Yamazaki, K. Fast Li-ion conduction in poly (ethylene carbonate)-based electrolytes and composites filled with TiO2 nanoparticles. Chem. Commun. 2014, 50, 4448–4450. [Google Scholar] [CrossRef]
- Timachova, K.; Sethi, G.K.; Bhattacharya, R.; Villaluenga, I.; Balsara, N.P. Ion diffusion across a disorder-to-order phase transition in a poly(ethylene oxide)-b-poly(silsesquioxane) block copolymer electrolyte. Mol. Syst. Des. Eng. 2019, 4, 357–364. [Google Scholar] [CrossRef]
- Cheung, I.; Chin, K.; Greene, E.; Smart, M.; Abbrent, S.; Greenbaum, S.; Prakash, G.; Surampudi, S. Electrochemical and solid state NMR characterization of composite PEO-based polymer electrolytes. Electrochim. Acta 2003, 48, 2149–2156. [Google Scholar] [CrossRef]
- Martynov, A.; Sushama, L.; Laprise, R. Simulation of temperate freezing lakes by one-dimensional lake models: Perfor-mance assessment for interactive coupling with regional climate models. Boreal Environ. Res. 2010, 15, 143–164. [Google Scholar] [CrossRef]
- Gorecki, W. NMR, DSC and conductivity study of the polymer solid electrolytes P(EO) (LiCp+1F2p+3SO3)x. Solid State Ion. 1988, 28–30, 1018–1022. [Google Scholar] [CrossRef]
- Al-Salih, H.; Huang, A.; Yim, C.-H.; Freytag, A.I.; Goward, G.R.; Baranova, E.; Abu-Lebdeh, Y. A Polymer-Rich Quaternary Composite Solid Electrolyte for Lithium Batteries. J. Electrochem. Soc. 2020, 167, 070557. [Google Scholar] [CrossRef]
- Zhang, Z.; Madsen, L.A. Observation of separate cation and anion electrophoretic mobilities in pure ionic liquids. J. Chem. Phys. 2014, 140, 084204. [Google Scholar] [CrossRef] [PubMed]
- Stilbs, P.; Furó, I. Electrophoretic NMR. Curr. Opin. Colloid Interface Sci. 2006, 11, 3–6. [Google Scholar] [CrossRef]
- Pettersson, E.; Furó, I.; Stilbs, P. On experimental aspects of electrophoretic NMR. Concepts Magn. Reson. Part A 2004, 22, 61–68. [Google Scholar] [CrossRef]
- Rosenwinkel, M.P.; Schönhoff, M. Lithium Transference Numbers in PEO/LiTFSA Electrolytes Determined by Electrophoretic NMR. J. Electrochem. Soc. 2019, 166, A1977–A1983. [Google Scholar] [CrossRef]
- Harris, R.K. Nuclear Magnetic Resonance Spectroscopy; Pitman Publishing Inc.: Marshfield, MA, USA, 1983. [Google Scholar]
- Foerster, H.; Struppe, J.; Steuernager, S. Solid State NMR AVANCE Solids User Manual; Bruker Biospin GmBH: Rheinstetten, Germany, 2009. [Google Scholar]
- Jeon, J.-D.; Kwak, S.-Y. Variable-Temperature7Li Solid-State NMR Investigation of Li-Ion Mobility and Its Correlation with Conductivity in Pore-Filling Polymer Electrolytes for Secondary Batteries. Macromolecules 2006, 39, 8027–8034. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Clarendon Press: Oxford, UK, 1983. [Google Scholar]
- Stone, N. Table of nuclear magnetic dipole and electric quadrupole moments. At. Data Nucl. Data Tables 2005, 90, 75–176. [Google Scholar] [CrossRef]
- Kentgens, A.P.M. A practival guide to solid-state NMR of half-integer quadrupolar nuclei with some applications to disordered systems. Geoderma 1997, 80, 271–306. [Google Scholar] [CrossRef]
- Lin, C.-L.; Kao, H.-M.; Wu, R.-R.; Kuo, P.-L. Multinuclear Solid-State NMR, DSC, and Conductivity Studies of Solid Polymer Electrolytes Based on Polyurethane/Poly (dimethylsiloxane) Segmented Copolymers. Macromolecules 2002, 35, 3083–3096. [Google Scholar] [CrossRef]
- Stallworth, P.; Greenbaum, S.; Croce, F.; Slane, S.; Salomon, M. Lithium-7 NMR and ionic conductivity studies of gel electrolytes based on poly (methylmethacrylate). Electrochim. Acta 1995, 40, 2137–2141. [Google Scholar] [CrossRef]
- Kumar, B.; Marsh, R.A. Polymer Batteries. SAE Tech. Pap. Ser. 1991, 100, 408–412. [Google Scholar] [CrossRef]
- Chen-Yang, Y.; Chen, H.; Lin, F.; Chen, C. Polyacrylonitrile electrolytes 1. A novel high-conductivity composite polymer electrolyte based on PAN, LiClO4 and α-Al2O3. Solid State Ion. 2002, 150, 327–335. [Google Scholar] [CrossRef]
- Diederichsen, K.M.; Buss, H.G.; McCloskey, B.D. The Compensation Effect in the Vogel–Tammann–Fulcher (VTF) Equation for Polymer-Based Electrolytes. Macromolecules 2017, 50, 3831–3840. [Google Scholar] [CrossRef]
- Keeler, J. Chapter 8. Relaxation. Available online: http://www-keeler.ch.cam.ac.uk/lectures/understanding/chapter_8.pdf (accessed on 26 February 2019).
- Peng, J.; Xiao, Y.; Clarkson, D.A.; Greenbaum, S.G.; Zawodzinski, T.A.; Chen, X.C. A Nuclear Magnetic Resonance Study of Cation and Anion Dynamics in Polymer–Ceramic Composite Solid Electrolytes. ACS Appl. Polym. Mater. 2020, 2, 1180–1189. [Google Scholar] [CrossRef]
- Schmutzler, R.; Reddy, G.S.; An, N.M.R. Study of some Tertiary Butyl—Phosphorus Compounds. Z. Nat. B 1965, 20, 832–835. [Google Scholar] [CrossRef]
- Pak, Y.; Adamic, K.; Greenbaum, S.; Wintersgill, M.; Fontanella, J.; Coughlin, C. Complex impedance and multifrequency 23Na NMR study of poly (propylene oxide) complexed with NaB (C6H5)4. Solid State Ion. 1991, 45, 277–284. [Google Scholar] [CrossRef]
- Forsyth, M.; Macfarlane, D.; Meakin, P.; Smith, M.; Bastow, T. An nmr investigation of ionic structure and mobility in plasticized solid polymer electrolytes. Electrochim. Acta 1995, 40, 2343–2347. [Google Scholar] [CrossRef]
- Rahman, A.; Choudhary, M.I.; Wahab, A. Spin-Echo and Polarization Transfer. In Solving Problems with NMR Spectroscopy, 2nd ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 133–190. [Google Scholar]
- Forsyth, M.; Jiazeng, S.; Macfarlane, D.R. Novel high salt content polymer electrolytes based on high Tg polymers. Electrochim. Acta 2000, 45, 1249–1254. [Google Scholar] [CrossRef]
- Carr, H.Y.; Purcell, E.M. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D.R. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef] [Green Version]
- Donoso, J.; Cavalcante, M.; Bonagamba, T.; Nascimento, O.; Panepucci, H. Magnetic resonance study of water absorption in some peo-lithium salt polymer electrolytes. Electrochim. Acta 1995, 40, 2357–2360. [Google Scholar] [CrossRef]
- Franco, R. NMR and DSC study of polymer electrolyte–Carbon Black composites. Solid State Ion. 2000, 136-137, 1181–1187. [Google Scholar] [CrossRef]
- Kwak, G.-H.; Tominaga, Y.; Asai, S.; Sumita, M. Effect of reaction kinetics of polymer electrolyte on the ion-conductive behavior for poly (oligo oxyethylene methacrylate)-LiTFSI mixtures. J. Appl. Polym. Sci. 2003, 89, 2149–2156. [Google Scholar] [CrossRef]
- unterion Transport in an Electrospun Polymer-Gel Electrolyte. Macromolecules 2015, 48, 4481–4490. [CrossRef]
- Poinsignon, C. Polymer electrolytes. Mater. Sci. Eng. B 1989, 3, 31–37. [Google Scholar] [CrossRef]
- Chung, S.; Wang, Y.; Persi, L.; Croce, F.; Greenbaum, S.; Scrosati, B.; Plichta, E. Enhancement of ion transport in polymer electrolytes by addition of nanoscale inorganic oxides. J. Power Sources 2001, 97–98, 644–648. [Google Scholar] [CrossRef]
- Liu, T.-M.; Saikia, D.; Ho, S.-Y.; Chen, M.-C.; Kao, H.-M. High ion-conducting solid polymer electrolytes based on blending hybrids derived from monoamine and diamine polyethers for lithium solid-state batteries. RSC Adv. 2017, 7, 20373–20383. [Google Scholar] [CrossRef] [Green Version]
- Munoz, S.; Greenbaum, S. Review of Recent Nuclear Magnetic Resonance Studies of Ion Transport in Polymer Electrolytes. Membranes 2018, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.; Zax, D.B. What do NMR linewidths tell us? Dynamics of alkali cations in a PEO-based nanocomposite polymer electrolyte. Electrochim. Acta 1997, 42, 3513–3518. [Google Scholar] [CrossRef]
- Forsyth, M.; Smith, M.E.; Meakin, P.; Macfarlane, D.R. 23Na NMR in urethane cross-linked polyether solid polymer electrolytes. J. Polym. Sci. Part B 1994, 32, 2077–2084. [Google Scholar] [CrossRef]
- Berman, M.B.; Greenbaum, S.G. NMR Studies of Solvent-Free Ceramic Composite Polymer Electrolytes—A Brief Review. Membranes 2015, 5, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Menkin, S.; Lifshitz, M.; Haimovich, A.; Goor, M.; Blanga, R.; Greenbaum, S.; Goldbourt, A.; Golodnitsky, D. Evaluation of ion-transport in composite polymer-in-ceramic electrolytes. Case study of active and inert ceramics. Electrochim. Acta 2019, 304, 447–455. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, T.; Chu, X.; Su, H.; Wang, Z.; Chen, N.; Gu, B.; Zhang, H.; Deng, W.; Zhang, H. Strong Lewis Acid–Base and Weak Hydrogen Bond Synergistically Enhancing Ionic Conductivity of Poly(ethylene oxide)@SiO2 Electrolytes for a High Rate Capability Li-Metal Battery. ACS Appl. Mater. Interfaces 2020, 12, 10341–10349. [Google Scholar] [CrossRef]
- Zheng, J.; Hu, Y.-Y. New Insights into the Compositional Dependence of Li-Ion Transport in Polymer–Ceramic Composite Electrolytes. ACS Appl. Mater. Interfaces 2018, 10, 4113–4120. [Google Scholar] [CrossRef]
- Lago, N.; Garcia-Calvo, O.; Del Amo, J.M.L.; Rojo, T.; Armand, M. All-Solid-State Lithium-Ion Batteries with Grafted Ceramic Nanoparticles Dispersed in Solid Polymer Electrolytes. ChemSusChem 2015, 8, 3039–3043. [Google Scholar] [CrossRef]
- Spindler, R.; Shriver, D.F. Investigations of a siloxane-based polymer electrolyte employing carbon-13, silicon-29, lithium-7, and sodium-23 solid-state NMR spectroscopy. J. Am. Chem. Soc. 1988, 110, 3036–3043. [Google Scholar] [CrossRef]
- Bain, A.D. Chemical exchange in NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2003, 43, 63–103. [Google Scholar] [CrossRef]
- Bain, A.D. Chemical Exchange. Annu. Rep. NMR Spectrosc. 2008, 63, 23–48. [Google Scholar] [CrossRef]
- Smiley, D.L.; Goward, G.R. Ex Situ 23Na Solid-State NMR Reveals the Local Na-Ion Distribution in Carbon-Coated Na2FePO4F during Electrochemical Cycling. Chem. Mater. 2016, 28, 7645–7656. [Google Scholar] [CrossRef]
- Autschbach, J.; Zheng, S.; Schurko, R.W. Analysis of electric field gradient tensors at quadrupolar nuclei in common structural motifs. Concepts Magn. Reson. Part A 2010, 36, 84–126. [Google Scholar] [CrossRef]
- Foran, G.Y.; Goward, G.R. Site-Specific Proton Dynamics in Indium-Doped Tin Pyrophosphate. J. Phys. Chem. C 2020, 124, 28407–28416. [Google Scholar] [CrossRef]
- Liu, Q.; Li, C.; Wei, L.; Shen, M.; Yao, Y.; Hu, B.; Chen, Q. The phase structure, chain diffusion motion and local reorientation motion: 13C Solid-state NMR study on the highly-crystalline solid polymer electrolytes. Polymer 2014, 55, 5454–5459. [Google Scholar] [CrossRef]
- Yang, L.-Y.; Wei, D.-X.; Xu, M.; Yao, Y.-F.; Chen, Q. Transferring Lithium Ions in Nanochannels: A PEO/Li+Solid Polymer Electrolyte Design. Angew. Chem. Int. Ed. 2014, 53, 3631–3635. [Google Scholar] [CrossRef]
- Zheng, J.; Tang, M.; Hu, Y.-Y. Lithium Ion Pathway within Li7 La3 Zr2 O12 -Polyethylene Oxide Composite Electrolytes. Angew. Chem. 2016, 128, 12726–12730. [Google Scholar] [CrossRef]
- Zagórski, J.; Del Amo, J.M.L.; Cordill, M.J.; Aguesse, F.; Buannic, L.; Llordés, A. Garnet–Polymer Composite Electrolytes: New Insights on Local Li-Ion Dynamics and Electrodeposition Stability with Li Metal Anodes. ACS Appl. Energy Mater. 2019, 2, 1734–1746. [Google Scholar] [CrossRef]
- Kolodziejski, W.; Klinowski, J. Kinetics of Cross-Polarization in Solid-State NMR: A Guide for Chemists. Chem. Rev. 2002, 102, 613–628. [Google Scholar] [CrossRef]
- Levitt, M.H. Spin Dynamics: Basics of Nuclear Magnetic Resonance, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Hartmann, S.R.; Hahn, E.L. Nuclear Double Resonance in the Rotating Frame. Phys. Rev. 1962, 128, 2042–2053. [Google Scholar] [CrossRef]
- Lim, K.; Grey, C. Triple Quantum Cross-Polarization Nmr of 1 H/27 Al and 19 F/23 Na Spin Systemsin Solid. Chem. Phys. Lett. 1999, 312, 45–56. [Google Scholar] [CrossRef]
- Puls, S.P.; Eckert, H. Site Discrimination in Mixed-Alkali Glasses Studied by Cross-Polarization NMR. J. Phys. Chem. B 2006, 110, 14253–14261. [Google Scholar] [CrossRef]
- Fu, X.-B.; Yang, G.; Wu, J.-Z.; Wang, J.-C.; Chen, Q.; Yao, Y.-F. Fast Lithium-Ion Transportation in Crystalline Polymer Electrolytes. ChemPhysChem 2018, 19, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Foran, G.Y.; Brouwer, D.H.; Goward, G.R. Quantifying Site-Specific Proton Dynamics in Phosphate Solid Acids by 1H Double Quantum NMR Spectroscopy. J. Phys. Chem. C 2017, 121, 25641–25650. [Google Scholar] [CrossRef]
- Gullion, T.; Schaefer, J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1989, 81, 196–200. [Google Scholar] [CrossRef]
- Eckert, H. Short and Medium Range Order in Ion-Conducting Glasses Studied by Modern Solid State NMR Techniques. Z. Phys. Chem. 2010, 224, 1591–1654. [Google Scholar] [CrossRef]
- Garbow, J.R.; Gullion, T. Improvements in REDOR NMR spectroscopy. Minimizing resonance-offset effects. J. Magn. Reson. 1991, 95, 442–445. [Google Scholar] [CrossRef]
- Bertmer, M.; Eckert, H. Dephasing of spin echoes by multiple heteronuclear dipolar interactions in rotational echo double resonance NMR experiments. Solid. State. Nucl. Magn. Reson. 1999, 15, 139–152. [Google Scholar] [CrossRef]
- Chan, J.C.; Eckert, H. Dipolar Coupling Information in Multispin Systems: Application of a Compensated REDOR NMR Approach to Inorganic Phosphates. J. Magn. Reson. 2000, 147, 170–178. [Google Scholar] [CrossRef]
- Voigt, N.; van Wüllen, L. The mechanism of ionic transport in PAN-based solid polymer electrolytes. Solid State Ion. 2012, 208, 8–16. [Google Scholar] [CrossRef]
- Voigt, N.; Van Wüllen, L. The effect of plastic-crystalline succinonitrile on the electrolyte system PEO:LiBF4: Insights from solid state NMR. Solid State Ion. 2014, 260, 65–75. [Google Scholar] [CrossRef]
- Wu, N.; Chien, P.-H.; Li, Y.; Dolocan, A.; Xu, H.; Xu, B.; Grundish, N.S.; Jin, H.; Hu, Y.-Y.; Goodenough, J.B. Fast Li+ Conduction Mechanism and Interfacial Chemistry of a NASICON/Polymer Composite Electrolyte. J. Am. Chem. Soc. 2020, 142, 2497–2505. [Google Scholar] [CrossRef]
- Liu, M.; Cheng, Z.; Ganapathy, S.; Wang, C.; Haverkate, L.A.; Tułodziecki, M.; Unnikrishnan, S.; Wagemaker, M. Tandem Interface and Bulk Li-Ion Transport in a Hybrid Solid Electrolyte with Microsized Active Filler. ACS Energy Lett. 2019, 4, 2336–2342. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Chien, P.-H.; Shi, J.; Li, Y.; Wu, N.; Liu, Y.; Hu, Y.-Y.; Goodenough, J.B. High-performance all-solid-state batteries enabled by salt bonding to perovskite in poly(ethylene oxide). Proc. Natl. Acad. Sci. USA 2019, 116, 18815–18821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Zheng, J.; Cheng, Q.; Hu, Y.-Y.; Chan, C.K. Composite Polymer Electrolytes with Li7La3Zr2O12 Garnet-Type Nanowires as Ceramic Fillers: Mechanism of Conductivity Enhancement and Role of Doping and Morphology. ACS Appl. Mater. Interfaces 2017, 9, 21773–21780. [Google Scholar] [CrossRef]
- Yang, H.; Bright, J.; Chen, B.; Zheng, P.; Gao, X.; Liu, B.; Kasani, S.; Zhang, X.; Wu, N. Chemical interaction and enhanced interfacial ion transport in a ceramic nanofiber–polymer composite electrolyte for all-solid-state lithium metal batteries. J. Mater. Chem. A 2020, 8, 7261–7272. [Google Scholar] [CrossRef]
- Martini, F.; Carignani, E.; Nardelli, F.; Rossi, E.; Borsacchi, S.; Cettolin, M.; Susanna, A.; Geppi, M.; Calucci, L. Glassy and Polymer Dynamics of Elastomers by 1H Field-Cycling NMR Relaxometry: Effects of Cross-Linking. Macromolecules 2020, 53, 10028–10039. [Google Scholar] [CrossRef]
- Jayakody, N.K.; Fraenza, C.C.; Greenbaum, S.G.; Ashby, D.S.; Dunn, B.S. NMR Relaxometry and Diffusometry Analysis of Dynamics in Ionic Liquids and Ionogels for Use in Lithium-Ion Batteries. J. Phys. Chem. B 2020, 124, 6843–6856. [Google Scholar] [CrossRef]
- Rachocki, A.; Andrzejewska, E.; Dembna, A.; Tritt-Goc, J. Translational dynamics of ionic liquid imidazolium cations at solid/liquid interface in gel polymer electrolyte. Eur. Polym. J. 2015, 71, 210–220. [Google Scholar] [CrossRef]




















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foran, G.; Verdier, N.; Lepage, D.; Malveau, C.; Dupré, N.; Dollé, M. Use of Solid-State NMR Spectroscopy for the Characterization of Molecular Structure and Dynamics in Solid Polymer and Hybrid Electrolytes. Polymers 2021, 13, 1207. https://doi.org/10.3390/polym13081207
Foran G, Verdier N, Lepage D, Malveau C, Dupré N, Dollé M. Use of Solid-State NMR Spectroscopy for the Characterization of Molecular Structure and Dynamics in Solid Polymer and Hybrid Electrolytes. Polymers. 2021; 13(8):1207. https://doi.org/10.3390/polym13081207
Chicago/Turabian StyleForan, Gabrielle, Nina Verdier, David Lepage, Cédric Malveau, Nicolas Dupré, and Mickaël Dollé. 2021. "Use of Solid-State NMR Spectroscopy for the Characterization of Molecular Structure and Dynamics in Solid Polymer and Hybrid Electrolytes" Polymers 13, no. 8: 1207. https://doi.org/10.3390/polym13081207