
Diffusion Coefficients and Activation Energies of Diffusion of Organic Molecules in Polystyrene below and above Glass Transition Temperature


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Materials and Model Compounds for Desorption Testing
2.1.1. Manufacturing of Polystyrene Sheets Spiked with Model Compounds
2.1.2. Quantification of Spiking Levels in the Polystyrene Sheets
2.1.3. Determination of Diffusion Coefficients
2.2. Sample Materials and Model Compounds for Permeation Testing
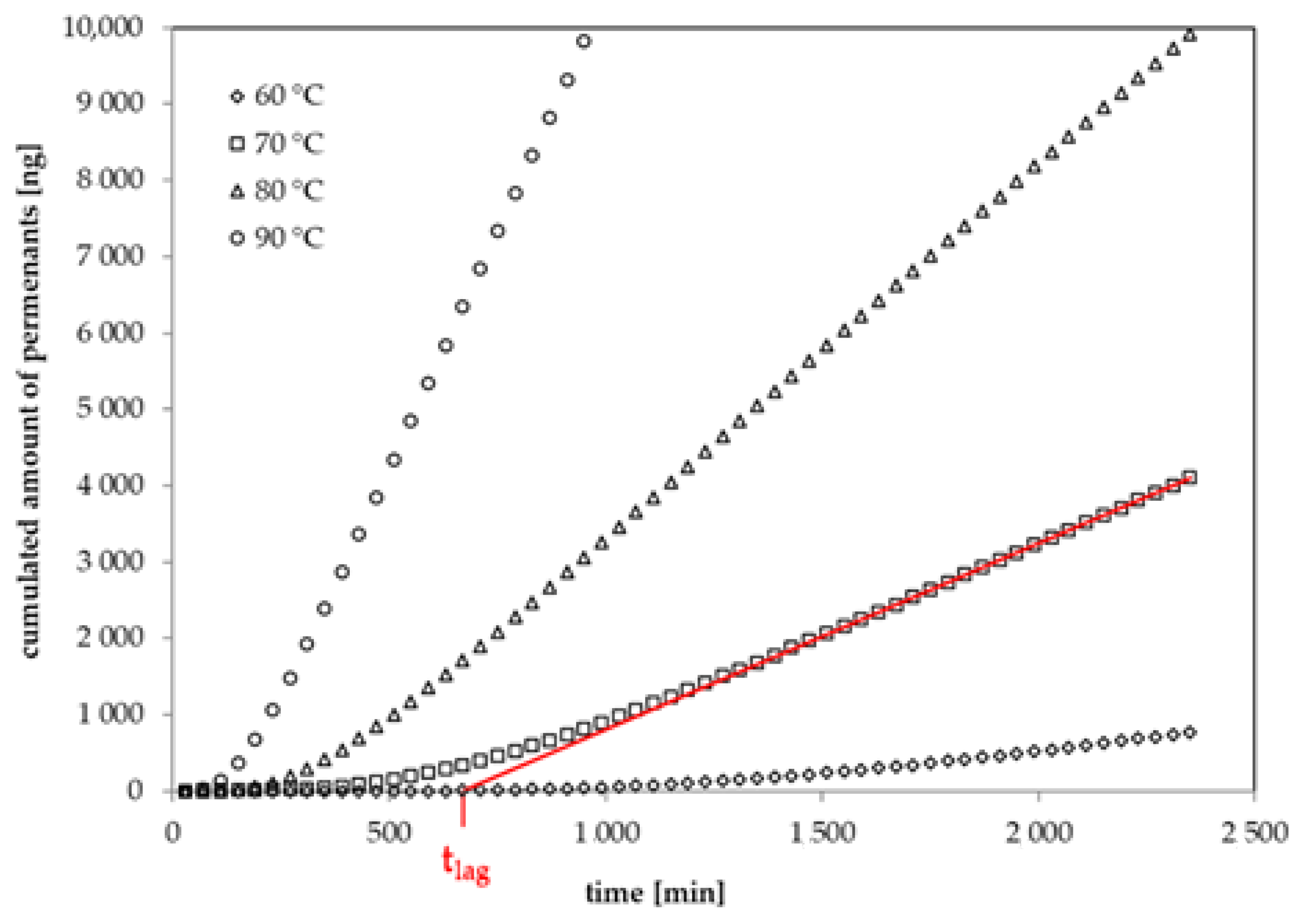
2.3. Permeation Measurements
2.4. Calculation of Molecular Volumes
3. Results and Discussion
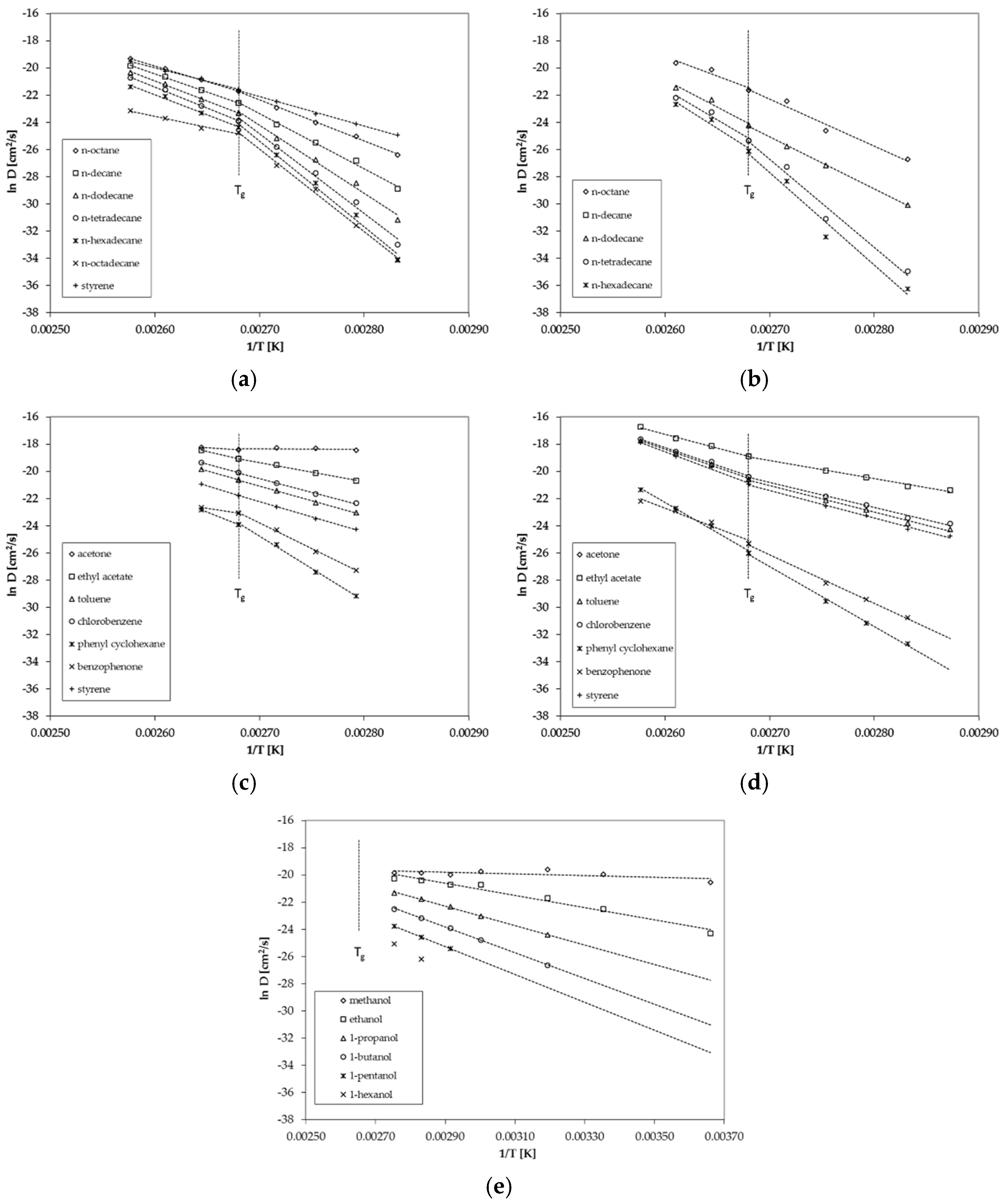
3.1. Diffusion Coefficients from Desorption Kinetics
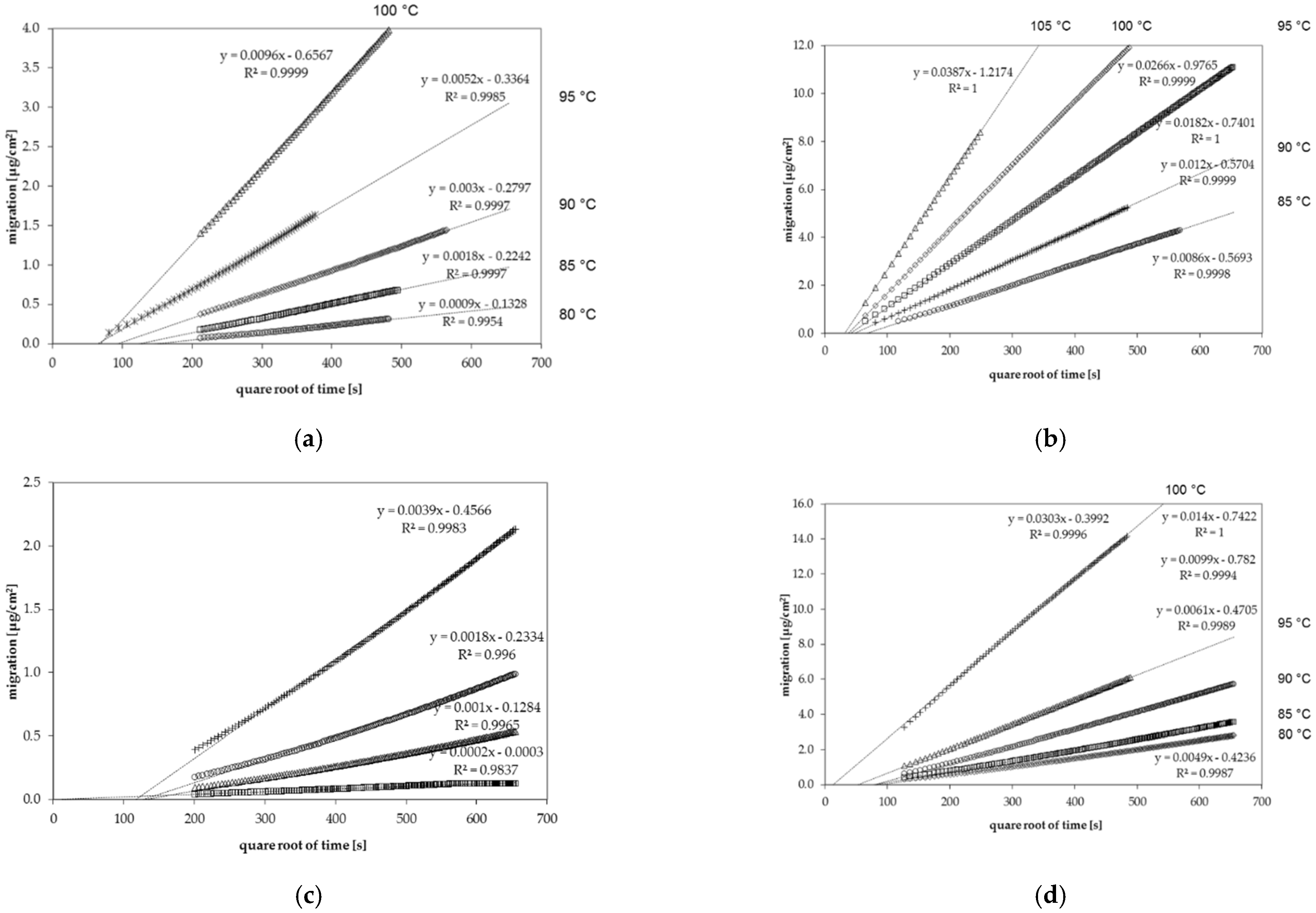
3.2. Diffusion Coefficients from Permeation Kinetics
3.3. Activation Energies of Diffusion
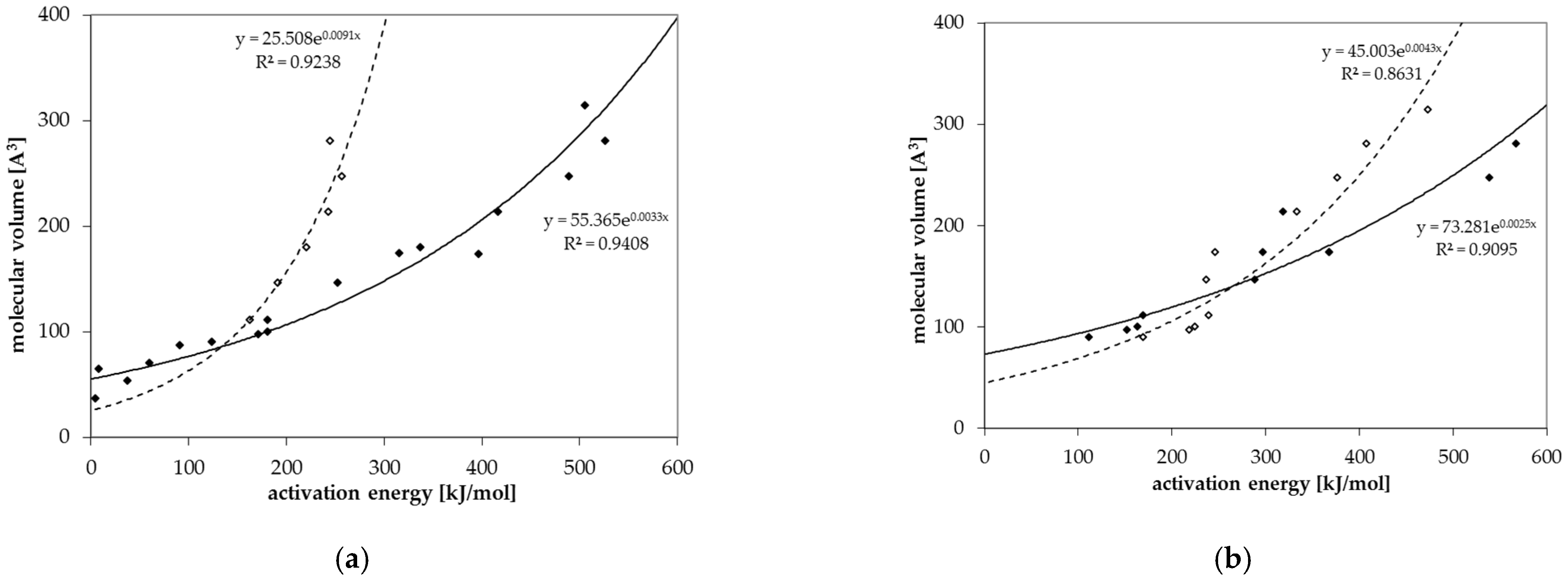
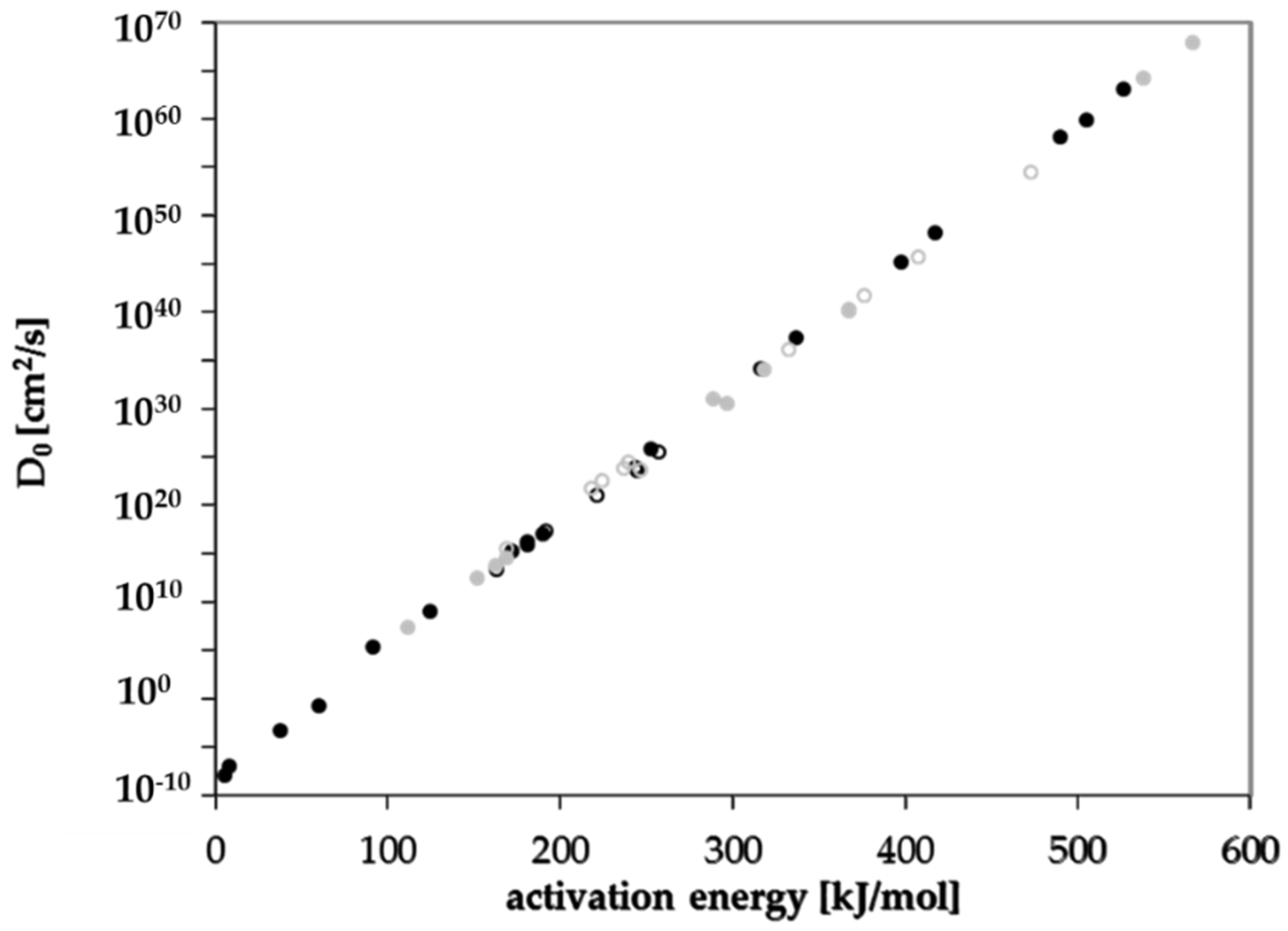
3.4. Prediction of Diffusion Coefficients
3.5. Prediction of Migration into Food and Food Simulants
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Block, C.; Brands, B.; Gude, T. Packaging Materials 2. Polystyrene for Food Packaging Applications—Updated Version; International Life Sciences Institute: Brussels, Belgium, 2017; ISBN 9789078637448. [Google Scholar]
- Briston, J. Advances in Plastics Packaging Technology; Pira International: Leatherhead, UK, 1992; Chapter 6; pp. 45–47. [Google Scholar]
- Ozaki, A.; Kishi, E.; Kaneko, R.; Ooshima, T.; Shimizu, M.; Kawamura, Y. Validation Study on Headspace-GC Analytical Method for Residual Volatile Substances in Food Contact Polystyrene and Its Application for Surveillance (1998–2014). J. Food Hyg. Soc. Jpn. (Shokuhin Eiseigaku Zasshi) 2015, 56, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Paraskevopoulou, D.; Achilias, D.S.; Paraskevopoulou, A. Migration of styrene from plastic packaging based on polystyrene into food simulants. Polym. Int. 2011, 61, 141–148. [Google Scholar] [CrossRef]
- Ramesh, N.; Duda, J. Predicting migration of trace amounts of styrene in poly(styrene) below the glass transition temperature. Food Chem. Toxicol. 2001, 39, 355–360. [Google Scholar] [CrossRef]
- Tawfik, M.S.; Huyghebaert, A. Polystyrene cups and containers: Styrene migration. Food Addit. Contam. 1998, 15, 592–599. [Google Scholar] [CrossRef]
- Linssen, J.; Reitsma, J. Comparison of migration of styrene monomer from high impact polystyrene in oil in water emulsions and fatty foods. J. Polym. Eng. 1995, 15, 133–138. [Google Scholar] [CrossRef]
- O’Neill, E.T.; Tuohy, J.J.; Franz, R. Comparison of milk and ethanol/water mixtures with respect to monostyrene migration from a polystyrene packaging material. Int. Dairy J. 1994, 4, 271–283. [Google Scholar] [CrossRef]
- Begley, T.; Castle, L.; Feigenbaum, A.; Franz, R.; Hinrichs, K.; Lickly, T.; Mercea, P.; Milana, M.R.; O’Brien, A.; Rebre, S.R.; et al. Evaluation of migration models that might be used in support of regulations for food-contact plastics. Food Addit. Contam. 2005, 22, 73–90. [Google Scholar] [CrossRef] [Green Version]
- Nakai, M.; Tsubokura, M.; Suzuki, M.; Fujishima, S.; Watanabe, Y.; Hasegawa, Y.; Oyama, K.; Ogura, S. Genotoxicity of styrene oligomers extracted from polystyrene intended for use in contact with food. Toxicol. Rep. 2014, 1, 1175–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.O.; Jitsunari, F.; Asakawa, F.; Lee, D.S. Migration of styrene monomer, dimers and trimers from polystyrene to food simulants. Food Addit. Contam. 2005, 22, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Klärner, P.; Klenz, R.; Eder, R.; Volz, W.E.; Schnell, H.W.; Leyendecker, D.; Güntner, A.; Nieβner, N.; Morris, C.R.; Christian, M.S. Preparation and Analysis of Styrene Oligomers Containing Migrates from Various Polystyrenes Used in Food Packaging. Drug Chem. Toxicol. 1998, 21, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, Y.; Nishi, K.; Sasaki, H.; Yamada, T. Determination Method of Styrene Dimers and Trimers in Instant Noodles Contained in Polystyrene Cups. J. Food Hyg. Soc. Jpn. (Shokuhin Eiseigaku Zasshi) 1998, 39, 310–314_1. [Google Scholar] [CrossRef] [Green Version]
- Khaksar, M.-R.; Ghazi-Khansari, M. Determination of migration monomer styrene from GPPS (general purpose polystyrene) and HIPS (high impact polystyrene) cups to hot drinks. Toxicol. Mech. Methods 2009, 19, 257–261. [Google Scholar] [CrossRef]
- Genualdi, S.; Ntim, S.A.; Begley, T. Suitability of polystyrene as a functional barrier layer in coloured food contact materials. Food Addit. Contam. Part A 2015, 32, 1–8. [Google Scholar] [CrossRef]
- Dole, P.; Feigenbaum, A.E.; De La Cruz, C.; Pastorelli, S.; Paseiro, P.; Hankemeier, T.; Voulzatis, Y.; Aucejo, S.; Saillard, P.; Papaspyrides, C. Typical diffusion behaviour in packaging polymers – application to functional barriers. Food Addit. Contam. 2006, 23, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Welle, F. Is PET bottle-to-bottle recycling safe? Evaluation of post-consumer recycling processes according to the EFSA guidelines. Resour. Conserv. Recycl. 2013, 73, 41–45. [Google Scholar] [CrossRef]
- Welle, F. Simulation of the Decontamination Efficiency of PET Recycling Processes based on Solid-state Polycondensation. Packag. Technol. Sci. 2012, 27, 141–148. [Google Scholar] [CrossRef]
- Ewender, J.; Welle, F. Determination of the activation energies of diffusion of organic molecules in poly(ethylene terephthalate). J. Appl. Polym. Sci. 2012, 128, 3885–3892. [Google Scholar] [CrossRef]
- Welle, F. Activation energies of diffusion of organic migrants in cyclo olefin polymer. Int. J. Pharm. 2014, 473, 510–517. [Google Scholar] [CrossRef]
- The Molecular Volumes Were Calculated from the Free Internet Program Molinspiration. Available online: http://www.molinspiration.com (accessed on 10 May 2018).
- Coulson, C.A.; Crank, J. The Mathematics of Diffusion. Math. Gaz. 1958, 42, 165. [Google Scholar] [CrossRef] [Green Version]
- Ewender, J.; Welle, F. Determination and Prediction of the Lag Times of Hydrocarbons through a Polyethylene Terephthalate Film. Packag. Technol. Sci. 2014, 27, 963–974. [Google Scholar] [CrossRef]
- Ewender, J.; Welle, F. Functional Barrier Performance of a Polyamide-6 Membrane Towards n -Alkanes and 1-Alcohols. Packag. Technol. Sci. 2016, 29, 277–287. [Google Scholar] [CrossRef]
- Welle, F. A new method for the prediction of diffusion coefficients in poly(ethylene terephthalate). J. Appl. Polym. Sci. 2013, 129, 1845–1851. [Google Scholar] [CrossRef]
- Ewender, J.; Welle, F. Diffusion Coefficients of n-Alkanes and 1-Alcohols in Polyethylene Naphthalate (PEN). Int. J. Polym. Sci. 2019, 2019, 2748649. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, K.M.A.; Ewender, J.; Welle, F. Recyclable Multilayer Packaging by Means of Thermoreversibly Crosslinking Adhesive in the Context of Food Law. Polymer 2020, 12, 2988. [Google Scholar] [CrossRef] [PubMed]
- Arrhenius, S. Über die Dissoziationswärme und den Einfluss der Temperatur auf den Dissoziationsgrad der Elektrolyte. Zeitschrift für Physikalische Chemie 1889, 4, 96–116. [Google Scholar] [CrossRef] [Green Version]
- Overington, A.R.; Wong, M.; Harrison, J.A.; Ferreira, L.B. Estimation of Mass Transfer Rates through Hydrophobic Pervaporation Membranes. Sep. Sci. Technol. 2009, 44, 787–816. [Google Scholar] [CrossRef]
- Meyer, W.; Neldel, H. Über die Beziehung zwischen der Energiekonstanten und der Mengenkonstanten a in der Leitwerts-Temperaturformel bei oxydischen Halbleitern. Zeitschrift für Technische Physik 1937, 12, 588–593. [Google Scholar]
- Regulation (EU) No 10/2011. Commission Regulation (EU) on Plastic Materials and Articles Intended to Come into Contact with Food. 2011. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32011R0010 (accessed on 16 April 2021).
- Franz, R.; Huber, M.; Piringer, O. Presentation and experimental verification of a physico-mathematical model describing the migration across functional barrier layers into foodstuffs. Food Addit. Contam. 1997, 14, 627–640. [Google Scholar] [CrossRef]
- Guazzotti, V.; Ebert, A.; Gruner, A.; Welle, F. Migration from acrylonitrile butadiene styrene (ABS) polymer: Swelling effect of food simulants compared to real foods. J. Consum. Prot. Food Saf. 2021, 16, 19–33. [Google Scholar] [CrossRef]
- Gehring, C.; Welle, F. Migration Testing of Polyethylene Terephthalate: Comparison of Regulated Test Conditions with Migration into Real Food at the End of Shelf Life. Packag. Technol. Sci. 2018, 31, 771–780. [Google Scholar] [CrossRef]
- Genualdi, S.; Nyman, P.; Begley, T. Updated evaluation of the migration of styrene monomer and oligomers from polystyrene food contact materials to foods and food simulants. Food Addit. Contam. Part A 2014, 31, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, B.; Gontard, N.; Peyron, S. Worst case prediction of additives migration from polystyrene for food safety purposes: A model update. Food Addit. Contam. Part A 2017, 35, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, G.; Choudhury, R.P.; Beckham, H.W. Diffusivity of small molecules in polymers: Carboxylic acids in polystyrene. Polymer 2012, 53, 976–983. [Google Scholar] [CrossRef]
- Bernardo, G. Diffusivity of alcohols in amorphous polystyrene. J. Appl. Polym. Sci. 2012, 127, 1803–1811. [Google Scholar] [CrossRef]
- Bernardo, G. Diffusivity of alkanes in polystyrene. J. Polym. Res. 2012, 19, 9836. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sheet | Substance | Spiked Concentration (mg/kg) | |
|---|---|---|---|
| General Purpose Polystyrene (GPPS) | High Impact Polystyrene (HIPS) | ||
| sheet 1 | n-Octane | 422 ± 5 | 474 ± 37 |
| n-Decane | 486 ± 4 | 354 ± 4 | |
| n-Dodecane | 518 ± 3 | 610 ± 8 | |
| n-Tetradecane | 531 ± 4 | 692 ± 15 | |
| n-Hexadecane | 538 ± 5 | 709 ± 19 | |
| n-Octadecane | 522 ± 5 | 674 ± 19 | |
| Styrene 1 | 627 ± 22 | 354 ± 2 | |
| sheet 2 | Acetone | 180 2 | / |
| Ethyl acetate | 270 2 | 500 2 | |
| Toluene | 464 ± 3 | 763 ± 11 | |
| Chlorobenzene | 521 ± 4 | 821 ± 13 | |
| Phenyl cyclohexane | 627 ± 6 | 1229 ± 33 | |
| Benzophenone | 538 ± 7 | 1002 ± 27 | |
| Styrene 1 | 595 ± 5 | 363 ± 6 | |
| Temperature (°C) | Concentration (µg/L) | |||||
|---|---|---|---|---|---|---|
| Methanol | Ethanol | 1-Propanol | 1-Butanol | 1-Pentanol | 1-Hexanol | |
| 0 | 9.16 | 12.8 | 9.29 | 7.49 | 5.65 | 3.76 |
| 25 | 27.5 | 28.3 | 27.9 | 22.5 | 17.0 | 11.3 |
| 40 | 54.9 | 76.7 | 55.8 | 45.0 | 33.9 | 45.0 |
| 60 | 201 | 281 | 204 | 165 | 124 | 82.8 |
| 70 | 330 | 460 | 335 | 270 | 203 | 135 |
| 80 | 549 | 767 | 558 | 450 | 339 | 226 |
| 90 | 916 | 1280 | 929 | 749 | 565 | 376 |
| Polymer | Temperature | Diffusion Coefficient (cm2/s) | ||||||
|---|---|---|---|---|---|---|---|---|
| (°C) | n-Octane | n-Decane | n-Dodecane | n-Tetradecane | n-Hexadecane | n-Octadecane | Styrene | |
| GPPS | 80 | 3.4 × 10−12 | 2.8 × 10−13 | 3.0 × 10−14 | 4.7 × 10−15 | 1.5 × 10−15 | 1.6 × 10−15 | 1.5 × 10−11 |
| 85 | 1.4 × 10−11 | 2.2 × 10−12 | 4.3 × 10−13 | 1.1 × 10−13 | 4.1 × 10−14 | 1.9 × 10−14 | 3.3 × 10−11 | |
| 90 | 3.8 × 10−11 | 8.6 × 10−12 | 2.4 × 10−12 | 8.9 × 10−13 | 4.3 × 10−13 | 2.8 × 10−13 | 7.1 × 10−11 | |
| 95 | 1.1 × 10−10 | 3.3 × 10−11 | 1.2 × 10−11 | 6.3 × 10−12 | 3.4 × 10−12 | 1.6 × 10−12 | 1.8 × 10−10 | |
| 100 | 3.8 × 10−10 | 1.6 × 10−10 | 7.5 × 10−11 | 4.2 × 10−11 | 2.6 × 10−11 | 1.8 × 10−11 | 4.1 × 10−10 | |
| 105 | 8.6 × 10−10 | 4.0 × 10−10 | 2.1 × 10−10 | 1.3 × 10−10 | 7.6 × 10−11 | 9.7 × 10−10 | ||
| 110 | 2.0 × 10−9 | 1.1 × 10−9 | 6.3 × 10−10 | 4.1 × 10−10 | 2.6 × 10−10 | 1.6 × 10−9 | ||
| 115 | 4.0 × 10−9 | 2.4 × 10−9 | 1.5 × 10−9 | 9.9 × 10−10 | 5.2 × 10−10 | 3.3 × 10−9 | ||
| HIPS | 80 | 2.5 × 10−12 | artefacts 1 | 8.6 × 10−14 | 6.7 × 10−16 | 1.8 × 10−16 | ||
| 90 | 2.0 × 10−11 | 1.6 × 10−12 | 3.1 × 10−14 | 8.1 × 10−15 | ||||
| 95 | 1.8 × 10−10 | 6.5 × 10−12 | 1.4 × 10−12 | 5.0 × 10−13 | ||||
| 100 | 4.0 × 10−10 | 3.0 × 10−11 | 9.7 × 10−12 | 4.6 × 10−12 | ||||
| 105 | 1.9 × 10−9 | 2.0 × 10−10 | 8.2 × 10−11 | 4.6 × 10−11 | ||||
| 110 | 2.9 × 10−9 | 4.9 × 10−10 | 2.3 × 10−10 | 1.4 × 10−10 | ||||
| Polymer | Temperature | Diffusion Coefficient (cm2/s) | ||||||
|---|---|---|---|---|---|---|---|---|
| (°C) | Acetone | Ethyl Acetate | Toluene | Chlorobenzene | Phenyl Cyclohexane | Benzophenone | Styrene | |
| GPPS | 85 | 9.8 × 10−9 | 1.1 × 10−9 | 1.0 × 10−10 | 2.0 × 10−10 | 2.1 × 10−13 | 1.4 × 10−12 | 2.9 × 10−11 |
| 90 | 1.1 × 10−8 | 1.8 × 10−9 | 2.1 × 10−10 | 3.9 × 10−10 | 1.2 × 10−12 | 5.7 × 10−12 | 6.4 × 10−11 | |
| 95 | 1.2 × 10−8 | 3.3 × 10−9 | 4.9 × 10−10 | 8.8 × 10−10 | 9.5 × 10−12 | 2.8 × 10−11 | 1.5 × 10−10 | |
| 100 | 1.0 × 10−8 | 5.2 × 10−9 | 1.1 × 10−9 | 1.9 × 10−9 | 4.1 × 10−11 | 9.6 × 10−11 | 3.5 × 10−10 | |
| 105 | 1.2 × 10−8 | 9.8 × 10−9 | 2.4 × 10−9 | 4.0 × 10−9 | 1.2 × 10−10 | 1.5 × 10−10 | 8.3 × 10−10 | |
| HIPS | 75 | artefacts 1 | 5.1 × 10−10 | 2.9 × 10−11 | 4.5 × 10−11 | 1.8 × 10−11 | ||
| 80 | 6.9 × 10−10 | 4.6 × 10−11 | 6.9 × 10−11 | 6.4 × 10−15 | 4.5 × 10−14 | 2.9 × 10−11 | ||
| 85 | 1.3 × 10−9 | 1.2 × 10−10 | 1.7 × 10−10 | 4.0 × 10−14 | 1.7 × 10−13 | 7.9 × 10−11 | ||
| 90 | 2.2 × 10−9 | 2.4 × 10−10 | 3.3 × 10−10 | 1.5 × 10−13 | 5.5 × 10−13 | 4.0 × 10−11 | ||
| 100 | 6.3 × 10−9 | 1.1 × 10−9 | 1.4 × 10−9 | 5.1 × 10−12 | 1.0 × 10−11 | 8.2 × 10−10 | ||
| 105 | 1.3 × 10−8 | 3.6 × 10−9 | 4.2 × 10−9 | 3.4 × 10−11 | 4.9 × 10−11 | 2.7 × 10−9 | ||
| 110 | 2.3 × 10−8 | 7.8 × 10−9 | 8.7 × 10−9 | 1.4 × 10−10 | 1.1 × 10−10 | 6.2 × 10−9 | ||
| 115 | 5.4 × 10−8 | 2.0 × 10−8 | 2.2 × 10−8 | 5.3 × 10−10 | 2.3 × 10−10 | 1.7 × 10−8 | ||
| Temperature (°C) | Diffusion Coefficient (cm2/s) | |||||
|---|---|---|---|---|---|---|
| Methanol | Ethanol | 1-Propanol | 1-Butanol | 1-Pentanol | 1-Hexanol | |
| 0 | 1.2 × 10−9 | 2.8 × 10−11 | ||||
| 25 | 2.2 × 10−9 | 1.7 × 10−10 | ||||
| 40 | 3.1 × 10−9 | 3.8 × 10−10 | 2.5 × 10−11 | 2.7 × 10−12 | ||
| 60 | 2.7 × 10−9 | 1.0 × 10−9 | 1.0 × 10−10 | 1.7 × 10−11 | ||
| 70 | 2.1 × 10−9 | 1.0 × 10−9 | 2.0 × 10−10 | 4.1 × 10−11 | 9.3 × 10−12 | |
| 80 | 2.4 × 10−9 | 1.4 × 10−9 | 3.5 × 10−10 | 8.5 × 10−11 | 2.1 × 10−11 | 4.2 × 10−12 |
| 90 | 2.4 × 10−9 | 1.6 × 10−9 | 5.6 × 10−10 | 1.7 × 10−10 | 4.8 × 10−11 | 1.3 × 10−11 |
| Substance | Molecular Volume | Molecular Weight | Temperature Range | Activation Energy | Pre-Exponential Factor D0 | Method |
|---|---|---|---|---|---|---|
| (Å3) | (g/mol) | (°C) | (kJ/mol) | (cm2/s) | ||
| Methanol | 37.2 | 32.0 | 0–90 | 4.9 | 1.4 × 10−8 | Permeation, below Tg |
| Ethanol | 54.0 | 46.1 | 0–90 | 37.3 | 5.1 × 10−4 | Permeation, below Tg |
| Acetone | 64.7 | 58.1 | 85–105 | 7.6 | 1.3 × 10−7 | Desorption |
| 1-Propanol | 70.8 | 60.1 | 40–90 | 59.6 | 2.2 × 10−1 | Permeation, below Tg |
| 1-Butanol | 87.6 | 74.1 | 40–90 | 91.1 | 2.6 × 105 | Permeation, below Tg |
| Ethyl acetate | 90.6 | 88.1 | 85–105 | 123.9 | 1.2 × 109 | Desorption |
| Chlorobenzene | 97.6 | 112.6 | 85–105 | 171.3 | 1.8 × 1015 | Desorption |
| Toluene | 100.6 | 92.1 | 85–105 | 180.6 | 2.1 × 1016 | Desorption |
| Styrene | 111.8 | 104.2 | 80–100 | 180.9 | 8.2 × 1015 | Desorption, below Tg |
| Styrene | 111.8 | 104.2 | 100–115 | 162.6 | 2.5 × 1013 | Desorption, above Tg |
| Styrene | 111.8 | 104.2 | 85–105 | 189.8 | 1.3 × 1017 | Desorption |
| n-Octane | 146.6 | 114.2 | 80–100 | 252.1 | 7.0 × 1025 | Desorption, below Tg |
| n-Octane | 146.6 | 114.2 | 100–115 | 191.3 | 2.3 × 1017 | Desorption, above Tg |
| Phenyl cyclohexane | 174.0 | 160.3 | 85–105 | 396.8 | 1.6 × 1045 | Desorption |
| Benzophenone | 174.4 | 182.2 | 85–105 | 315.5 | 1.5 × 1034 | Desorption |
| n-Decane | 180.2 | 142.3 | 80–100 | 336.7 | 2.2 × 1037 | Desorption, below Tg |
| n-Decane | 180.2 | 142.3 | 100–115 | 220.6 | 1.2 × 1021 | Desorption, above Tg |
| n-Dodecane | 213.8 | 170.3 | 80–100 | 416.4 | 1.6 × 1048 | Desorption, below Tg |
| n-Dodecane | 213.8 | 170.3 | 100–115 | 243.1 | 8.1 × 1023 | Desorption, above Tg |
| n-Tetradecane | 247.3 | 198.4 | 80–100 | 489.1 | 1.5 × 1058 | Desorption, below Tg |
| n-Tetradecane | 247.3 | 198.4 | 100–115 | 256.4 | 3.4 × 1025 | Desorption, above Tg |
| n-Hexadecane | 281.0 | 226.5 | 80–100 | 525.8 | 1.4 × 1063 | Desorption, below Tg |
| n-Hexadecane | 281.0 | 226.5 | 100–115 | 244.3 | 4.4 × 1023 | Desorption, above Tg |
| n-Octadecane | 314.6 | 254.5 | 80–100 | 504.9 | 8.5 × 1059 | Desorption, below Tg |
| Substance | Molecular Volume | Molecular Weight | Temperature Range | Activation Energy | Pre-Exponential Factor D0 | Method |
|---|---|---|---|---|---|---|
| (Å3) | (g/mol) | (°C) | (kJ/mol) | (cm2/s) | ||
| Ethyl acetate | 90.6 | 88.1 | 75–100 | 111.4 | 2.4 × 107 | Desorption, below Tg |
| Ethyl acetate | 90.6 | 88.1 | 100–115 | 169.3 | 3.1 × 1015 | Desorption, above Tg |
| Chlorobenzene | 97.6 | 112.6 | 75–100 | 152.2 | 2.6 × 1012 | Desorption, below Tg |
| Chlorobenzene | 97.6 | 112.6 | 100–115 | 218.1 | 5.1 × 1021 | Desorption, above Tg |
| Toluene | 100.6 | 92.1 | 75–100 | 162.8 | 6.7 × 1013 | Desorption, below Tg |
| Toluene | 100.6 | 92.1 | 100–115 | 224.4 | 3.2 × 1022 | Desorption, above Tg |
| Styrene | 111.8 | 104.2 | 75–100 | 169.2 | 3.7 × 1014 | Desorption, below Tg |
| Styrene | 111.8 | 104.2 | 100–115 | 239.3 | 6.9 × 1023 | Desorption, above Tg |
| n-Octane | 146.6 | 114.2 | 80–100 | 288.5 | 1.0 × 1031 | Desorption, below Tg |
| n-Octane | 146.6 | 114.2 | 100–110 | 236.8 | 6.7 × 1023 | Desorption, above Tg |
| Phenyl cyclohexane | 174.0 | 160.3 | 80–100 | 367.4 | 1.2 × 1040 | Desorption, below Tg |
| Phenyl cyclohexane | 174.0 | 160.3 | 100–115 | 367.7 | 1.8 × 1040 | Desorption, above Tg |
| Benzophenone | 174.4 | 182.2 | 80–100 | 297.0 | 3.4 × 1030 | Desorption, below Tg |
| Benzophenone | 174.4 | 182.2 | 100–115 | 246.3 | 3.9 × 1023 | Desorption, above Tg |
| n-Dodecane | 213.8 | 170.3 | 80–100 | 318.7 | 1.4 × 1034 | Desorption, below Tg |
| n-Dodecane | 213.8 | 170.3 | 100–115 | 233.0 | 1.4 × 1036 | Desorption, above Tg |
| n-Tetradecane | 247.3 | 198.4 | 80–100 | 538.4 | 2.1 × 1064 | Desorption, below Tg |
| n-Tetradecane | 247.3 | 198.4 | 100–110 | 376.1 | 5.1 × 1041 | Desorption, above Tg |
| n-Hexadecane | 281.0 | 226.5 | 80–100 | 567.2 | 9.3 × 1067 | Desorption, below Tg |
| n-Hexadecane | 281.0 | 226.5 | 100–110 | 407.6 | 6.3 × 1045 | Desorption, above Tg |
| n-Octadecane | 314.6 | 254.5 | 100–110 | 472.8 | 3.7 × 1054 | Desorption, above Tg |
| Parameter | GPPS | HIPS | ||
|---|---|---|---|---|
| below Tg | above Tg | below Tg | above Tg | |
| a (1/K) | 2.59 × 10−3 | 2.44 × 10−3 | 2.55 × 10−3 | 2.46 × 10−3 |
| b (cm2/s) | 7.38 × 10−9 | 6.46 × 10−8 | 9.21 × 10−9 | 2.07 × 10−7 |
| c (Å3) | 55.71 | 25.51 | 73.28 | 45.00 |
| d (1/K) | 2.73 × 10−5 | 7.55 × 10−5 | 2.04 × 10−5 | 3.57 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welle, F. Diffusion Coefficients and Activation Energies of Diffusion of Organic Molecules in Polystyrene below and above Glass Transition Temperature. Polymers 2021, 13, 1317. https://doi.org/10.3390/polym13081317
Welle F. Diffusion Coefficients and Activation Energies of Diffusion of Organic Molecules in Polystyrene below and above Glass Transition Temperature. Polymers. 2021; 13(8):1317. https://doi.org/10.3390/polym13081317
Chicago/Turabian StyleWelle, Frank. 2021. "Diffusion Coefficients and Activation Energies of Diffusion of Organic Molecules in Polystyrene below and above Glass Transition Temperature" Polymers 13, no. 8: 1317. https://doi.org/10.3390/polym13081317
APA StyleWelle, F. (2021). Diffusion Coefficients and Activation Energies of Diffusion of Organic Molecules in Polystyrene below and above Glass Transition Temperature. Polymers, 13(8), 1317. https://doi.org/10.3390/polym13081317