
Dynamics & Spectroscopy with Neutrons—Recent Developments & Emerging Opportunities


1
Materials Physics Center, CSIC-UPV/EHU, Paseo Manuel de Lardizabal 5, 20018 Donostia-San Sebastian, Spain
2
Polish Academy of Sciences, Center of Molecular and Macromolecular Studies, Sienkiewicza 112, 90-363 Lodz, Poland
3
Elettra—Sincrotrone Trieste S.C.p.A., S.S. 14 km 163.5 in Area Science Park, 34149 Trieste, Italy
4
Donostia International Physics Center (DIPC), Paseo Manuel de Lardizabal 4, 20018 Donostia-San Sebastian, Spain
5
Department of Physics and Astronomy, University College London, Gower Street, London WC1E 6BT, UK
6
IKERBASQUE, Basque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Spain
*
Author to whom correspondence should be addressed.
Polymers 2021, 13(9), 1440; https://doi.org/10.3390/polym13091440
Submission received: 12 April 2021
/
Accepted: 27 April 2021
/
Published: 29 April 2021
(This article belongs to the Special Issue Polymers and Soft Matter: From Synthesis to Structure & Dynamics)
Abstract
:This work provides an up-to-date overview of recent developments in neutron spectroscopic techniques and associated computational tools to interrogate the structural properties and dynamical behavior of complex and disordered materials, with a focus on those of a soft and polymeric nature. These have and continue to pave the way for new scientific opportunities simply thought unthinkable not so long ago, and have particularly benefited from advances in high-resolution, broadband techniques spanning energy transfers from the meV to the eV. Topical areas include the identification and robust assignment of low-energy modes underpinning functionality in soft solids and supramolecular frameworks, or the quantification in the laboratory of hitherto unexplored nuclear quantum effects dictating thermodynamic properties. In addition to novel classes of materials, we also discuss recent discoveries around water and its phase diagram, which continue to surprise us. All throughout, emphasis is placed on linking these ongoing and exciting experimental and computational developments to specific scientific questions in the context of the discovery of new materials for sustainable technologies.
1. Overview
The use of the neutron as an exquisite probe of the structure and dynamics of condensed matter is not only well-established but also continues to evolve in exciting directions. Currently under construction in Lund (Sweden), the European Spallation Source (ESS) seeks to offer order-of-magnitude gains in capability relative to the state-of-the-art, as presented in the recent overview of its forthcoming instrument suite by Andersen et al. [1]. Equally, recent advances towards the design and construction of increasingly more compact neutron sources across the globe are pushing the boundaries of the discipline into entirely uncharted territory [2,3,4,5,6,7,8,9], as well as it is providing fertile new ground and a plethora of fresh opportunities for further developments, much in the same way as it happened when X-ray sources entered the laboratory some decades ago [10].
Progress in neutron science over the past few decades and up until a few years ago has been extensively reviewed in a series of three recent monographs [11,12,13]. Among the wide range of techniques available at present, Neutron Spin-Echo (NSE) and QuasiElastic Neutron Scattering (QENS) have been the most-commonly used to explore the dynamics of soft matter [14,15,16,17,18,19,20,21]. Recent highlights in QENS include the implementation of neutron polarization analysis on a wide-angle time-of-flight spectrometer at a pulsed neutron source, allowing a clean separation of coherent and incoherent contributions with sub-meV resolution and over a wide momentum-transfer (Q) range [22]. In contrast, the widespread use of Inelastic Neutron Scattering (INS) can still be regarded in its infancy compared to the above [23,24,25], although its potential to explore a number of important and relevant phenomena has been recognized for some time now, including: crystallinity [26,27,28]; methyl [29,30] and short-chain dynamics [31]; the vibrational dynamics of polymer glasses [18,32,33,34,35,36]; or even (although more rarely) polymer thin-films [37,38,39]. For the purposes of this review, INS would refer to the study of well-defined modes at energy transfers (E) ranging from a few meV all the way up to ca. 500 meV, the highest energy characterizing the vibration of the lightest molecule, H.
Beyond the aforementioned upper bound for INS, the use of epithermal (electron-volt) neutrons leads to Neutron Compton Scattering (NCS), also referred to as Deep Inelastic Neutron Scattering (DINS). NCS gives access to mass-resolved nuclear mean kinetic energies and the underlying momentum distributions, of relevance to the study of quantum effects in condensed-matter systems [40]. Traditionally, NCS has been particularly useful in the study of hydrogenous matter [41], yet at the same time the technique continues to be extended to heavier elements under the wider umbrella of MAss-resolved Neutron SpEctroscopic (MANSE) techniques [42,43,44,45,46,47,48,49]. Neutron spectrometers in a so-called inverted-geometry configuration at a pulsed source represent the most natural way to implement NCS and MANSE, as exemplifed by the current science programme on the VESUVIO spectrometer (ISIS, UK) [50,51,52,53,54,55,56,57,58,59], although it is also to be noted that other approaches have been attempted in the past [60,61,62].
The primary purpose of this contribution is to highlight recent advances and applications of INS and NCS in the context of soft matter. For the benefit of both novice and expert, emphasis is placed on the new tools and methodologies that have become available to interrogate the dynamical behavior of a growing range of complex materials of relevance to sustainable applications, particularly those of a soft and polymeric nature. In doing so, we also seek to bring to the fore the unrivalled ability of neutron spectroscopic techniques to probe dynamical phenomena over a wide range (decades) in timescales, from slow stochastic and relaxation phenomena in disordered media to the ultimate effects of quantum mechanics on properties and function (see Figure 1). To this end, we have chosen a limited number of areas of research that serve to illustrate the most salient points and take-home messages that we would like to convey to the reader, particularly to those who have shied away until now from the use of these superb tools of scientific inquiry.
2. Soft Media for Photovoltaics & Photonics
A class of solids currently dubbed Organometal Halide Perovskites (hereafter OHPs) were nothing more than a scientific curiosity not so long ago, practically unknown to most but a handful of materials scientists interested in their rather unusual properties as soft ‘plastic’ crystals [64]. This situation has changed dramatically since 2009 [65], following the use of the OHP methylammonium lead iodide (molecular formula CHNHPbI, hereafter denoted MAPbI) as a sensitizer in solar cells [66,67]. A decade on, top-performing OHP-based devices have attained record conversion efficiencies of solar light into electricity of 20–25%, a figure well above the (rather-shy) value of 3.8% of a decade ago, and comparable to far-more-costly silicon-based technologies [68]. Notwithstanding this progress, critical issues remain, primarily associated with their physico-chemical stability and their propensity to degrade [69,70]. Circumventing these difficulties requires a robust and coherent understanding of OHPs at the atomic and molecular levels, and this task has and continues to be a challenge to both experiment and theory. This is where recent developments in neutron spectroscopy come into the picture.
Chemically speaking, OHPs arise from the insertion of an organic cation into an inorganic perovskite lattice, replacing the more traditional metal ion. Such a substitution gives rise to an incredibly rich and complex structural diversity, as well as to new properties associated with the additional degrees of freedom introduced by the organic cation. Among the OHPs studied to date, the ones containing methylammonium (MA+) and formamidinium (FA+) moieties confined in iodo- and bromoplumbate frameworks are the most archetypal ones, and as such they will be used as a guide for further discussion below. This hybrid organic-inorganic chemical composition also translates into an unusual softness and flexibility, which gives rise to a rich polymorphism [71]. In terms of elastic properties, Figure 2 serves to illustrate where OHPs sit relative to other classes of materials [72,73], essentially at the cross-roads between the ‘soft’ and ‘hard’ worlds, and comparable to typical polymer or macromolecular systems of an organic nature.
The structural complexity of the iodoplumbates is further illustrated in the compositional phase diagram of solid solutions (Figure 3) [75]. In this Figure, the arrows next to ‘Pressure’ indicate additional phase transitions as a function of this parameter for the parent compound [76]. Other phenomena illustrated in the figure include: strong isotope effects as a function of pressure (left) [77]; thermodynamic metastability leading to disproportionation into non-perovskite phases with poor photophysical response (right) [78]; or the emergence of orientational-glassy phases as a result of the dynamical arrest of the organic moiety (bottom) [79,80]. In addition, both MAPbI3 and FAPbI3 reveal a very complex polymorphism, closely linked to the progressive ‘melting’ of the organic cations within the solid. At ambient pressure, MAPbI3 shows three different crystal structures as a function of temperature: a low-temperature orthorhombic phase for T < 160 K; a tetragonal phase for 160 K < T < 330 K; and a high-temperature cubic phase for T > 330 K. For FAPbI3, the thermodynamically stable phase at ambient conditions is a non-perovskite hexagonal phase (yellow -polymorph). However, a metastable cubic perovskite phase (black -phase) can be stabilized at RT for hours to weeks via heating above the hexagonal-to-cubic phase transition at 410 K. Upon cooling from the cubic phase, a tetragonal phase (-phase) is formed below 285 K, and yet another structure is found below 140 K (-phase).
A substantial part of our current understanding of the structural properties of these materials stems from Neutron Diffraction (ND) [82,83,84,85,86,87,88,89,90,91,92,93,94]. Most importantly, the use of neutron Laue diffractometers, operating both at pulsed (e.g., SXD at ISIS, UK [95]; TOPAZ at SNS [96], US; SENJU at J-PARC, JP [97]) and continuous neutron sources (e.g., KOALA at ANSTO, AU [98]; FALCON at BER-2, DE [99]—currently transferred to the spallation source SINQ, CH) provide the means to precisely explore OHP single-crystal samples from a crystallographic perspective. Notwithstanding these efforts, the structure of both MAPbI3 and FAPbI3 remains elusive to a great extent, progress being hindered by the presence of both static and dynamical disorder, as well as by the formation of domain structures of multiple origins [75], or the propensity of these crystals to twin [100]. As a result, the assignment of the space group of each perovskite phase of both MAPbI3 and FAPbI3 discussed above has been debated from the outset [75]. The strong tendency of MAPbX3 to twinning, for example, arises from the ferroelastic relations between the high- and low-temperature phases, where the samples first nucleate and then crystallize, respectively [100]. This phenomenon has been recently explored by Breternitz et al. using single-crystal ND on the state-of-the-art instrument FALCON [94]. Similarly, the high-resolution total-scattering ND experiments on powder samples of FAPbI3 have brought to the fore the importance of partial disorder and the formation of an orientational glass to account for the metastability of the perovskite phases in this material. Extensive studies of the structural properties of FAPbI3 at ambient pressure can be found in Refs. [87,91].
Pressure studies of the structure of OHPs have been primarily carried out using synchrotron X-rays, with some ongoing debates on the space-group assignment of the high-pressure phases [76,100,101,102,103,104]. Parallel studies using neutrons continue to be quire sparse, largely limited by the large incoherent cross section from hydrogen and the need for per-deuterated specimens. Structural changes upon compression in (MA-d6)PbBr3 have been studied with powder ND up to ca. 2.8 GPa by Swainson and co-authors [105], yet even a fundamental understanding of the structural properties of this material at ambient conditions remains elusive [106]. Isotope effects have also been largely ignored in most high-pressure OHP research. In a recent paper, Kong and co-workers [107] used both high-pressure synchrotron and neutron diffraction (PLANET beamline, J-PARC, JP) and found that H/D substitution plays a critical role in suppressing lattice disorder in MAPbI3. Furthermore, per-deuteration gives rise to a large enhancement of light emission and structural robustness, acompanied by a slower degradation of photovoltaic performance.
Beyond structural studies, work to date on INS has been briefly reviewed by Mozur et al. [108]. These activities can be conveniently subdivided into coherent and incoherent INS experiments, the latter focused on the hydrogenous organic cations. The coherent phonon response brings information on the collective dynamics of the OHP framework, which can be effectively explored by neutron scattering. A number of neutron scattering instruments have been used so far for the study of OHPs. These instruments can be roughly classified as triple-axis spectrometers (TAS), operating at reactor sources (e.g., HB3, HFIR, ORNL, US [109]; BT4 and SPINS at NIST [110,111,112], US; IN12 and IN22 at ILL, FR. [112,113]; 4F1, 4F2 and 1T at LLB, FR [113,114,115,116]); and the time-of-flight (TOF) instruments available at spallation (e.g., MAPS and MARI at ISIS, UK [117,118,119]; 4SEASONS and AMATERAS at J-PARC, JP [120,121]; or ARCS at SNS, ORNL, US [120]) and reactor sources (IN4 and IN5 at ILL, FR [116,117,122]). However, as with ND, the incoherent scattering from hydrogen masks and obscures the coherent response. As a result, there have also been several reports on the successful application of Inelastic X-ray Scattering (IXS) to elucidate the collective dynamics of hydrogenous samples [88,123,124]. Overall, these studies of coherent lattice dynamics continue to provide essential information on the mechanical properties of the soft inorganic frameworks (mediated by acoustic phonons). In addition, they have facilitated a better understanding of charge-transport properties since the frontier electronic states of OHPs couple primarily to low-energy optical phonons [108]. The complementarity between inelastic neutron and X-ray scattering is illustrated in Figure 4a, showing phonon-dispersion relations for both per-deuterated and hydrogenous single crystals of MAPbI3 by means of INS-TAS and IXS, respectively. As clearly shown in these data, H/D substitution can lead to substantial shifts of the low-energy modes and the resulting elastic properties, thus serving as a timely cautionary note in future studies.
The first INS experiments on OHPs were reported by Swainson and co-workers, dealing with the case of MAPbBr3 [117]. This work identified the mode softening associated with rich polymorphic transitions in this system. Soon after, Létoublon et al. presented an exhaustive analysis of the low-frequency dynamics in MAPbBr3 with INS-TAS experiments combined with Raman and Brillouin scattering, and ultrasound measurements [115]. This line of research has been continued by the authors, providing the most comprehensive analysis of the mechanical properties of both MAPbX3 and FAPbX3 published to date [113]. INS has the advantage of providing microscopic information on the elastic properties of OHPs, since the acoustic phonons possess wavelengths which are considerably shorter than crystal dimensions. Moreover, it allows measurements in the THz range, where relaxation effects are negligible [125]. These works have built a picture of OHPs as materials with an exceptionally low shear elastic constant C44. For instance, the C44 constants reported for FAPbBr3 and FAPbI3 by Ferreira et al. amount to 3.1 ± 0.1 GPa and 2.7 ± 0.3 GPa, respectively [113]. These values are comparable to 2.1 ± 0.3 GPa in crystalline polyethylene, measured with INS-TAS by Heyer et al. [125]. Furthermore, an analysis of the elastic properties of the cubic perovskite phase of FAPbI3 provides important clues on its metastability. The softening of the shear modulus with temperature also confirms the aforementioned ferroelastic relations in MAPbX3 perovskites, a pivotal finding to understand their strong tendency to exhibit twinning [100]. Twinning has been also evidenced by Li et al. in high-resolution experiments on a hydrogenous MAPbI3 single-crystal, performed with the chopper TOF spectrometer AMATERAS [121]. These experiments combined with extensive QENS and MD analysis of the underlying cation dynamics, allowed a clean distinction between the individual contributions from acoustic and optical phonons to the (exceedingly low) thermal conductivities of MAPbX3 perovskites. The authors proposed that the nanoscale mean-free-paths of acoustic modes together with lower velocities are responsible for such a small thermal conductivity and suggested a marginal influence of optical phonons on heat-transport properties [121]. These conclusions have been corroborated and explored further by Gold-Parker et al. [110] with INS-TAS experiments on per-deuterated MAPbI3, providing high-precision momentum-resolved measurements of acoustic phonon lifetimes, and demonstrating that acoustic phonons are unable to dissipate heat efficiently. These results highlight the importance of the so-called ‘phonon bottleneck’ effect, a phenomenon that opens the possibility of using OHPs in hot-carrier solar cell devices. In these circumstances, the loss of photogenerated charge carriers can be mitigated, thereby boosting device efficiency. Normally, carrier thermalization and cooling mechanisms rely on the coupling to optical phonons which, however, must further decay into acoustic ones and then dissipate. However, in some cases these phonons are produced at very high density so that they cannot decay away fast enough, allowing to scatter back and reheat the carrier ensemble. This phenomenon was examined in the aforementioned paper by Gold-Parker et al. [110], showing a pronounced inefficiency of acoustic-phonon-mediated heat-transport mechanisms in the MAPbI3 lattice. Yang et al. have explained the ‘phonon bottleneck’ effect in OHPs by acoustic-phonon up-conversion, which leads to a LO phonon emission rate ten times slower in FAPbI3 compared to the all-inorganic counterpart CsPbI3 [126]. The most-recent research on MAPbI3 further addresses the isotope effect revealed earlier by Kong et al. [77]. Manley et al. [109] have extended their previous INS-TAS experiments on a per-deuterated MAPbI3 [110,113] (see Figure 4a), revealing that isotope substitution in MA+ causes a large (20–50%) softening of the longitudinal acoustic (LA) phonons near zone boundaries, reduces thermal conductivity by nearly 50%, and slows carrier relaxation kinetics. These significant effects serve to emphasize once more out the importance of a coupling of LA phonons to the librational modes of the methylammonium molecule, which are strongly mass-dependent [109].
INS-TAS in combination with INS-TOF experiments also bring to the fore the role of optical phonons in the photophysics of these materials, as discussed in Refs. [109,127,128,129,130]. As noted by Mozur et al., optical phonons are responsible for over half of the polarizability in the dielectric response of MAPbI3 [108]. As deduced from the characteristic line shape broadening of the photoluminescence (PL), the main mechanism limiting the mobility of free carriers and excitons in OHPs is via the Fröhlich interaction between charge carriers and Longitudinal Optical (LO) phonon modes, which is, hence, the most important form of electron-phonon coupling in OHPs [108,116]. This phonon scattering mechanism is believed to be the key fundamental factor in establishing the intrinsic limit of charge carrier mobility [116]. State-of-the art INS experiments on optical phonons across the entire frequency range and over a wide Q-range have been recently presented and discussed by Ferreira et al. [116] (MAPX3 and FAPX3) and Zhang et al. [120] (MAPI3). Ferreira et al. have established the dispersionless character of optical phonons, overlapping with the upper part of the acoustic branches. The authors also highlight a significant anharmonic behavior, manifested as phonon overdamping at temperatures well below ambient. These results raise some question marks on commonly accepted ways of describing charge-carrier mobilities based on a quasi-particle picture for low-energy optical lattice modes [116]. Phonon damping was also examined by Zhang et al., probing the spatial and temporal coherence of the optical modes [120]. Moreover, the above phenomena do not appear to be restricted to OHPs. The very recent and elegant work by Lanigan-Atkins et al. [131] on CsPbBr3 show unambiguous signatures of liquid-like damping of Br-dominated phonons of a two-dimensional nature directly impacting the electronic gap-edge states.
Beyond the above, INS studies focusing on the organic moiety by exploiting the incoherent scattering from protons address another set of fundamental questions on the nature and coupling of cation and framework motions, of direct relevance to electron-phonon coupling and the overall photophysics of these materials (see, e.g., the discussion presented in Ref. [108]). The organic cation is known to accelerate the formation of large polarons that interact with both excitons and charge carriers, thereby extending the lifetime of the latter [108,132]. In addition, they participate in dynamical processes that reduce phonon lifetimes in these systems through a ‘rattling’ effect [133], which further reduces the macroscopic thermal conductivity. There is also mounting evidence on mode-specific phonon-bandgap coupling in OHPs [134], well beyond the seminal works on organic-rotor dynamics in the QENS domain [121,135]. The above-discussed papers have used both INS-TAS and direct-geometry INS-TOF instruments, with a focus on the low-frequency regime. A great advantage of TAS is their flexibility, allowing access to basically any Q-point, yet it can suffer from limited resolution and count rate. On the other hand, direct-geometry chopper TOF instruments offer simultaneous access across a wide range of both energy-transfers and scattering vectors, having large detector areas and so offering very-high sensitivity. On the other hand, for a direct-geometry instrument there is a significant resolution broadening, and the best resolution is only obtained when the energy of a given phonon is close to the incident energy selected by a mechanical chopper. Therefore, it is not possible to collect a high-resolution spectrum over the entire spectral range in one go, and merging different spectral regions is difficult because of the varying resolution functions [136]. This can become a serious limitation in molecular systems, where vibrational modes are typically spread out over a wide spectral range, including the celebrated ´fingerprint´ region. Indirect-geometry neutron spectrometers offer a means of circumventing these issues. Optimized to use short neutron pulses, the prime example is TOSCA, operating at ISIS, UK [137,138,139,140,141]. The key difference between TOSCA and direct-geometry instruments is the fact that it operates at specific and narrow energy-momentum-transfer kinematic trajectories optimized for the study of hydrogenous systems, thus providing a high and nearly constant relative resolution over a broad energy-transfer range. TOSCA has been subjected to significant upgrades over the last decade [137,138,139,140,141], which are still in progress and will result in even a higher detected flux. Capitalizing from this success, two broadband instruments of the same geometry have been developed at pulsed sources: VISION (SNS, ORNL, US [142]; in operation) and VESPA (ESS, SE [1]; under construction). LAGRANGE (ILL, FR) also shares the same layout to attain a high resolution, yet the steady-state nature of the source requires that the incident energy needs to be scanned [143]. Contrary to TAS or direct-geometry TOF spectrometers, this kind of instrumentation is characterized by a very high efficiency, allowing to study small samples within experimental time-scales comparable to standard optical vibrational spectroscopy techniques, i.e., infrared and Raman. These merits open the path for, e.g., in-situ and operando experiments in catalysis, or the use of high-pressure and gas-handling systems, opening new and exciting opportunities for chemistry and materials science. A limitation that needs to be kept in mind is the lack of access to small Qs at high Es, which results in a damping of the intensity of fundamental phonon excitations with energy transfer, accompanied by a concomitant increase of the intensity of overtones. Notwithstanding this limitation, this review amply demonstrates the versatility and power of this novel instrumentation in the study of molecular systems, where a high spectral resolution across a broad energy-transfer range becomes the key figure of merit.
Figure 5 serves to illustrate the above, with an example of the orthorhombic phase of MAPbI studied at cryogenic conditions using indirect-geometry (TOSCA and VISION), TAS (1T) [116], and chopper-TOF (AMATERAS, Ei = 54 meV) [121] instruments. The spectral range has been limited to 50 meV, as, in this case, this range is key to understand the librational dynamics and the local structure in the vicinity of the organic cation. The first high-resolution INS studies of MAPbI over a wide spectral range were reported by Drużbicki et al. [144] using the previous incarnation of TOSCA (see the black curve in Figure 5). More recently, Manley et al. have reported the INS spectrum recorded on VISION [109] (blue curve in the figure), with a considerably better signal-to-noise ratio. These latter data are of comparable quality to those of a more recent study by Drużbicki et al. [75], obtained on an upgraded TOSCA, hereafter TOSCA+ [138,139,140] (red curve). While both VISION and TOSCA+ cannot offer the resolution at low-frequencies accessible with TAS or chopper-TOF instruments, recent developments on the state-of-the-art instrument OSIRIS, operating also at ISIS, provide unprecedented opportunities for THz-neutron spectroscopy [145].
The work of Drużbicki et al. [144] on the cation dynamics in MAPbI was heavily underpinned by the use of computational materials modeling to interpret the INS data, which is exquisitely sensitive to the local structure around the organic cation and the underlying vibrational motions. Contrary to crystallographic techniques providing information on time- and spatially averaged structure, spectroscopy enables access to atomic and molecular correlation functions in both space and time and, thus, furnish additional insights into the local structure and ensuing dynamics [75]. Moreover, these capabilities also find a natural ally in high-resolution solid-state NMR (ss-NMR) spectroscopy [146,147,148]. As illustrated by Drużbicki et al. [149], INS spectroscopy offers high potential to supplement ss-NMR in solving structural problems of a soft nature, particularly at cryogenic conditions which are hardly accessible to high-resolution NMR experiments (see, e.g., Ref. [150]). These synergies pave the way for the study of a wide variety of systems, including those disordered or nano-confined [151]. Drużbicki et al. [144] also highlight the mismatch between the averaged structures of OHPs defined with diffraction techniques and the local structure unveiled with spectroscopy. In particular, this work shows unequivocally that the commonly accepted Pnma model of the low-temperature phase of MAPbI is insufficient to describe the experimental spectroscopic data, pointing at the importance of octahedral deformations [144]. As extensively discussed in Ref. [75], the space-group assignment of each phase in this material has been debatable from the very beginning, and the origin of this discrepancy is linked to the dynamics and orientational disorder present at length scales commensurate with unit-cell dimensions. In this case, INS spectroscopy is particularly powerful.
The above work also illustrates the increasingly important role of first-principles modeling in linking the INS data to local structure. In this context, a very significant amount of progress has been made over the last decade with the development of advanced tools for the ab initio modeling of materials. These enable accurate and efficient Harmonic Lattice-Dynamics (HLD) calculations and makes more demanding, time-evolved modeling from first-principles (ab initio Molecular Dynamics, AIMD) feasible. A discussion on the current trends and practical aspects of modeling the low-frequency vibrational spectra can be found in Ref. [152]. Usually, electronic-structure calculations are performed using several implementations of density functional theory (DFT), relying either on the use of plane waves (e.g., CASTEP [153,154], QUANTUM ESPRESSO [155], ABINIT [156]), projector augmented-waves (e.g., VASP [157], GPAW [158]), and localized numerical (FHI-aims [159,160], SIESTA [161]) or Gaussian (CRYSTAL [162], TURBOMOLE [163]) basis sets, a hybrid plane-wave / Gaussian approach (CP2K [164]), or imposing all-electron full-potential Linearised Augmented Plane Waves (LAPW; WIEN2K [165] and ELK [166] programs). These codes offer calculations of phonon properties by means of finite-displacement methods (FD), typically using the Parlinski-Kawazoe Direct Method (DM) [167], or via perturbative treatments of ionic displacements through Density Functional Perturbation Theory (DFPT) [168,169,170]. Furthermore, the development of PHONOPY [171,172], an open source package for phonon calculations at the harmonic and quasi-harmonic levels with input from external codes (e.g., all the first-principles calculation codes listed above), should be emphasized. While the DM is straightforward to determine the total energy as a function of atomic displacements, phonon frequencies can be accurately calculated only at the wave vectors that are commensurate with the supercell geometry [173]. On the contrary, DFPT can calculate analytically the dynamical matrix at any arbitrary wave vector, but extensive programming might be required depending on the exchange-correlation (XC) functional and the basis-set definition used in the specific DFT implementation at hand [173]. The most extensive implementations of DFPT for lattice dynamics using a reciprocal-space formalism are offered by ABINIT [156], QUANTUM ESPRESSO [155] and CASTEP [154]. Recently, Scheffler and co-workers have released an efficient implementation using a real-space formalism, available in the FHI-aims code [160]. As an alternative, Lloyd-Williams and Monserrat [174] have proposed an extension of the FD method. This Non-diagonal Supercell Method (NCM) takes advantage of the periodicity of the system allowing a calculation of the dynamical matrix using supercells, which are much smaller than those used in DM calculations and, hence, more efficient. To date, a number of codes have also been developed to simulate neutron observables directly from HLD outputs [162,171,175,176,177,178,179,180].
Following the first high-resolution INS study of MAPbI by Drużbicki et al. [144], subsequent works have used TOSCA and VISION to interrogate the cation dynamics in OHPs. Using TOSCA, Kieslich et al. [181] investigated the relevance of the subtle balance between vibrational entropy and hybrogen bonding in the phase behavior of MAPbBr. Mozur et al. have employed VISION to study the effects of Cs+ substitution in MAPbBr and FAPbBr perovskites [81]. These efforts are driven by the need to find new ways to improve the long-term stability and reliability of OHPs, cation mixing being an important route to this end. More recently, Drużbicki et al. [75] have used INS heavily supported by computational materials modeling to interrogate the mechanism behind phase stabilization in solid mixed-cation compositions. This work provides a detailed and quantitative view of the local environment and vibrational dynamics of FA+ cations in this important OHP. In addition to confirming the formation of an orientational glass in FAPbI, these results link the dynamical behavior of the cations in [MA/FA]PbI to the structural stabilization of the photophysically active perovskite phase via a novel locking mechanism. Detailed simulations validated by the INS data show the emergence of synergistic dynamics involving MA+ and FA+ cations, directly affecting the geometry of the surrounding inorganic framework. This work also illustrates the advantages of the joint use of INS and AIMD simulations to unravel the behavior of a massively disordered soft-matter system of technological relevance.
Figure 6 further illustrates the power of such a joint experimental-computational approach by presenting more recent time-evolved simulations, relying on an extensive model of [MA/FA]PbI solid solutions. MA+ cations are trapped inside the tetragonal perovskite framework of the parent FAPbI and are modeled and compared to the INS spectrum of the [MA/FA]PbI solid solution measured on TOSCA+. The analysis of the difference spectrum obtained by substracting the INS signal of the parent FAPbI from the [MA/FA]PbI mixture evinces strong changes in the vibrational dynamics of both MA+ and FA+ cations (the latter analyzed in detail in Ref. [75]). An excellent match between the experimental and simulated spectra gives us sufficient confidence to explore further the local structure around the MA+ cations. Such structure is characterized by a weakening of the hydrogen-bonding (H-bonding) motifs confining methylammonium cations in the surrounding iodoplumbate cages. As a result, MA+ undergoes a pronounced rattling behavior, locking the formamidinium cations in the neighboring voids, and preventing the transformation of the metastable perovskite framework to the thermodynamically stable hexagonal phase. Establishing the mechanism of stabilization of the perovskite phase (which from the outset stems from the softness of the iodoplumbate framework) can therefore facilitate the design of efficient routes for the further optimization of these solid solutions. In closing this discussion, we also note that to date a number of tools has been developed to support neutron scattering experiments with AIMD simulations, including calculations of the Vibrational Densities of States or VDOS (TRAVIS [182], MDANSE [183], OCLIMAX [184], DYNASOR [185]), as well as anharmonic phonon-dispersion relations at finite temperatures (DYNASOR [185], DynaPhoPy [186], phq [187]). Further discussion on the application of AIMD as a route to explore the vibrational dynamics of condensed matter is presented in Section 4.
3. From Confined Polymers to Soft Supramolecular Frameworks
The use of INS in the study of polymers has experienced a steady rise over the past decade, certainly facilitated by recent developments in the requisite instrumentation. Particular emphasis has been placed on exploring the consequences of spatial confinement at the nanoscale. The use of graphitic materials as confining media represents a nice illustration of the merits and strengths of INS to isolate the spectral response of the (hydrogenous) polymeric phase relative to that of the carbon-based substrate. Owing to the increased sensitivity of the new TOSCA+ spectrometer, for example, it has been recently shown that INS can be exquisitely sensitive to chemical composition and the underlying (and disordered) structure of graphene-related materials [188]. Considerable attention has also been given to the study of other carbon-based substrates with INS [189,190,191,192,193], including hydrogen spillover mechanisms [194,195] or the uptake and intercalation of atomic and molecular species, particularly water [196,197,198,199,200]. In this context, Romanelli et al. has used NCS to show that the confinement of water molecules within Graphite Oxide (GO) membranes has a ‘soft’ character, with the predominance of non-specific and weak interactions between water and the underlying nanostructured substrate [199]. In contrast, studies of organic polymers confined within carbon-based substrates reveals an intrinsically different picture [201]. Poly(Ethylene Oxide) (PEO) under extreme confinement was first studied using GO as a substrate [202]. The insights brought forward by the use of INS are evident from the data shown in Figure 7. This study served to establish the preferred conformation of the confined PEO phase, as well as to relate these structural changes to the strong suppression of collective phenomena, including crystallization and dynamical relaxation processes. Subsequent studies have exploited similar methodologies to investigate in detail a broad range of aspects linked to the above: molecular-size effects [203], intercalation versus adsorption [204], or the morphology and composition of the underlying substrate [205].
The study of hydrogen dynamics and associated confinement phenomena in Proton Exchange Membranes (PEMs) represents another timely example of the use of INS in the field of sustainable materials and associated technologies [206,207,208]. In a similar vein, there is also incipient activity in the development of hydrogen and other gas-storage materials. Carbon-based materials including fullerenes, nanohorns, or intercalation compounds constitute good examplars of soft media of increasing relevance for applications as gas stores, particularly hydrogen. In this regard, there are emerging opportunities for INS to provide unique insights into the mechanisms behind their hydrogen uptake at the atomic level [209,210], as well as for NCS as a unique analytical means to assess hydrogen levels quantitatively, as recently illustrated by Krzystyniak et al. [211].
Among the carbon-based materials that have been considered for hydrogen-storage, fullerene adducts continue to offer untapped opportunities. The full hydrogenation of the celebrated Buckminsterfullene molecule C, for example, would lead to record-breaking hydrogen-storage capacities around 8 wt.% [212]. Several strategies have been attempted to ensure the reversibility of this process, either by intercalation [213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232], or through the use of intermediate hydrogen carriers, like ammonia [233,234,235]. The already excellent sorption properties of alkali-doped fullerides can be further improved via decoration with transition-metal nanoparticles [227,236]. An interesting feature of fullerides is also the possibility of their polymerization at extreme conditions [237,238,239,240], which opens new synthetic strategies for obtaining carbon allotropes with entirely new physico-chemical properties. Polymerization can also be promoted by the intercalation of a wide range of electron donors (e.g., metals including Li, Na, K, Rb, or Mg), by entering the voids of the host fullerene lattice to form fully fledged solids [237,241,242,243,244,245,246].
As an illustration of this line of work, the structure and dynamics of the fullerene polymer Li4C60 was studied with INS spectroscopy by Rols et al. [246,247]. Representative results are illustrated in Figure 8. The use of ND and INS, combined with extensive ab initio modeling allowed the authors to solve its solid-state structure. Figure mattiaa–b show the rather exotic structure of this polymeric material, leading to the rich temperature dependence of INS features displayed in Figure 8c. Further, it has found that the intercalated ions exhibit partial disorder, as further corroborated by the analysis of the INS data. The strength and the nature of the peculiar polymeric bonding between the sub-units rely on a three-electron charge transfer from the Li atoms to every C60 molecule. In addition to its good hydrogen-storage capabilities, the system shows superionic conductivity at low temperature, which is an exceptional property for a solid material, therefore hinting at new applications [248]. The calculations presented in this work also predict an unstable Li sub-lattice, a finding that is consistent with the aforementioned ionic conductivity. The temperature dependence of INS features depicted in Figure 8 c is also sensitive to the depolymerization transition, characterized by a (rather counterintuitive) transfer of spectral weight to lower frequencies in the monomeric phase [246].
Nanoporous coordination compounds and their polymeric counterparts constitute yet another family of soft media with new properties and great potential for applications. To date, Metal-Organic Frameworks (MOFs) and Covalent-Organic Frameworks (COFs) have been studied in greater depth. Owing to its superb sensitivity to hydrogen, INS has become a powerful probe of the structural and gas-sorption properties of these materials, and its use has certainly proliferated in recent years. Table 1 provides a comprehensive summary of work to date. Following the seminal papers from Eckert and co-workers [249,250], INS has evolved as a leading technique providing insights on the binding and dynamics of molecular hydrogen (primarily in its para form, p–H2) absorbed by MOFs. As illustrated by a growing number of works [209,210,251,252,253,254,255,256,257], the use of intense beams of low-energy neutrons is ideally suited for the study of ro-vibrational excitations in H2. In particular, the last decade has witnessed unprecedented progress in MOF research using INS, along with a considerable broadening in scope beyond H2 uptake, which is still ongoing (see, e.g., Ref. [258]). Alongside, instrument developments at neutron sources have continued to support these research programmes, including: (i) a new Target-Station 2 (TS2) at ISIS, which has considerably improved the flux on TOSCA at low-energies; (ii) developments in sample environment equipment, including advanced gas-handling capabilities (see, e.g., Ref. [259]); (iii) ongoing developments in indirect-geometry instruments, reducing the requisite sample quantities considerably [137,138,139,140,141]; (iv) the commissioning of new instruments, like VISION at the SNS and LAGRANGE at the ILL [143], or the design of VESPA for the ESS [1]; and (v) progress in the field of ab initio modeling and its integration with experiments—as shown in Table 1, we underline the substantial increase over time in the number of publications using solid-state DFT. Altogether, these have enabled the study of the vibrational dynamics over a wide energy-transfer range and, more importantly, to place a fresh focus on the structural properties and flexibility of these soft materials, key to understand their rich polymorphism and sorption behavior [74,260]. As shown by Ortiz et al. [261], some flexible MOFs (e.g., MIL-53 or DMOF-1) show highly anisotropic elastic properties, with deformation directions exhibiting very low Young’s and shear moduli, approaching a 400:1 ratio between the most rigid and the softest direction. Negative compressibilities as a function of temperature or pressure also seem to be the norm rather than the exception in these materials.
The outcome of the above-discussed developments can be illustrated with the family of Zeolitic Imidazolate Frameworks (ZIFs). ZIFs are nanoporous materials comprised of tetrahedrally coordinated nodes and imidazole-derived organic linkers. The linkage between the metal and imidazole resembles the Si–O–Si angles present in zeolites, which explains their name. Their facile synthesis, high stability under ambient conditions, and commercial availability of the building blocks make them one of the most studied classes of MOFs regarding their mechanical properties [260]. For instance, ZIF-8 has a remarkably low shear modulus below 1 GPa [262], claimed to be the lowest value known for a crystalline solid. It is also interesting to note that some of the OHPs discussed earlier [113] have very similar values of C44 as those seen in ZIF-8. Further, ZIF-4 is also known to exhibit a remarkably rich polymorphic behavior at ambient and high pressures, and this phenomenology is certainly linked to the flexibility of its network structure at the nanoscale [260,263]. Figure 9 presents the INS spectra of a ZIF series [264], obtained on a previous incarnation of TOSCA, along with the results of accurate (optimized for the THz range) HLD calculations performed with the CRYSTAL code. This figure illustrates the synergy between the high-resolution INS experiments and theoretical predictions, allowing the full and quantitative interpretation of a rather complex vibrational spectrum over the entire range [264]. The low-energy part of the INS data is of particular relevance to understand the unexpected sorption properties of these materials. The large cavities interconnected by narrow and flexible channels characteristic of these ZIFs lead to novel molecular-sieving phenomena driven by low-energy modes beyond those at play with small molecules like H2 or CH4. These dynamical processes can explain their rather unexpected ability to absorb larger molecules which would not ‘fit-in’ by considering the rigid structure of the substrate [265]. At an atomic level, this so-called ‘gate-opening’ is related to the swing of the imidazolate linkers, thereby increasing the intrinsic porosity. Developments in gas-handling systems have also paved the way to probe gate-opening and related network-breathing phenomena arising from the unique elastic properties of MOFs—see entries in Table 1 and Refs. [151,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288,289,290].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of INS studies on MOFs and COFs. The first column indicates the acronym of the material, as commonly used in the literature. The second column indicates whether the focus of the investigation was on the bare material or on the uptake of the chemical species shown. The third and fourth columns show the energy-transfer range and INS spectrometer used (QENS, IPNS; FANS, NIST; FOCUS, SINQ; TOSCA and MARI, ISIS; TOFTOF, MLZ; IN5, ILL; VISION and SEQUOIA, SNS). Asterisks on the latter column indicate that solid-state DFT was used to interpret the INS data. Works are presented in chronological order, with the year of publication given along with its bibliographic reference in the last column.
Table 1.
Summary of INS studies on MOFs and COFs. The first column indicates the acronym of the material, as commonly used in the literature. The second column indicates whether the focus of the investigation was on the bare material or on the uptake of the chemical species shown. The third and fourth columns show the energy-transfer range and INS spectrometer used (QENS, IPNS; FANS, NIST; FOCUS, SINQ; TOSCA and MARI, ISIS; TOFTOF, MLZ; IN5, ILL; VISION and SEQUOIA, SNS). Asterisks on the latter column indicate that solid-state DFT was used to interpret the INS data. Works are presented in chronological order, with the year of publication given along with its bibliographic reference in the last column.
| Acronym | Investigation | Range | Instrument | Reference |
|---|---|---|---|---|
| MOF-5 | p–H2 | 0–20 meV | QENS | 2003 [249] |
| IRMOF-X (X = 1, 8, 11, 177) | p–H2 | 0–20 meV | QENS | 2005 [250] |
| MOF-5 | Bare material | 20–170 meV | FANS * | 2006 [291] |
| NaNi3(OH)(SIP)2 | p–H2 | 0–20 meV | QENS | 2006 [292] |
| HKUST-1 | p–H2 | 5–45 meV | FANS | 2007 [293] |
| TMBB | p–H2 | 0–30 meV | QENS | 2007 [294] |
| MIL-53 | Bare material | 50–180 meV | FANS * | 2008 [295] |
| MOF-74 | p–H2 | 5–25 meV | FANS | 2008 [296] |
| PCN-12 | p–H2 | 0–20 meV | QENS | 2008 [297] |
| PCN-6; PCN-6’ | p–H2 | 0–20 meV | QENS | 2008 [298] |
| HKUST-1 | p–H2 | 5–200 meV | FANS * | 2009 [299] |
| ZMOFs | p–H2 | 0–25 meV | QENS | 2009 [300] |
| CPO-27-M (M = Ni, Co, Mg) | p–H2 | 0–15 meV | FOCUS | 2010 [301] |
| Cr3(BTC)2 | p–H2 | 5–45 meV | FANS | 2011 [302] |
| Mg2(dobdc) | p–H2 | 5–45 meV | FANS | 2011 [303] |
| MOF-324 | p–H2 | 0–25 meV | QENS | 2012 [304] |
| rht-MOF-1 and rht-MOF-4a | p–H2 | 0–20 meV | FOCUS | 2012 [305] |
| Fe2(dobdc) and Fe2(O2)(dobdc) | p–H2 | 0–125 meV | FANS / TOSCA | 2012 [306] |
| NU-301 and NU-302 | p–H2 | 0–20 meV | FOCUS | 2013 [307] |
| HKUST-1 | p–H2 | 0–50 meV | TOSCA / MARI | 2013 [308] |
| MIL-53(Fe) | CH3OH | 0–250 meV | TOSCA * | 2013 [309] |
| rht-MOF-7 | p–H2 | 0–20 meV | TOFTOF | 2013 [310] |
| CPO-27–M (M = Mn, Cu) | p–H2 | 0–20 meV | TOFTOF / FOCUS | 2014 [311] |
| SIFSIX-2-Cu and SIFSIX-2-Cu-i | p–H2 | 0–20 meV | TOFTOF / FOCUS | 2014 [312] |
| Y-FTZB | p–H2 | 0–20 meV | TOFTOF | 2014 [313] |
| NOTT-300 | p–H2 | 0–250 meV | TOSCA * | 2014 [314] |
| ZIF-4, ZIF-7, and ZIF-8 | Bare material | 0–400 meV | TOSCA * | 2014 [264] |
| a-[Mg3(O2CH)6] | p–H2 | 0–20 meV | QENS | 2015 [315] |
| Zn(trz)(tftph) | p–H2 | 0–20 meV | TOFTOF | 2015 [316] |
| M-MOF-74 (M = Mg, Ni, Co, Zn) | p–H2 | 0–20 meV | FOCUS | 2015 [317] |
| NOTT-300 | C2H6, C2H4, and C2H2 | 0–250 meV | TOSCA * | 2015 [314] |
| M-soc-MOF-1-X (M = Fe, In; X = a, b) | p–H2 | 0–20 meV | IN5 | 2016 [318] |
| ZIF-8; ZIF-8@AC | N2 | 0–150 meV | VISION * | 2016 [266] |
| Cu(I)-MFU-4l | H2 and D2 | 0–40 meV | VISION * | 2016 [267] |
| MFM-300(In) | H2 and CH4 | 0–250 meV | TOSCA * | 2016 [268] |
| MFM-300(In) | N2, CO2, and SO2 | 0–250 meV | TOSCA * | 2016 [269] |
| ZIF-8 | D2O and CH4 | 0–200 meV | TOSCA | 2016 [266] |
| COF-1; COF-2 | p–H2 | 0–20 meV | QENS | 2017 [270] |
| ZIF-7 | N2 | 0–200 meV | VISION * | 2017 [271] |
| ZIF-8 | N2, O2, Ar, and CO | 0–150 meV | VISION | 2017 [272] |
| MFM-300(Sc) | I2 | 0–250 meV | TOSCA * | 2017 [273] |
| MFM-300(V) | CO2 | 0–250 meV | TOSCA * | 2017 [274] |
| MIL-140A | Bare material | 0–250 meV | TOSCA * | 2017 [275] |
| MFM-305 and MFM-305-CH3 | CO2 | 0–250 meV | VISION * | 2018 [276] |
| MFM-102-NO2 | C2H2 | 0–250 meV | VISION * | 2018 [277] |
| MFM-520 | NO2 | 0–250 meV | VISION * | 2019 [276] |
| Zn(MeIm)2 | Bare material | 0–500 meV | SEQUOIA | 2019 [319] |
| MFM-102-NO2 and MFM-102-NH2 | CO2 | 0–250 meV | VISION * | 2019 [278] |
| ZIF-4 | Bare material | 0–250 meV | VISION * | 2019 [279] |
| CO2 | 0–250 meV | TOSCA | 2019 [280] | |
| ZIF-4(Zn) | Bare material | 0–250 meV | TOSCA * | 2019 [281] |
| ZIF-7 | CO2 | 0–150 meV | TOSCA | 2019 [282] |
| HKUST-1 | Drug encapsulation | 0–250 meV | TOSCA * | 2019 [151] |
| MFM-126 | CO2 | 0–400 meV | TOSCA* | 2019 [283] |
| MFM-100 | Benzyl alcohol | 0–250 meV | TOSCA* | 2019 [284] |
| Pd@OX-1 | Catalytic properties | 0–400 meV | TOSCA* | 2019 [285] |
| MFM-170 | H2O and SO2 | 0–150 meV | TOSCA * | 2019 [286] |
| Y-shp-MOF-5 and Cr-soc-MOF-1 | D2O and CH4 | 0–300 meV | VISION * | 2020 [287] |
| MIL-100 (Fe) | Drug Encapsulation | 0–250 meV | TOSCA | 2020 [288] |
| MFM-520 | D2O, CO2, and SO2 | 0–250 meV | TOSCA * | 2020 [289] |
| MFM-300(M) (M = Al, Fe, VIII, VIV) | NH3 | 0–200 meV | TOSCA / VISION * | 2021 [290] |
The aforementioned developments in instrumentation, sample environment, and ab initio modeling can be further illustrated by considering the polymorphism exhibited by ZIF-4 at ambient pressure. Figure 10 presents the INS spectra as a function of temperature, measured on the TOSCA+ spectrometer [138,139,140]. Owing to a greatly improved sensitivity, it is now possible to explore phase diagrams across physical space, alongside theoretical modeling. While extremely rare a decade ago [291], phonon calculations are now becoming an inseparable part of structural studies on MOFs using INS (see Table 1), allowing to decode complex vibrational information delivered by these metal-organic frameworks. However, as noted by Vanpoucke et al., the computations with periodic DFT are still challenged by the flexibility of these materials, and the guidelines from experience with standard solid-state calculations cannot be simply transferred to flexible porous frameworks [320]. As shown by Butler et al. [281] (see Figure 10), variable-temperature high-resolution INS experiments give us a direct handle on changes to the lattice dynamics across the closed-to-open-pore phase transition in ZIF-4. Moreover, these data allow for a robust validation of the calculations performed within the Quasi-Harmonic Approximation (QHA), one of the methods of choice in the study of thermal expansion from first-principles. Using this methodology, the authors were able to identify the key structural and dynamical features that govern the free-energy landscape of the material, including the central role played by the vibrational entropy as a driving parameter for their exceptional flexibility.
4. Back to Basics: Water
Water continues to be the focus of attention of many, and some recent works have capitalized from developments in neutron spectroscopy to move beyond the state-of-the-art. Following the discovery of ice XV (-XV) by Salzmann et al. [321], the recent identification of ice XIX [322] confirms that the condensed phases of water are far from being a solved challenge [323], and have also stimulated developments in both neutron scattering [324] and computational modeling from time immemorial [325]. In this regard, neutron scattering constitutes a priceless tool for the ultimate validation of the theoretical approaches developed to describe both structure and dynamics, from this seemingly simple triatomic system to the complex materials described earlier in this review. One of the most celebrated examples is perhaps the total-scattering experiments by Soper et al., providing the Radial Distribution Functions (RDFs) of water and ice [326,327] to validate a countless number of theoretical approximations. In addition to diffraction data, calculations of the VDOSs based on empirical or ab initio approaches can be considered as the most effective and direct way of testing our description of interatomic and intermolecular interactions, indispensable to build increasingly accurate models.
The study of water with INS has a long and distinguished history, providing insights into the nuclear dynamics of a large fraction of the known ice phases [328,329,330,331], water at extreme conditions [332], and under ultra-confinement [333,334]. Figure ice-polymorphisma shows the phase diagram of water, highlighting both crystalline and amorphous phases. An exhaustive discussion of this phase diagram (including metastable forms of ice not shown in this figure) can be found in Ref. [323]. Ice XV can be considered as a hydrogen-ordered polymorph of form VI, revealing an antiferroelectric ordering of protons. However, it has been recently shown that for the unit cell of ice VI, a total of forty-five different ferroelectric or antiferroelectric types of order are possible within the remit of the ice rules, clearly illustrating that there are more new phases expected to be discovered in the future [335]. The discovery of form XIX (-XV) tends to confirm these notions, yet not without debate [336,337]. Rosu-Finsen and Salzmann have suggested that ice -XV is not a crystalline form of ice, but that it rather contains an immobile disordered network of hydrogen atoms, called a deep-glassy state [336,337,338]. Using per-deuterated ice XV, Gasser et al. has argued in favor of the existence of -XV [322]. However, INS experiments performed on hydrogenous samples of ice VI, ice XV, and deep-glassy ice VI (or ice -XV above) by Rosu-Finsen et al. [328] question this conclusion. Figure ice-polymorphismb displays the INS spectra recorded for each phase, unequivocally illustrating striking similarities between the spectral envelopes of ice VI and deep-glassy ice VI. This debate also raises further questions on the importance of isotopic effects and their relation to the stability of the phase of interest. The most-recent INS experiments on ice polymorphs by Rosu-Finsen et al. [328] also illustrate the state-of-the-art developments on the TOSCA+ instrument (see Figure 11b–d), providing up to a two-order-of-magnitude increase in count rates, and enabling parametric neutron-spectroscopic studies alongside simultaneous ND measurements [137,138,139,140,141]. Figure ice-polymorphism also confirms that the ND profiles of both ice VI and deep-glassy ice VI are remarkably similar and both data sets could be fitted well using the same model of the long-range order as ice VI, strikingly different from the ND profile of the XV phase. Finally, an overwhelming increase of the sensitivity of TOSCA+ allows to complement the calorimetric studies by real-time INS measurements as a function of temperature (see Figure 11d.) upon heating deep-glassy ice VI from 80 to 138 K, followed by cooling to 80 K. These in-situ experiments provide further insights into the thermodynamic relation between ice polymorphs and it is anticipated that a similar approach will be deployed in the future to resolve remaining and emerging discrepancies across the phase diagram of water.
The superb ability of INS to elucidate local structure is not limited solely to fully or partially ordered solids, as illustrated by recent state-of-the art studies of supercritical water (SCW). SCW is formed at 647 K and 0.0221 GPa, and its thermophysical properties are quite different from water at normal conditions [147,339]. There is an increasing interest in this exotic state, of promise for many sustainable and industrial applications, including gasification of biomass [340], construction of new-generation Supercritical-Water-Cooled Reactors (SCWRs) [341], or as a novel medium for soft matter research [342]. However, a detailed picture of the structure of water at these extreme conditions, as well as its link to unique physico-chemical properties remain unclear. Parrinello and co-authors have studied water at supercritical conditions from first-principles [343], showing that contrary to the ordinary liquid state, the H-bond network is destabilized to various extents. There is a continuous breaking and forming of anomalous H-bonded structures and no continuous network per se, significantly affecting the dielectric response. This fundamental study has been recently extended by Andreani et al. [332], providing a detailed structural picture of this fascinating system. This study capitalized from a combination of high-resolution experiments as a function of temperature and state-of-the-art AIMD, as presented in Figure 12. The work by Andreani et al. [332] has two important implications. First, it features INS and NCS as powerful probes of the temporal and spatial characteristics of H-bonding interactions. And second, it puts state-of-the-art AIMD to the test using the latest experimental and computational methodologies.
In addition to challenging INS experiments, NCS has been used to provide additional and much-needed information on the local potential around water molecules in the supercritical state [40,344,345,346,347,348,349]. Andreani et al. has identified short-lived water clusters in SCW [332], thus demonstrating that the technique is sensitive to local dynamics in the femtosecond time scale. On the basis of combined experiments and simulations, the authors have been able to show how H-bonds decrease in number as a function of temperature under constant pressure, and have revealed a pronounced distortion of H-bonds in these short-lived water clusters, which increases the coupling of intermolecular and intramolecular vibrations at supercritical conditions. This coupling modifies the H-bonding potential exerted on hydrogen, making it less anisotropic in the molecular plane compared with water at ambient conditions. This microscopic picture of the local environment of water molecules in SCW is displayed in Figure 13.
Figure 11.
(a) Phase diagram of HO in the range T = 4–300 K and p = 0.01–10 GPa, highlighting the thermodynamically stable forms of ice of relevance to our discussion (crystal phases VI and XV). Ice XV is the hydrogen-ordered counterpart of ice VI and is thermodynamically stable below 130 K over the pressure interval 0.8–1.5 GPa [321]. The phase diagram also includes the ranges of observation of metastable amorphous ices (Low-, High-, and very-High-Density Amorphous forms are denoted as LDA, HDA, and vHDA, respectively). The LDA phase is mostly found in the low-pressure region of the phase diagram (0–0.2 GPa), whereas HDA and VHDA occur at intermediate pressures (0.2–2 GPa). At high pressures (>2 GPa), only crystalline phases are stable [350]. (b) INS spectra of ice VI, deep-glassy ice VI, and ice XV, collected at T = 15 K on TOSCA+. The gray-shaded areas highlight the spectral range where major differences between ice VI and XV are observed. The spectra are shifted vertically for clarity. Difference spectra are shown in the lower part of the panel [328]. (c) ND patterns of (1) ice VI, (2) deep-glassy ice VI, and (3) ice XV collected simultaneously on TOSCA+ at T = 15 K. The experimental diffraction data are shown as light data points and the associated Rietveld fits as darker solid lines. Tick-marks indicate the expected positions of Bragg reflections. [328]. (d) Contour plot of the librational region upon heating deep-glassy ice VI from 80 to 138 K, followed by cooling back to 80 K [328]. Adapted with permission from Refs. [328,350]. Copyright (2021) American Chemical Society.
Figure 11.
(a) Phase diagram of HO in the range T = 4–300 K and p = 0.01–10 GPa, highlighting the thermodynamically stable forms of ice of relevance to our discussion (crystal phases VI and XV). Ice XV is the hydrogen-ordered counterpart of ice VI and is thermodynamically stable below 130 K over the pressure interval 0.8–1.5 GPa [321]. The phase diagram also includes the ranges of observation of metastable amorphous ices (Low-, High-, and very-High-Density Amorphous forms are denoted as LDA, HDA, and vHDA, respectively). The LDA phase is mostly found in the low-pressure region of the phase diagram (0–0.2 GPa), whereas HDA and VHDA occur at intermediate pressures (0.2–2 GPa). At high pressures (>2 GPa), only crystalline phases are stable [350]. (b) INS spectra of ice VI, deep-glassy ice VI, and ice XV, collected at T = 15 K on TOSCA+. The gray-shaded areas highlight the spectral range where major differences between ice VI and XV are observed. The spectra are shifted vertically for clarity. Difference spectra are shown in the lower part of the panel [328]. (c) ND patterns of (1) ice VI, (2) deep-glassy ice VI, and (3) ice XV collected simultaneously on TOSCA+ at T = 15 K. The experimental diffraction data are shown as light data points and the associated Rietveld fits as darker solid lines. Tick-marks indicate the expected positions of Bragg reflections. [328]. (d) Contour plot of the librational region upon heating deep-glassy ice VI from 80 to 138 K, followed by cooling back to 80 K [328]. Adapted with permission from Refs. [328,350]. Copyright (2021) American Chemical Society.

Figure 12.
(a) Hydrogen-projected VDOSs as a function of temperature at a pressure of 0.025 GPa. These data have been normalized to unity and vertically offset to facilitate visual comparison. (b) Simulated VDOSs of H2O at subcritical (bottom) and supercritical (top) conditions. In each case, the full spectrum (solid line) is compared with the projected VDOS (dashed line), after removing translational contributions. The shaded spectra correspond to intramolecular vibrational spectra, with librational and rotational motions projected out. Adapted with permission from Ref. [332]. Copyright (2021) American Chemical Society.
Figure 12.
(a) Hydrogen-projected VDOSs as a function of temperature at a pressure of 0.025 GPa. These data have been normalized to unity and vertically offset to facilitate visual comparison. (b) Simulated VDOSs of H2O at subcritical (bottom) and supercritical (top) conditions. In each case, the full spectrum (solid line) is compared with the projected VDOS (dashed line), after removing translational contributions. The shaded spectra correspond to intramolecular vibrational spectra, with librational and rotational motions projected out. Adapted with permission from Ref. [332]. Copyright (2021) American Chemical Society.

In addition to these experimental efforts, one also needs to note the accurate computational modeling using AIMD, building upon the seminal work of Car-Parinello [351]. Recent and exciting developments include those from Artificial Intelligence (AI) and the use of Deep Neural Networks (DNNs) to represent the interatomic potential [352]. Such an approach, as well as that by Behler and Parinello [353], allows for a description of highly dimensional potential-energy hypersurfaces in systems of arbitrary size whilst retaining the accuracy of high-end electronic-structure calculations. The end result is a speed-up of an otherwise very costly AIMD simulation by several orders of magnitude relative to the high-end reference method from which the artificial neural network is trained. These calculations of interatomic potentials and forces are fully sufficient to deal with the dynamic information encoded in neutron-scattering experiments. Hence, it also paves the way for the use of these methodologies to describe the longer timescales probed with QENS or NSE. INS, with its direct link to nuclear dynamics, becomes the method of choice for the validation of such theoretical developments. In addition, we note that recent progress in the development of Machine Learning (ML) ML-based simulations includes the calculation of charges, dipole moments and polarizability tensors, of direct relevance to the description of experimental observables accessible with optical spectroscopies [354,355]. Another area of increased interest relates to the use of ML algorithms in the calculation of molecular wavefunctions [356].
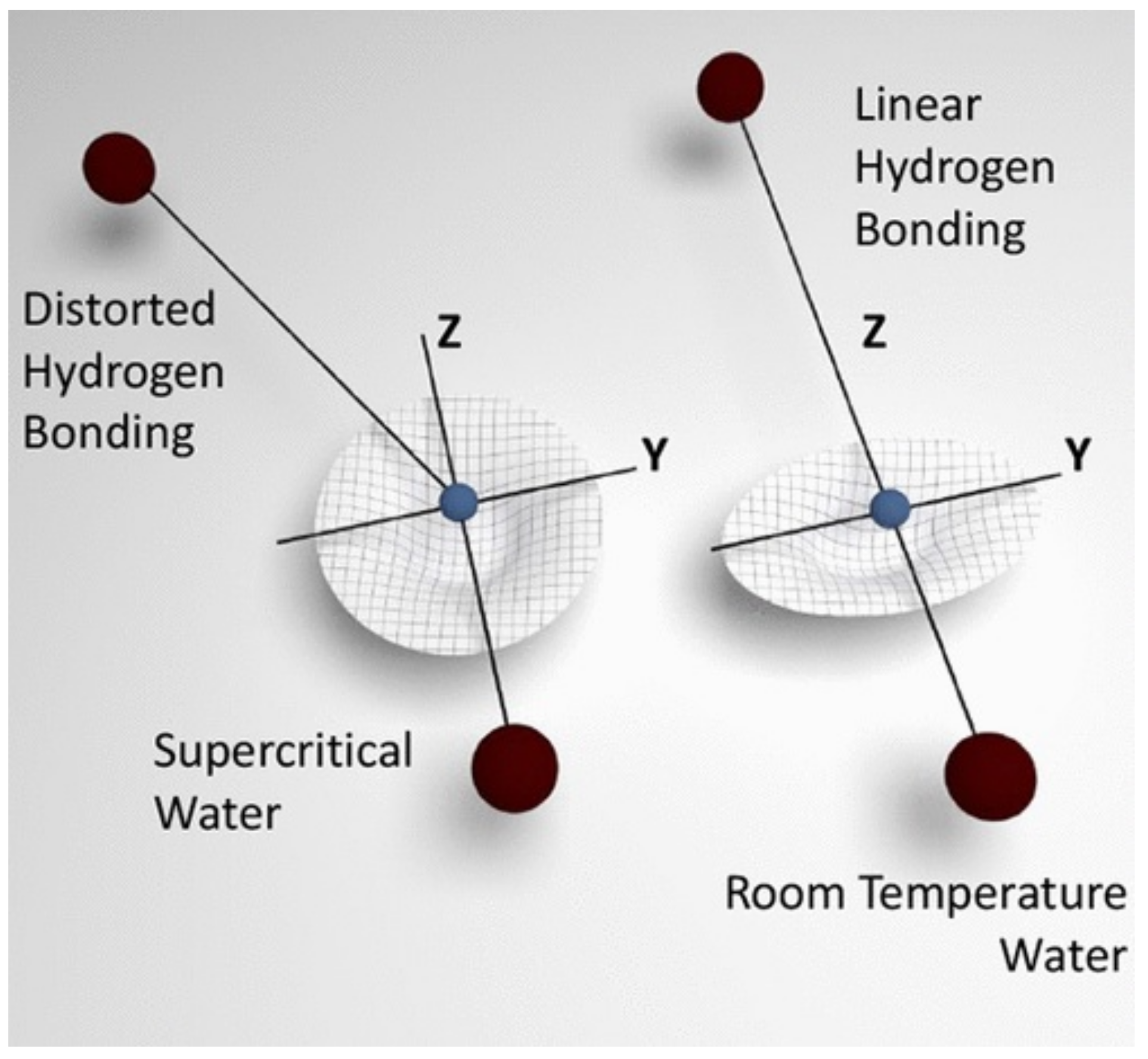
Figure 13.
Pictorial representation of the local H-bond geometry for H2O at supercritical (left) and ambient conditions (right). The figure depicts the coordinate frames used in the interpretation of NCS data, and the corresponding (partially isotropic and anisotropic) proton momentum distributions. Reprinted with permission from Ref. [332]. Copyright (2021) American Chemical Society.
Figure 13.
Pictorial representation of the local H-bond geometry for H2O at supercritical (left) and ambient conditions (right). The figure depicts the coordinate frames used in the interpretation of NCS data, and the corresponding (partially isotropic and anisotropic) proton momentum distributions. Reprinted with permission from Ref. [332]. Copyright (2021) American Chemical Society.
The spectacular speed-up of MD simulations with ML-trained Force-Fields (ML-FF) also helps us formulate and tackle important questions concerning which parent electronic-structure method is most appropriate to describe structural and dynamical properties. And, in this context, water serves as an obvious benchmark. Recent advances employ the gold-standard of quantum chemistry, i.e., Coupled-Cluster (CC) theory, yet still limited to small-molecules, like water and molecular clusters [357,358]. To date, DFT calculations on extended systems still remain as the work-horse. In the aforementioned work on SCW by Parinello and co-authors [343], the authors used the Generalized-Gradient Approximation (GGA) to the XC functional, which, to date, remains the most-commonly used approach in simulations of condensed-matter systems. Although considerable progress has been made with the construction of hybrid XC functionals or parallel developments in advanced semi-empirical corrections accounting for van-der-Waals (vdW) contributions, progress in constructing a better functional obeying a number of required constrains and norms for semi-local DFT has been far more modest. The development of a Strongly-Constrained and Appropriately Normed (SCAN) functional of a meta-GGA form [359] certainly offers a way forward, significantly improving accuracy when calculating the properties of different types of bonding [359,360,361]. SCAN provides an accuracy comparable to hybrid functionals at a considerably reduced cost, while still remaining a pure DFT functional by definition [362]. Both SCAN and hybrid functionals may be, therefore, be regarded as the most-advanced and still computationally feasible approximations to describe extended condensed matter systems. In the case of the structure of liquid water, its underlying H-bond geometry, and associated Mean Square Displacements (MSDs), Chen et al. [363] have shown that SCAN outperforms GGA. LaCount and Gygi have further confirmed the superb performance of SCAN in the description of the VDOSs of liquid water at ambient conditions [364]. This finding has been corroborated by AIMD simulations of liquid water using high-quality meta-GGA functionals presented by Ruiz-Pestana et al. [365]. The work by Andreani et al. [332] relied on SCAN-trained DNN potentials, fully confirming their robustness and high reliability to study in a realistic manner the structure and dynamics of water at supercritical conditions where—to first order—nuclear quantum effects (NQEs) may be ignored in these high-temperature simulations. The role of NQEs cannot be, however, neglected when discussing water at ambient conditions and below. Due to the small mass of the proton, NQEs, such as spatial nuclear delocalization, zero-point energy (ZPE), and proton tunneling, modify the H-bond geometry and strength in a significant manner [366]. In recent years, we are witnessing a pronounced interest in these effects, which can the key for a better understanding of fundamental phenomena and macroscopic properties. An exhaustive discussion can be found, e.g., in Refs. [367,368,369,370,371].
To illustrate the above in the particular case of water, calculations of vibrational properties using the GGA approximation seem to provide the ‘right’ answer, yet it has been found that this agreement is caused by a fortuitous cancellation of fairly large intrinsic errors in the description of the potential energy surface by these functionals [372,373]. In addition, most simulations to date do not incorporate NQEs from the outset, including ZPE contributions to the free energy which may be particularly pronounced for light species. These effects have been illustrated by Ruiz-Pestana et al. [365], showing that well-justified approximations, like high-quality meta-GGA or hybrid functionals, give a worse description of vibrational properties than the simpler GGA scheme when NQEs are not taken into account [365]. More simple scaling corrections have been proposed for practical reasons [374], yet these should always be assessed carefully to undertake physically meaningful simulations, particularly in those systems where NQEs are expected to play a significant role. This cautionary note is confirmed by the works of Ruiz-Pestana et al. [375] and Marsalek et al. [376], showing that the inclusion of NQEs using advanced meta-GGA DFT functionals can reproduce the properties of bulk water including RDFs, MSDs, and vibrational spectra (IR and Raman) with an accuracy comparable to those of far-more-costly hybrid functionals. Figure 14 serves to confirm the above considerations, with an example of the infrared spectrum of water simulated with both classical and approximate quantum MD simulations using the vdW-corrected hybrid functional (revPBE0-D3) [376]. While classical simulations are unable to describe satisfactorily the bending (ca. 1600 cm−1/200 meV) and stretching regions (ca. 3500 cm−1/450 meV) of H-bonded water, the quantum simulations provide the results with an excellent match to the experimental spectrum at ambient conditions. The cartoons in this Figure illustrate the competing quantum effects at play in the H-bonding between two water molecules. These affect the bending and stretching modes, with two qualitatively different contributions to the vibrational ZPE [367]. The left-upper panel in the Figure depict the in-plane bending vibrational mode. The right cartoon shows the O–H stretch. These contributions weaken and strengthen the H-bond, respectively, further refining the structural picture drawn from the RDFs accessible from diffraction. In addition, the results from IR spectroscopy (middle panel) are confronted with the INS spectrum of ambient water measured with the SEQUOIA spectrometer (see bottom panel). A small difference in the peak position of the OH-stretch band probed with IR and INS can be noticed. In this regard, INS spectroscopy should be considered as a better suited approach for the validation of theoretical predictions, as it is directly related to the underlying VDOS. On the contrary, accurate simulations of both the position and the shape of a band observed in IR spectroscopy requires an accurate treatment of dielectric or other charge-derived properties, including long-range dipole couplings [377] and electrical anharmonicities [378]. As illustrated in the Figure, NQEs can be successfully accounted for using quantum simulations. In its simplest formulation, well-established classical methods can be used to properly map the properties of a quantum particle, e.g., a proton, by means of the Feynman Path-Integral formalism. In the simplest case, a classical system composed of a closed-ring polymer with adjacent replicas connected with harmonic springs is isomorphic to the quantum one. Some of the approximate approaches to deal with dynamical observables (e.g., Thermostatted Ring Polymer MD, TRPMD; or Partially Adiabatic Centroid MD, PACMD, both used in the above-presented work by Marsalek et al. [376]) provide access to the time-evolved properties by propagating the centroid motion in real time. These approximate approaches may suffer from issues, like the curvature problem in centroid MD [379], which is particularly severe at low temperatures [380], or ZPE-leakage [381] and spurious resonances occurring in TRPMD [381,382]. Recently, Althorpe and co-workers have proposed an alternative approach called Quasi-Centroid MD (QCMD) which circumvents these limitations [380,383]. More rigorous Path-Integral MD (PIMD) approaches have also been developed, yet at a clear computational cost [384,385,386,387].
When NQEs are at the center of attention, experimental access to nuclear mean kinetic energies <EK> and associated momentum distributions n(p) via NCS becomes particularly relevant. As shown in Figure 14, local bending and stretching modes can be considered as descriptors of the interplay between NQEs arising from ZPE and the strength of H-bonds [389]. As discussed by Senesi et al. [390], the above-mentioned observables probed with NCS allow for a direct characterization of H-bonded systems. For example, it is now well-established that the strengthening the O–H covalent bonds broadens the associated proton momentum width probed with NCS. The tail of the n(p) distribution can be associated with the momentum along the O-H bond direction, and a large momentum width reflects a tightness of the proton binding, providing information about the strength and anharmonicity of the associated local vibrations. Upon increasing the H-bond strength, the effective proton potential will soften and, consequently, the proton <EK> and the O-H stretch frequency will decrease [390]. These considerations have been used by Parmentier et al. [391] to follow the evolution of the H-bond in amorphous forms of ice as a function of their density, clearly illustrating the complementarity between INS and NCS to characterize these systems (see Figure 15). Within a simplified (one-dimensional) model of the potential along the OH stretch, the cartoon in this Figure illustrates the primary differences between INS and NCS. INS probes the 0→ 1 excitation of a given mode from its vibrational ground state (an energy difference), whereas NCS measures directly the total mean kinetic energy associated with ZPEs, by operating within the so-called Impulse Approximation (IA). The bottom panel in Figure 15 displays the INS spectra measured on MARI (ISIS, UK) for three types of amorphous ice (LDA, uHDA, and vHDA, listed in order of increasing density). Three distinct bands in each INS spectrum can be associated to the water libration, bending, and OH-stretch modes. Similarly, NCS can provide information on directional mean kinetic energies, which can be further associated with these three types of vibrations and the associated ZPE contributions, for further comparison with INS (see Reference [391] for an extensive discussion). For a perfectly harmonic system, the value of the ZPE measured by both techniques would be, in principle, indistinguishable. The combination of both INS and NCS experiments allows, therefore, a further quantification of the degree of anharmonicity. The authors have focused on the OH-stretch mode dominating the total mean kinetic energy of the proton and, in so doing, have determined effective anharmonic constants for each amorphous phase. The mean kinetic energies were found to increase with increasing density, indicating the weakening of H-bonds, as well as a trend toward a steeper and more harmonic vibrational manifold.
The work by Parmentier et al. [391] highlights the power of NCS to retrieve ground-state kinetic energies without any underlying assumptions, which can be used as an accurate reference for testing and developing advanced theoretical approaches to account for NQEs. Since path-integral simulations converge slowly with the number of replicas used describe quantum behavior, the cost associated with their use alongside AIMD continues to be prohibitively high in most situations. The above-discussed use of neural networks to approximate the potentials with ab initio accuracy paves the way for obtaining fully converged quantum simulations [392], as well as opens new opportunities for making these approaches more widespread. This trend is also corroborated by considerable improvements in stochastic thermostatting [393,394], new methods to accelerate these simulations [395,396], as well as advanced tools now available to the wider community [397,398]. Such developments are providing synergies across experiment and theory, to understand and quantify NQEs at an unprecedented level of detail [399,400,401,402,403].
All of these advances can be illustrated further by returning to the first question posed in this Section, relating to how many structures of ice remain to be discovered. One way to tackle this challenge is the the calculation of accurate water phase diagrams from first-principles. Recent work demonstrates that progress to date looks promising, in spite of the difficulties associated with deficiencies in commonly used DFT approximations, as well as the need to incorporate NQEs from the outset—e.g., GGA predicts that ice sinks in water [363,404]. The SCAN functional is providing promising results, as it gives the observed trend in density across liquid water and hexagonal ice [363]. Following the achievements with neural-network simulations of SCW discussed earlier in this Section, Zhang et al. have presented a model that reproduces accurately the potential energy surface of the SCAN approximation of water, from low temperatures and pressures to about 2400 K and 50 GPa [405]. Using this potential and classical MD simulations, agreement with experimental data is generally satisfactory, correctly predicting the fluid, molecular and ionic phases of water, and almost all stable ice polymorphs, both ordered and disordered. Ice III and ice XV remain notable exceptions to the above, as they are stable but are found metastable in the simulations [406]. This result is quite intriguing, considering what is known on the polymorphs of ice XV. Nonetheless, the work from Zhang et al. [406] seems to be the first attempt with proven success to describe water over such a wide range of thermodynamic conditions without the inclusion of NQEs. Using ML-FF trained at the hybrid-functional level, Cheng et al. [405] have demonstrated that taking into account NQEs, anharmonic fluctuations, and proton disorder, it is possible to describe the structural and thermodynamic properties of liquid water, as well as hexagonal (Ih) and cubic (Ic) ice [405], in excellent agreement with experiments. This work also reports reliable estimates of the melting points of light and heavy water from first-principles, and has established that the increased stability of ice Ih over ice Ic stems from NQEs. Following this strategy, Reinhardt and Cheng have recently reported the phase diagram of water over the range T = 150–300 K and p = 0.01–1 GPa at the hybrid-DFT level, including NQEs [407]. These exciting results are shown in Figure 16. Starting from liquid water and a comprehensive set of fifty hypothetical ice structures waiting to be discovered, they show that none of them is thermodynamically stable, at least on a computer. This result suggests the completeness of the experimental phase diagram of water within these thermodynamic conditions. Figure 16 clearly evinces the importance of accounting for NQEs in the description of the phase diagram: forms III and V would not exist; and the proton-ordered ice-XV phase would be more stable than its disordered analogue ice VI [407]. In addition, the inclusion of NQEs improves the quantitative agreement with experimental data, including the not-so-trivial calculation of chemical potentials, particularly for ice III. This powerful computational framework is already being applied beyond water, with proven success in simulations of other systems discussed in this review. Such is the case of the application of the SCAN functional to the study of OHPs, including: its validation against accurate many-body theories (Random Phase Approximation, RPA) [408]; its superb performance when applied to the study of finite-temperature effects [409]; or in the description of their phase diagrams [410]. As recently demonstrated by Kapil et al. [411], the introduction of accelerated simulation techniques with the use of ML-FF are now becoming affordable to tackle phenomena, such as the aforementioned loading of guest molecules into extremely flexible MOF scaffolds. Furthermore, it opens new opportunities to address challenges across soft matter and polymer science. These range from dynamical phenomena and glassy behavior exhibiting significant NQEs [368,412,413,414], macromolecular thermophysics [415], isotope effects on polymer crystallization [416,417] or the chemistry of polymerization, including extreme conditions [418].
Figure 15.
(a) Pictorial representation of the anharmonic potential of the OH-stretch mode of H2O. The vibrational ZPE measured with NCS (DINS), as well as the fundamental 0 → 1 transition probed with INS, are highlighted. (b) Experimental INS spectra of different forms of amorphous ice (LDA, red; unannealed HDA, green; and vHDA, blue) measured at T = 80 K on MARI. The spectra were obtained after averaging over the indicated Q-range. (c) Mean kinetic energies (<EK>) from NCS (orange) and corresponding values obtained from INS using the harmonic model discussed in Ref. [399] (blue). (d) Energies of the OH-stretch, bend, and librational modes of H2O, obtained from INS (blue), as well as the corresponding values derived from NCS (orange). Adapted with permission from Ref. [391]. Copyright (2021) American Chemical Society.
Figure 15.
(a) Pictorial representation of the anharmonic potential of the OH-stretch mode of H2O. The vibrational ZPE measured with NCS (DINS), as well as the fundamental 0 → 1 transition probed with INS, are highlighted. (b) Experimental INS spectra of different forms of amorphous ice (LDA, red; unannealed HDA, green; and vHDA, blue) measured at T = 80 K on MARI. The spectra were obtained after averaging over the indicated Q-range. (c) Mean kinetic energies (<EK>) from NCS (orange) and corresponding values obtained from INS using the harmonic model discussed in Ref. [399] (blue). (d) Energies of the OH-stretch, bend, and librational modes of H2O, obtained from INS (blue), as well as the corresponding values derived from NCS (orange). Adapted with permission from Ref. [391]. Copyright (2021) American Chemical Society.

Figure 16.
Experimental (a) and computational (b,c) phase diagrams of H2O in the range T = 50–300 K and p = 0.02–1 GPa. The computational phase diagrams have been obtained with classical (b) and PIMD simulations (c) using a vdW-corrected hybrid-DFT (revPBE0-D3). See Ref. [407] for more details. Adapted with permission from Ref. [407]
Figure 16.
Experimental (a) and computational (b,c) phase diagrams of H2O in the range T = 50–300 K and p = 0.02–1 GPa. The computational phase diagrams have been obtained with classical (b) and PIMD simulations (c) using a vdW-corrected hybrid-DFT (revPBE0-D3). See Ref. [407] for more details. Adapted with permission from Ref. [407]

5. Outlook
The past decade has witnessed very significant advances in neutron spectroscopy, and this work has made an attempt to provide a timely overview of the use of this tool in the study of complex materials, with a focus on those of a soft and polymeric nature. Our choices in the selection of the areas of research and scientific drivers described herein have been unavoidably colored by our personal preferences, appreciation, and knowledge of an otherwise vast and rapidly growing area of activity. The field of OHPs constitutes a good case-in-point, as it is by now abundantly clear that the degree of complexity at the atomic scale associated with these soft solids requires the insights brought forward by the technique, as highlighted in Section 2 above. In this regard, INS stands out as the method of choice to scrutinize their behavior, particularly if coupled to first-principles computational materials modeling. Similar remarks would equally apply to the study of confined soft matter and supramolecular frameworks, as covered in Section 3. In this case, progress has also required parallel developments in the requisite sample-environment equipment in order to carry out in-situ, operando, or high-pressure studies, now possible owing to order-of-magnitude improvements in measurement speeds. The proliferation in the number of experiments on MOFs and COFs in the past years summarized in Table 1 is testament to the above. At a fundamental level, these studies have taught us that both structure and function in these nanoporous media are inextricably linked to dynamics and motion. Once more, a robust interpretation of INS data is predicated upon the use of computational modeling, and much progress has been achieved in parallel in the reliable description of these rather large systems with the requisite level of accuracy. Last but not least, going back to water in Section 4 has been intentional for several reasons. The level of complexity exhibited by this seemingly simple molecular system across its phase diagram continues to baffle us. State-of-the-art INS experiments as those illustrated in Figure 11 represent a novel means of resolving ongoing (and often heated) controversies on this pivotal system, simply thought unthinkable not so long ago. Moreover, the role played by NQEs can now be probed across the phase diagram with NCS, and these experiments are also motivating further and exciting theoretical developments. The spectacular shifts in phase boundaries shown in Figure 16 as NQEs are switched ‘on’ and ‘off’ are a tantalizing reminder of how much remains to be explored, taking water as a starting point in an otherwise long and fruitful journey into other classes of materials.
It is certainly tempting (perhaps foolhardy) to end with a few words on the road ahead. As qualitative scientific advances generally follow developments in our tools and methods of inquiry, the advent of next-generation neutron facilities, like the European Spallation Source, will certainly take us to a whole new level over the next few years. As such, we very much hope that this review provides a spring board for the community to capitalize from these in new and original ways.
Author Contributions
This manuscript has been written with the contribution from all authors (K.D., M.G. and F.F.-A.). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Gipuzkoako Foru Aldundia under Grant Number 2020-CIEN-000009-01.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank E. Verde-Sesto and J. Maiz for their kind invitation to write this contribution to this Special Issue.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Andersen, K.; Argyriou, D.; Jackson, A.; Houston, J.; Henry, P.; Deen, P.; Toft-Petersen, R.; Beran, P.; Strobl, M.; Arnold, T.; et al. The Instrument Suite of the European Spallation Source. Nucl. Instrum. Methods Phys. Res. 2020, 957, 163402. [Google Scholar] [CrossRef]
- Carpenter, J.M. The Development of Compact Neutron Sources. Nat. Rev. Phys. 2019, 1, 177–179. [Google Scholar] [CrossRef]
- Ott, F.; Menelle, A.; Alba-Simionesco, C. The SONATE Project, a French CANS for Materials Sciences Research. EPJ Web Conf. 2020, 231, 01004. [Google Scholar] [CrossRef]
- Otake, Y. RIKEN Accelerator-driven Compact Neutron Systems. EPJ Web Conf. 2020, 231, 01009. [Google Scholar] [CrossRef]
- Zakalek, P.; Cronert, T.; Baggemann, J.; Doege, P.E.; Rimmler, M.; Voigt, J.; Mauerhofer, E.; Rücker, U.; Gutberlet, T.; Podlech, H.; et al. High-brilliance Neutron Source Project. J. Phys. Conf. Ser. 2020, 1401, 012010. [Google Scholar] [CrossRef]
- Anderson, I.; Andreani, C.; Carpenter, J.; Festa, G.; Gorini, G.; Loong, C.K.; Senesi, R. Research Opportunities with Compact Accelerator-driven Neutron Sources. Phys. Rep. 2016, 654, 1–58. [Google Scholar] [CrossRef] [Green Version]
- De Vicente, J.P.; Sordo, F.; Perlado, J.M.; Bermejo, F.J.; Fernandez-Alonso, F. Guiding Criteria for Instrument Design at Long-pulse Neutron Sources. J. Phys. Conf. Ser. 2015, 663, 012011. [Google Scholar] [CrossRef]
- Sordo, F.; Fernandez-Alonso, F.; Terrón, S.; Magán, M.; Ghiglino, A.; Martinez, F.; Bermejo, F.; Perlado, J. Baseline Design of a Low Energy Neutron Source at ESS-Bilbao. Phys. Procedia 2014, 60, 125–137. [Google Scholar] [CrossRef] [Green Version]
- De Vicente, J.; Fernandez-Alonso, F.; Sordo, F.; Bermejo, F. Neutrons at ESS-Bilbao: From Production to Utilisation; Technical Report RAL-TR-2013-016; Rutherford Appleton Laboratory: Oxfordshire, UK, 2013. [Google Scholar]
- Skarzynski, T. Collecting Data in the Home Laboratory: Evolution of X-ray Sources, Detectors and Working Practices. Acta Crystallogr. D 2013, 69, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Alonso, F.; Price, D. (Eds.) Neutron Scattering—Fundamentals; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Fernandez-Alonso, F.; Price, D. (Eds.) Neutron Scattering—Magnetic and Quantum Phenomena; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Fernandez-Alonso, F.; Price, D. (Eds.) Neutron Scattering—Applications in Biology, Chemistry, and Materials Science; Academic Press: Cambridge, MA, USA, 2017. [Google Scholar]
- Richter, D.; Monkenbusch, M.; Arbe, A.; Colmenero, J. Neutron Spin Echo in Polymer Systems; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar] [CrossRef]
- Krutyeva, M.; Wischnewski, A.; Monkenbusch, M.; Willner, L.; Maiz, J.; Mijangos, C.; Arbe, A.; Colmenero, J.; Radulescu, A.; Holderer, O.; et al. Effect of Nanoconfinement on Polymer Dynamics: Surface Layers and Interphases. Phys. Rev. Lett. 2013, 110. [Google Scholar] [CrossRef] [Green Version]
- Richter, D.; Monkenbusch, M.; Allgeier, J.; Arbe, A.; Colmenero, J.; Farago, B.; Bae, Y.C.; Faust, R. From Rouse Dynamics to Local Relaxation: A Neutron Spin Echo Study on Polyisobutylene Melts. J. Chem. Phys. 1999, 111, 6107–6120. [Google Scholar] [CrossRef]
- Colmenero, J.; Mukhopadhyay, R.; Alegría, A.; Frick, B. Quantum Rotational Tunneling of Methyl Groups in Polymers. Phys. Rev. Lett. 1998, 80, 2350–2353. [Google Scholar] [CrossRef]
- Zorn, R.; Frick, B.; Fetters, L.J. Quasielastic Neutron Scattering Study of the Methyl Group Dynamics in Polyisoprene. J. Chem. Phys. 2002, 116, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Colmenero, J.; Arbe, A. Segmental Dynamics in Miscible Polymer Blends: Recent Results and Open Questions. Soft Matter 2007, 3, 1474. [Google Scholar] [CrossRef]
- Colmenero, J.; Arbe, A. Recent Progress on Polymer Dynamics by Neutron Scattering: From Simple Polymers to Complex Materials. J. Polym. Sci. B Polym. Phys. 2012, 51, 87–113. [Google Scholar] [CrossRef] [Green Version]
- Bhowmik, D.; Pomposo, J.A.; Juranyi, F.; Sakai, V.G.; Zamponi, M.; Arbe, A.; Colmenero, J. Investigation of a Nanocomposite of 75 wt % Poly(methyl methacrylate) Nanoparticles with 25 wt % Poly(ethylene oxide) Linear Chains: A Quasielatic Neutron Scattering, Calorimetric, and WAXS Study. Macromolecules 2014, 47, 3005–3016. [Google Scholar] [CrossRef]
- Arbe, A.; Nilsen, G.J.; Stewart, J.R.; Alvarez, F.; Sakai, V.G.; Colmenero, J. Coherent Structural Relaxation of Water from Meso- to Intermolecular Scales Measured using Neutron Spectroscopy with Polarization Analysis. Phys. Rev. Res. 2020, 2. [Google Scholar] [CrossRef] [Green Version]
- Fischer, E.W. Studies of Structure and Dynamics of Solid Polymers by Elastic and Inelastic Neutron Scattering. Pure Appl. Chem. 1978, 50, 1319–1341. [Google Scholar] [CrossRef]
- Higgins, J. Neutron Scattering from Polymers: Five Decades of Developing Possibilities. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Berrod, Q.; Lagrené, K.; Ollivier, J.; Zanotti, J.M. Inelastic and Quasi-elastic Neutron Scattering. Application to Soft-matter. EPJ Web Conf. 2018, 188, 05001. [Google Scholar] [CrossRef]
- Araujo, C.F.; Nolasco, M.M.; Ribeiro-Claro, P.J.A.; Rudić, S.; Silvestre, A.J.D.; Vaz, P.D.; Sousa, A.F. Inside PEF: Chain Conformation and Dynamics in Crystalline and Amorphous Domains. Macromolecules 2018, 51, 3515–3526. [Google Scholar] [CrossRef]
- Lambri, O.A.; Giordano, E.D.V.; Bonifacich, F.G.; Jiménez-Ruiz, M.; Lambri, M.A.; Sánchez, F.A.; Pérez-Landazábal, J.I.; García, J.Á.; Boschetti, C.E.; Recarte, V.; et al. Changes in the Crystalline Degree in Neutron Irradiated EPDM Viewed Through Infrared Spectroscopy and Inelastic Neutron Scattering. Matéria (Rio de Janeiro) 2018, 23. [Google Scholar] [CrossRef]
- Nolasco, M.M.; Araujo, C.F.; Thiyagarajan, S.; Rudić, S.; Vaz, P.D.; Silvestre, A.J.D.; Ribeiro-Claro, P.J.A.; Sousa, A.F. Asymmetric Monomer, Amorphous Polymer? Structure–Property Relationships in 2, 4-FDCA and 2, 4-PEF. Macromolecules 2020, 53, 1380–1387. [Google Scholar] [CrossRef]
- Moreno, A.J.; Alegría, A.; Colmenero, J.; Frick, B. Methyl Group Dynamics in Poly(methyl methacrylate): From Quantum Tunneling to Classical Hopping. Macromolecules 2001, 34, 4886–4896. [Google Scholar] [CrossRef]
- Colmenero, J.; Moreno, A.J.; Alegría, A. Neutron Scattering Investigations on Methyl Group Dynamics in Polymers. Prog. Polym. Sci. 2005, 30, 1147–1184. [Google Scholar] [CrossRef] [Green Version]
- Goracci, G.; Arbe, A.; Alegría, A.; Sakai, V.G.; Rudić, S.; Schneider, G.J.; Lohstroh, W.; Juranyi, F.; Colmenero, J. Influence of Solvent on Poly(2-(Dimethylamino)Ethyl Methacrylate) Dynamics in Polymer-Concentrated Mixtures: A Combined Neutron Scattering, Dielectric Spectroscopy, and Calorimetric Study. Macromolecules 2015, 48, 6724–6735. [Google Scholar] [CrossRef] [Green Version]
- Annis, B.K.; Lohse, D.J.; Trouw, F. Observation of Boson Peaks by Inelastic Neutron Scattering in Polyolefins. J. Chem. Phys. 1999, 111, 1699–1704. [Google Scholar] [CrossRef]
- Etienne, S.; David, L.; Dianoux, A.; Saviot, L.; Duval, E. Effect of Aging on the Boson Peak and Relaxation Processes in a Glassy Polymer. J. Non-Cryst. Solids 2002, 307–310, 109–113. [Google Scholar] [CrossRef]
- Hong, L.; Begen, B.; Kisliuk, A.; Alba-Simionesco, C.; Novikov, V.N.; Sokolov, A.P. Pressure and Density Dependence of the Boson Peak in Polymers. Phys. Rev. B 2008, 78. [Google Scholar] [CrossRef]
- Zorn, R.; Yin, H.; Lohstroh, W.; Harrison, W.; Budd, P.M.; Pauw, B.R.; Böhning, M.; Schönhals, A. Anomalies in the Low Frequency Vibrational Density of States for a Polymer with Intrinsic Microporosity—The Boson Peak of PIM-1. Phys. Chem. Chem. Phys. 2018, 20, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomoshige, N.; Mizuno, H.; Mori, T.; Kim, K.; Matubayasi, N. Boson Peak, Elasticity, and Glass Transition Temperature in Polymer Glasses: Effects of the Rigidity of Chain Bending. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Inoue, R.; Kanaya, T.; Nishida, K.; Tsukushi, I.; Shibata, K. Inelastic Neutron Scattering Study of Low Energy Excitations in Polymer Thin Films. Phys. Rev. Lett. 2005, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaya, T.; Miyazaki, T.; Inoue, R.; Nishida, K. Thermal Expansion and Contraction of Polymer Thin Films. Phys. Status Solidi B 2005, 242, 595–606. [Google Scholar] [CrossRef]
- Kanaya, T.; Inoue, R.; Kawashima, K.; Miyazaki, T.; Tsukushi, I.; Shibata, K.; Matsuba, G.; Nishida, K.; Hino, M. Glassy Dynamics and Heterogeneity of Polymer Thin Films. J. Phys. Soc. Jpn. 2009, 78, 041004. [Google Scholar] [CrossRef]
- Andreani, C.; Krzystyniak, M.; Romanelli, G.; Senesi, R.; Fernandez-Alonso, F. Electron-volt Neutron Spectroscopy: Beyond Fundamental Systems. Adv. Phys. 2017, 66, 1–73. [Google Scholar] [CrossRef]
- Evans, A.C.; Mayers, J.; Timms, D.N.; Cooper, M.J. Deep Inelastic Neutron Scattering in the Study of Atomic Momentum Distributions. Z. Naturforsch. A 1993, 48, 425–432. [Google Scholar] [CrossRef]
- Krzystyniak, M.; Seel, A.G.; Richards, S.E.; Gutmann, M.J.; Fernandez-Alonso, F. Mass-selective Neutron Spectroscopy Beyond the Proton. J. Phys. Conf. Ser. 2014, 571, 012002. [Google Scholar] [CrossRef] [Green Version]
- Krzystyniak, M.; Drużbicki, K.; Fernandez-Alonso, F. Nuclear Dynamics in the Metastable Phase of the Solid Acid Caesium Hydrogen Sulfate. Phys. Chem. Chem. Phys. 2015, 17, 31287–31296. [Google Scholar] [CrossRef] [Green Version]
- Syrykh, G.F.; Stolyarov, A.A.; Krzystyniak, M.; Romanelli, G.; Sadykov, R.A. Temperature Dependence of the Kinetic Energy in the Zr40Be60 Amorphous Alloy. JETP Lett. 2017, 105, 591–594. [Google Scholar] [CrossRef]
- Krzystyniak, M.; Syrykh, G.; Stolyarov, A.; Sadykov, R.A.; Armstrong, J.; da Silva, I.; Romanelli, G.; Fernandez-Alonso, F. Mass-selective Neutron Spectroscopy of Glassy versus Polycrystalline Structures in Binary Mixtures of Beryllium and Zirconium. J. Phys. Conf. Ser. 2018, 1055, 012004. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.; Krzystyniak, M.; Romanelli, G.; Parker, S.F.; Drużbicki, K.; Fernandez-Alonso, F. Fractal Dimension as a Scaling Law for Nuclear Quantum Effects: A Neutron Compton Scattering Study on Carbon Allotropes. J. Phys. Conf. Ser. 2018, 1055, 012007. [Google Scholar] [CrossRef]
- Krzystyniak, M.; Gutmann, M.J.; Romanelli, G.; Trenikhina, Y.; Romanenko, A.; Fernandez-Alonso, F. Nitrogen Doping and the Performance of Superconducting Radio-frequency Niobium Cavities: Insights from Neutron Diffraction and Neutron Compton Scattering. J. Phys. Conf. Ser. 2018, 1055, 012006. [Google Scholar] [CrossRef]
- Ding, H.; Liu, Q.; Wang, X.; Fan, X.; Krzystyniak, M.; Glandut, N.; Li, C. Effects of Boron Addition on the Microstructure and Properties of in situ Synthesis TiC-Reinforced Cu Ti C Composites. J. Alloys Compd. 2018, 766, 66–73. [Google Scholar] [CrossRef]
- Krzystyniak, M.; Drużbicki, K.; Rudić, S.; Fabian, M. Positional, Isotopic Mass and Force Constant Disorder in Molybdate Glasses and Their Parent Metal Oxides as Observed by Neutron Diffraction and Compton Scattering. J. Phys. Commun. 2020, 4, 095027. [Google Scholar] [CrossRef]
- Senesi, R.; Andreani, C.; Bowden, Z.; Colognesi, D.; Degiorgi, E.; Fielding, A.; Mayers, J.; Nardone, M.; Norris, J.; Praitano, M.; et al. VESUVIO: A Novel Instrument for Performing Spectroscopic Studies in Condensed Matter With eV Neutrons at the ISIS Facility. Phys. B Condens. Matter 2000, 276–278, 200–201. [Google Scholar] [CrossRef]
- Andreani, C.; Pietropaolo, A.; Senesi, R.; Gorini, G.; Tardocchi, M.; Bracco, A.; Rhodes, N.; Schooneveld, E. Electron-volt Spectroscopy at a Pulsed Neutron Source Using a Resonance Detector Technique. Nucl. Instrum. Methods Phys. Res. 2002, 481, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, A.; Andreani, C.; D’Agelo, A.; Gorini, G.; Imberti, S.; Rhodes, N.; Schoneveld, E.M.; Senesi, R.; Tardocchi, M. The Resonance Detector Spectrometer for Neutron Spectroscopy in the eV Energy Region. In Capture Gamma-Ray Spectroscopy and Related Topics; World Scientific: Singapore, 2003. [Google Scholar] [CrossRef]
- Andreani, C.; Colognesi, D.; Degiorgi, E.; Filabozzi, A.; Nardone, M.; Pace, E.; Pietropaolo, A.; Senesi, R. Double Difference Method in Deep Inelastic Neutron Scattering on the VESUVIO Spectrometer. Nucl. Instrum. Methods Phys. Res. 2003, 497, 535–549. [Google Scholar] [CrossRef] [Green Version]
- Mayers, J.; Tomkinson, J.; Abdul-Redah, T.; Stirling, W.; Andreani, C.; Senesi, R.; Nardone, M.; Colognesi, D.; Degiorgi, E. VESUVIO—the Double Difference Inverse Geometry Spectrometer at ISIS. Phys. B Condens. Matter 2004, 350, E659–E662. [Google Scholar] [CrossRef] [Green Version]
- Imberti, S.; Andreani, C.; Garbuio, V.; Gorini, G.; Pietropaolo, A.; Senesi, R.; Tardocchi, M. Resolution of the VESUVIO Spectrometer for High-energy Inelastic Neutron Scattering Experiments. Nucl. Instrum. Methods Phys. Res. 2005, 552, 463–476. [Google Scholar] [CrossRef] [Green Version]
- Gorini, G.; Festa, G.; Andreani, C. Epithermal Neutron Instrumentation at ISIS. J. Phys. Conf. Ser. 2014, 571, 012005. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, G.; Krzystyniak, M.; Senesi, R.; Raspino, D.; Boxall, J.; Pooley, D.; Moorby, S.; Schooneveld, E.; Rhodes, N.J.; Andreani, C.; et al. Characterisation of the Incident Beam and Current Diffraction Capabilities on the VESUVIO Spectrometer. Meas. Sci. Technol. 2017, 28, 095501. [Google Scholar] [CrossRef]
- Ulpiani, P.; Romanelli, G.; Arcidiacono, L.; Onorati, D.; Festa, G.; Krzystyniak, M.; Schooneveld, E.; Fernandez-Alonso, F.; Andreani, C.; Senesi, R. Enhancement of Counting Statistics and Noise Reduction in the Forward-scattering Detectors on the VESUVIO Spectrometer. J. Phys. Conf. Ser. 2018, 1055, 012008. [Google Scholar] [CrossRef] [Green Version]
- Krzystyniak, M.; Romanelli, G.; Fabian, M.; Gutmann, M.; Festa, G.; Arcidiacono, L.; Gigg, M.; Drużbicki, K.; Andreani, C.; Senesi, R.; et al. VESUVIO: The Current Testbed for a Next-generation Epithermal Neutron Spectrometer. J. Phys. Conf. Ser. 2018, 1021, 012026. [Google Scholar] [CrossRef]
- Senesi, R.; Kolesnikov, A.I.; Andreani, C. Measurement of Proton Momentum Distributions Using a Direct Geometry Instrument. J. Phys. Conf. Ser. 2014, 571, 012007. [Google Scholar] [CrossRef] [Green Version]
- Stock, C.; Cowley, R.A.; Taylor, J.W.; Bennington, S.M. High-energy Neutron Scattering from Hydrogen Using a Direct Geometry Spectrometer. Phys. Rev. B 2010, 81. [Google Scholar] [CrossRef] [Green Version]
- Mayers, J.; Gidopoulos, N.I.; Adams, M.A.; Reiter, G.; Andreani, C.; Senesi, R. Comment on “High-energy Neutron Scattering from Hydrogen Using a Direct Geometry Spectrometer”. Phys. Rev. B 2011, 84. [Google Scholar] [CrossRef] [Green Version]
- ESS Technical Design Report. Available online: https://docdb01.esss.lu.se/DocDB/0002/000274/015/TDR_online_ver_all.pdf (accessed on 28 April 2021).
- Poglitsch, A.; Weber, D. Dynamic Disorder in Methylammoniumtrihalogenoplumbates (II) Observed by Millimeter-wave Spectroscopy. J. Chem. Phys. 1987, 87, 6373–6378. [Google Scholar] [CrossRef]
- Angelis, F.D. Celebrating 10 Years of Perovskite Photovoltaics. ACS Energy Lett. 2019, 4, 853–854. [Google Scholar] [CrossRef] [Green Version]
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef]
- Gonzalez-Pedro, V.; Juarez-Perez, E.J.; Arsyad, W.S.; Barea, E.M.; Fabregat-Santiago, F.; Mora-Sero, I.; Bisquert, J. General Working Principles of CH3NH3PbX3 Perovskite Solar Cells. Nano Lett. 2014, 14, 888–893. [Google Scholar] [CrossRef]
- Jena, A.K.; Kulkarni, A.; Miyasaka, T. Halide Perovskite Photovoltaics: Background, Status, and Future Prospects. Chem. Rev. 2019, 119, 3036–3103. [Google Scholar] [CrossRef]
- Bisquert, J.; Juarez-Perez, E.J. The Causes of Degradation of Perovskite Solar Cells. J. Phys. Chem. Lett. 2019, 10, 5889–5891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aranda, C.; Guerrero, A.; Bisquert, J. Crystalline Clear or Not: Beneficial and Harmful Effects of Water in Perovskite Solar Cells. ChemPhysChem 2019, 20, 2587–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaei, A.; Circelli, A.; Yuan, Y.; Yang, Y.; Lee, S.S. Polymorphism in Metal Halide Perovskites. Mater. Adv. 2021, 2, 47–63. [Google Scholar] [CrossRef]
- Sun, S.; Isikgor, F.H.; Deng, Z.; Wei, F.; Kieslich, G.; Bristowe, P.D.; Ouyang, J.; Cheetham, A.K. Factors Influencing the Mechanical Properties of Formamidinium Lead Halides and Related Hybrid Perovskites. ChemSusChem 2017, 10, 3740–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Fang, Y.; Kieslich, G.; White, T.J.; Cheetham, A.K. Mechanical Properties of Organic–inorganic Halide Perovskites, CH3NH3PbX3(X = I, Br and Cl), by Nanoindentation. J. Mater. Chem. A 2015, 3, 18450–18455. [Google Scholar] [CrossRef]
- Tan, J.C.; Cheetham, A.K. Mechanical Properties of Hybrid Inorganic–organic Framework Materials: Establishing Fundamental Structure–property Relationships. Chem. Soc. Rev. 2011, 40, 1059. [Google Scholar] [CrossRef]
- Drużbicki, K.; Lavén, R.; Armstrong, J.; Malavasi, L.; Fernandez-Alonso, F.; Karlsson, M. Cation Dynamics and Structural Stabilization in Formamidinium Lead Iodide Perovskites. J. Phys. Chem. Lett. 2021, 3503–3508. [Google Scholar] [CrossRef]
- Li, M.; Liu, T.; Wang, Y.; Yang, W.; Lü, X. Pressure Responses of Halide Perovskites with Various Compositions, Dimensionalities, and Morphologies. Matter Radiat. Extremes 2020, 5, 018201. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Gong, J.; Hu, Q.; Capitani, F.; Celeste, A.; Hattori, T.; Sano-Furukawa, A.; Li, N.; Yang, W.; Liu, G.; et al. Suppressed Lattice Disorder for Large Emission Enhancement and Structural Robustness in Hybrid Lead Iodide Perovskite Discovered by High-Pressure Isotope Effect. Adv. Funct. Mater. 2020, 31, 2009131. [Google Scholar] [CrossRef]
- Tan, S.; Yavuz, I.; Weber, M.H.; Huang, T.; Chen, C.H.; Wang, R.; Wang, H.C.; Ko, J.H.; Nuryyeva, S.; Xue, J.; et al. Shallow Iodine Defects Accelerate the Degradation of α-Phase Formamidinium Perovskite. Joule 2020, 4, 2426–2442. [Google Scholar] [CrossRef]
- Fabini, D.H.; Hogan, T.; Evans, H.A.; Stoumpos, C.C.; Kanatzidis, M.G.; Seshadri, R. Dielectric and Thermodynamic Signatures of Low-Temperature Glassy Dynamics in the Hybrid Perovskites CH3NH3PbI3 and HC(NH2)2PbI3. J. Phys. Chem. Lett. 2016, 7, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Fabini, D.H.; Siaw, T.A.; Stoumpos, C.C.; Laurita, G.; Olds, D.; Page, K.; Hu, J.G.; Kanatzidis, M.G.; Han, S.; Seshadri, R. Universal Dynamics of Molecular Reorientation in Hybrid Lead Iodide Perovskites. J. Am. Chem. Soc. 2017, 139, 16875–16884. [Google Scholar] [CrossRef] [Green Version]
- Francisco-López, A.; Charles, B.; Alonso, M.I.; Garriga, M.; Campoy-Quiles, M.; Weller, M.T.; Goñi, A.R. Phase Diagram of Methylammonium/Formamidinium Lead Iodide Perovskite Solid Solutions from Temperature-Dependent Photoluminescence and Raman Spectroscopies. J. Phys. Chem. C 2020, 124, 3448–3458. [Google Scholar] [CrossRef]
- Swainson, I.; Hammond, R.; Soullière, C.; Knop, O.; Massa, W. Phase transitions in the perovskite methylammonium lead bromide, CH3ND3PbBr3. J. Solid State Chem. 2003, 176, 97–104. [Google Scholar] [CrossRef]
- Baikie, T.; Barrow, N.S.; Fang, Y.; Keenan, P.J.; Slater, P.R.; Piltz, R.O.; Gutmann, M.; Mhaisalkar, S.G.; White, T.J. A Combined Single Crystal Neutron/X-ray Diffraction and Solid-state Nuclear Magnetic Resonance Study of the Hybrid Perovskites CH3NH3PbX3 (X = I, Br and Cl). J. Mater. Chem. A 2015, 3, 9298–9307. [Google Scholar] [CrossRef]
- Weller, M.T.; Weber, O.J.; Frost, J.M.; Walsh, A. Cubic Perovskite Structure of Black Formamidinium Lead Iodide, α-[HC(NH2)2]PbI3, at 298 K. J. Phys. Chem. Lett. 2015, 6, 3209–3212. [Google Scholar] [CrossRef]
- Weller, M.T.; Weber, O.J.; Henry, P.F.; Pumpo, A.M.D.; Hansen, T.C. Complete structure and cation orientation in the perovskite photovoltaic methylammonium lead iodide between 100 and 352 K. Chem. Commun. 2015, 51, 4180–4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitfield, P.S.; Herron, N.; Guise, W.E.; Page, K.; Cheng, Y.Q.; Milas, I.; Crawford, M.K. Structures, Phase Transitions and Tricritical Behavior of the Hybrid Perovskite Methyl Ammonium Lead Iodide. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Foley, B.J.; Park, C.; Brown, C.M.; Harriger, L.W.; Lee, J.; Ruff, J.; Yoon, M.; Choi, J.J.; Lee, S.H. Entropy-driven Structural Transition and Kinetic Trapping in Formamidinium Lead Iodide Perovskite. Sci. Adv. 2016, 2, e1601650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comin, R.; Crawford, M.K.; Said, A.H.; Herron, N.; Guise, W.E.; Wang, X.; Whitfield, P.S.; Jain, A.; Gong, X.; McGaughey, A.J.H.; et al. Lattice Dynamics and the Nature of Structural Transitions in Organolead Halide Perovskites. Phys. Rev. B 2016, 94. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Oswald, I.W.H.; Wang, X.; McCandless, G.T.; Chan, J.Y. Orientation of Organic Cations in Hybrid Inorganic–Organic Perovskite CH3NH3PbI3 from Subatomic Resolution Single Crystal Neutron Diffraction Structural Studies. Cryst. Growth Des. 2016, 16, 2945–2951. [Google Scholar] [CrossRef]
- Minns, J.L.; Zajdel, P.; Chernyshov, D.; van Beek, W.; Green, M.A. Structure and Interstitial Iodide Migration in Hybrid Perovskite Methylammonium Lead Iodide. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Weber, O.J.; Ghosh, D.; Gaines, S.; Henry, P.F.; Walker, A.B.; Islam, M.S.; Weller, M.T. Phase Behavior and Polymorphism of Formamidinium Lead Iodide. Chem. Mater. 2018, 30, 3768–3778. [Google Scholar] [CrossRef]
- Mozur, E.M.; Trowbridge, J.C.; Maughan, A.E.; Gorman, M.J.; Brown, C.M.; Prisk, T.R.; Neilson, J.R. Dynamical Phase Transitions and Cation Orientation-Dependent Photoconductivity in CH(NH2)2PbBr3. ACS Mater. Lett. 2019, 1, 260–264. [Google Scholar] [CrossRef]
- Franz, A.; Többens, D.M.; Lehmann, F.; Kärgell, M.; Schorr, S. The Influence of Deuteration on the Crystal Structure of Hybrid Halide Perovskites: A Temperature-dependent Neutron Diffraction Study of FAPbBr3. Acta Crystallogr. B 2020, 76, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breternitz, J.; Tovar, M.; Schorr, S. Twinning in MAPbI3 at Room Temperature Uncovered Through Laue Neutron Diffraction. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Keen, D.A.; Gutmann, M.J.; Wilson, C.C. SXD – the single-crystal diffractometer at the ISIS spallation neutron source. J. Appl. Crystallogr. 2006, 39, 714–722. [Google Scholar] [CrossRef]
- Koetzle, T.F.; Bau, R.; Hoffmann, C.; Piccoli, P.M.B.; Schultz, A.J. Topaz: A Single-crystal Diffractometer for the Spallation Neutron Source. Acta Crystallogr. A 2006, 62, s116. [Google Scholar] [CrossRef]
- Ohhara, T.; Kiyanagi, R.; Oikawa, K.; Kaneko, K.; Kawasaki, T.; Tamura, I.; Nakao, A.; Hanashima, T.; Munakata, K.; Moyoshi, T.; et al. SENJU: A new time-of-flight single-crystal neutron diffractometer at J-PARC. J. Appl. Crystallogr. 2016, 49, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.J. Neutron Diffraction – Recent Applications to Chemical Structure Determination. Aust. J. Chem. 2011, 64, 869. [Google Scholar] [CrossRef]
- Raventós, M.; Tovar, M.; Medarde, M.; Shang, T.; Strobl, M.; Samothrakitis, S.; Pomjakushina, E.; Grünzweig, C.; Schmidt, S. Laue Three Dimensional Neutron Diffraction. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Szafrański, M.; Katrusiak, A. Photovoltaic Hybrid Perovskites under Pressure. J. Phys. Chem. Lett. 2017, 8, 2496–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postorino, P.; Malavasi, L. Pressure-Induced Effects in Organic–Inorganic Hybrid Perovskites. J. Phys. Chem. Lett. 2017, 8, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Capitani, F.; Marini, C.; Caramazza, S.; Postorino, P.; Garbarino, G.; Hanfland, M.; Pisanu, A.; Quadrelli, P.; Malavasi, L. High-pressure Behavior of Methylammonium Lead Iodide (MAPbI3) Hybrid Perovskite. J. Appl. Phys. 2016, 119, 185901. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, A.; Lin, Y.; Beavers, C.M.; Voss, J.; Mao, W.L.; Karunadasa, H.I. High-Pressure Single-Crystal Structures of 3D Lead-Halide Hybrid Perovskites and Pressure Effects on their Electronic and Optical Properties. ACS Cent. Sci. 2016, 2, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Szafrański, M.; Katrusiak, A. Mechanism of Pressure-Induced Phase Transitions, Amorphization, and Absorption-Edge Shift in Photovoltaic Methylammonium Lead Iodide. J. Phys. Chem. Lett. 2016, 7, 3458–3466. [Google Scholar] [CrossRef] [Green Version]
- Swainson, I.P.; Tucker, M.G.; Wilson, D.J.; Winkler, B.; Milman, V. Pressure Response of an Organic-Inorganic Perovskite: Methylammonium Lead Bromide. Chem. Mater. 2007, 19, 2401–2405. [Google Scholar] [CrossRef]
- Wiedemann, D.; Breternitz, J.; Paley, D.W.; Schorr, S. Hybrid Perovskite at Full Tilt: Structure and Symmetry Relations of the Incommensurately Modulated Phase of Methylammonium Lead Bromide, MAPbBr3. J. Phys. Chem. Lett. 2021, 2358–2362. [Google Scholar] [CrossRef]
- Kong, L.; Gong, J.; Hu, Q.; Capitani, F.; Celeste, A.; Hattori, T.; Sano-Furukawa, A.; Li, N.; Yang, W.; Liu, G.; et al. Halide Perovskites: Suppressed Lattice Disorder for Large Emission Enhancement and Structural Robustness in Hybrid Lead Iodide Perovskite Discovered by High-Pressure Isotope Effect. Adv. Funct. Mater. 2021, 31, 2170057. [Google Scholar] [CrossRef]
- Mozur, E.M.; Neilson, J.R. Cation Dynamics in Hybrid Halide Perovskites. arXiv 2020, arXiv:2012.05115. [Google Scholar]
- Manley, M.E.; Hong, K.; Yin, P.; Chi, S.; Cai, Y.; Hua, C.; Daemen, L.L.; Hermann, R.P.; Wang, H.; May, A.F.; et al. Giant Isotope Effect on Phonon Dispersion and Thermal Conductivity in Methylammonium Lead Iodide. Sci. Adv. 2020, 6, eaaz1842. [Google Scholar] [CrossRef] [PubMed]
- Gold-Parker, A.; Gehring, P.M.; Skelton, J.M.; Smith, I.C.; Parshall, D.; Frost, J.M.; Karunadasa, H.I.; Walsh, A.; Toney, M.F. Acoustic Phonon Lifetimes Limit Thermal Transport in Methylammonium Lead Iodide. Proc. Natl. Acad. Sci. USA 2018, 115, 11905–11910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weadock, N.J.; Gehring, P.M.; Gold-Parker, A.; Smith, I.C.; Karunadasa, H.I.; Toney, M.F. Test of the Dynamic-Domain and Critical Scattering Hypotheses in Cubic Methylammonium Lead Triiodide. Phys. Rev. Lett. 2020, 125. [Google Scholar] [CrossRef]
- Songvilay, M.; Bari, M.; Ye, Z.G.; Xu, G.; Gehring, P.M.; Ratcliff, W.D.; Schmalzl, K.; Bourdarot, F.; Roessli, B.; Stock, C. Lifetime-shortened acoustic phonons and static order at the Brillouin zone boundary in the organic-inorganic perovskite CH3NH3PbCl3. Phys. Rev. Mater. 2018, 2. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Létoublon, A.; Paofai, S.; Raymond, S.; Ecolivet, C.; Rufflé, B.; Cordier, S.; Katan, C.; Saidaminov, M.I.; Zhumekenov, A.A.; et al. Elastic Softness of Hybrid Lead Halide Perovskites. Phys. Rev. Lett. 2018, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Even, J.; Paofai, S.; Bourges, P.; Letoublon, A.; Cordier, S.; Durand, O.; Katan, C. Carrier scattering processes and low energy phonon spectroscopy in hybrid perovskites crystals. In Physics, Simulation, and Photonic Engineering of Photovoltaic Devices V; Freundlich, A., Lombez, L., Sugiyama, M., Eds.; SPIE: Bellingham, WA, USA, 2016. [Google Scholar] [CrossRef] [Green Version]
- Létoublon, A.; Paofai, S.; Rufflé, B.; Bourges, P.; Hehlen, B.; Michel, T.; Ecolivet, C.; Durand, O.; Cordier, S.; Katan, C.; et al. Elastic Constants, Optical Phonons, and Molecular Relaxations in the High Temperature Plastic Phase of the CH3NH3PbBr3 Hybrid Perovskite. J. Phys. Chem. Lett. 2016, 7, 3776–3784. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.C.; Paofai, S.; Létoublon, A.; Ollivier, J.; Raymond, S.; Hehlen, B.; Rufflé, B.; Cordier, S.; Katan, C.; Even, J.; et al. Direct Evidence of Weakly Dispersed and Strongly Anharmonic Optical Phonons in Hybrid Perovskites. Commun. Phys. 2020, 3. [Google Scholar] [CrossRef]
- Swainson, I.P.; Stock, C.; Parker, S.F.; Eijck, L.V.; Russina, M.; Taylor, J.W. From Soft Harmonic Phonons to Fast Relaxational Dynamics in CH3NH3PbBr3. Phys. Rev. B 2015, 92. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.L.; Parker, S.F.; García, I.R.; Mukhopadhyay, S.; Sakai, V.G.; Stock, C. Molecular Orientational Melting Within a Lead-halide Octahedron Framework: The Order-disorder Transition in CH3NH3PbBr3. Phys. Rev. B 2017, 96. [Google Scholar] [CrossRef] [Green Version]
- Songvilay, M.; Wang, Z.; Sakai, V.G.; Guidi, T.; Bari, M.; Ye, Z.G.; Xu, G.; Brown, K.L.; Gehring, P.M.; Stock, C. Decoupled molecular and inorganic framework dynamics in CH3NH3PbCl3. Phys. Rev. Mater. 2019, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Hu, X.; Chen, T.; Abernathy, D.L.; Kajimoto, R.; Nakamura, M.; Kofu, M.; Foley, B.J.; Yoon, M.; Choi, J.J.; et al. Temporally Decoherent and Spatially Coherent Vibrations in Metal Halide Perovskites. Phys. Rev. B 2020, 102. [Google Scholar] [CrossRef]
- Li, B.; Kawakita, Y.; Liu, Y.; Wang, M.; Matsuura, M.; Shibata, K.; Ohira-Kawamura, S.; Yamada, T.; Lin, S.; Nakajima, K.; et al. Polar Rotor Scattering as Atomic-level Origin of Low Mobility and Thermal Conductivity of Perovskite CH3NH3PbI3. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Schuck, G.; Lehmann, F.; Ollivier, J.; Mutka, H.; Schorr, S. Influence of Chloride Substitution on the Rotational Dynamics of Methylammonium in MAPbI3–xClxPerovskites. J. Phys. Chem. C 2019, 123, 11436–11446. [Google Scholar] [CrossRef] [Green Version]
- Beecher, A.N.; Semonin, O.E.; Skelton, J.M.; Frost, J.M.; Terban, M.W.; Zhai, H.; Alatas, A.; Owen, J.S.; Walsh, A.; Billinge, S.J.L. Direct Observation of Dynamic Symmetry Breaking above Room Temperature in Methylammonium Lead Iodide Perovskite. ACS Energy Lett. 2016, 1, 880–887. [Google Scholar] [CrossRef]
- Ma, H.; Ma, Y.; Wang, H.; Slebodnick, C.; Alatas, A.; Urban, J.J.; Tian, Z. Experimental Phonon Dispersion and Lifetimes of Tetragonal CH3NH3PbI3 Perovskite Crystals. J. Phys. Chem. Lett. 2018, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Heyer, D.; Buchenau, U.; Stamm, M. Determination of elastic shear constants of polyethylene at room temperature by inelastic neutron scattering. J. Polym. Sci. Polym. Phys. Ed. 1984, 22, 1515–1527. [Google Scholar] [CrossRef]
- Yang, J.; Wen, X.; Xia, H.; Sheng, R.; Ma, Q.; Kim, J.; Tapping, P.; Harada, T.; Kee, T.W.; Huang, F.; et al. Acoustic-optical Phonon Up-conversion and Hot-phonon Bottleneck in Lead-halide Perovskites. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Handa, T.; Yamada, T.; Nagai, M.; Kanemitsu, Y. Phonon, Thermal, and Thermo-optical Properties of Halide Perovskites. Phys. Chem. Chem. Phys. 2020, 22, 26069–26087. [Google Scholar] [CrossRef]
- Sendner, M.; Nayak, P.K.; Egger, D.A.; Beck, S.; Müller, C.; Epding, B.; Kowalsky, W.; Kronik, L.; Snaith, H.J.; Pucci, A.; et al. Optical phonons in methylammonium lead halide perovskites and implications for charge transport. Mater. Horizons 2016, 3, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Herz, L.M. Charge-Carrier Mobilities in Metal Halide Perovskites: Fundamental Mechanisms and Limits. ACS Energy Lett. 2017, 2, 1539–1548. [Google Scholar] [CrossRef]
- Herz, L.M. How Lattice Dynamics Moderate the Electronic Properties of Metal-Halide Perovskites. J. Phys. Chem. Lett. 2018, 9, 6853–6863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanigan-Atkins, T.; He, X.; Krogstad, M.J.; Pajerowski, D.M.; Abernathy, D.L.; Xu, G.N.M.N.; Xu, Z.; Chung, D.Y.; Kanatzidis, M.G.; Rosenkranz, S.; et al. Two-dimensional Overdamped Fluctuations of the Soft Perovskite Lattice in CsPbBr3. Nat. Mater. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, F.; Meggiolaro, D.; Mosconi, E.; Angelis, F.D. Charge Localization, Stabilization, and Hopping in Lead Halide Perovskites: Competition between Polaron Stabilization and Cation Disorder. ACS Energy Lett. 2019, 4, 2013–2020. [Google Scholar] [CrossRef] [Green Version]
- Leguy, A.M.A.; Goñi, A.R.; Frost, J.M.; Skelton, J.; Brivio, F.; Rodríguez-Martínez, X.; Weber, O.J.; Pallipurath, A.; Alonso, M.I.; Campoy-Quiles, M.; et al. Dynamic Disorder, Phonon Lifetimes, and the Assignment of Modes to the Vibrational Spectra of Methylammonium Lead Halide Perovskites. Phys. Chem. Chem. Phys. 2016, 18, 27051–27066. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Hunger, J.; Cánovas, E.; Karakus, M.; Mics, Z.; Grechko, M.; Turchinovich, D.; Parekh, S.H.; Bonn, M. Direct observation of mode-specific phonon-band gap coupling in methylammonium lead halide perovskites. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Foley, B.J.; Ipek, B.; Tyagi, M.; Copley, J.R.D.; Brown, C.M.; Choi, J.J.; Lee, S.H. Rotational Dynamics of Organic Cations in the CH3NH3PbI3 Perovskite. Phys. Chem. Chem. Phys. 2015, 17, 31278–31286. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.F.; Ramirez-Cuesta, A.J.; Albers, P.W.; Lennon, D. The use of Direct Geometry Spectrometers in Molecular Spectroscopy. J. Phys. Conf. Ser. 2014, 554, 012004. [Google Scholar] [CrossRef]
- Parker, S.F.; Fernandez-Alonso, F.; Ramirez-Cuesta, A.J.; Tomkinson, J.; Rudic, S.; Pinna, R.S.; Gorini, G.; Castañon, J.F. Recent and future developments on TOSCA at ISIS. J. Phys. Conf. Ser. 2014, 554, 012003. [Google Scholar] [CrossRef]
- Pinna, R.S.; Rudić, S.; Capstick, M.J.; McPhail, D.J.; Pooley, D.E.; Howells, G.D.; Gorini, G.; Fernandez-Alonso, F. Detailed Characterisation of the Incident Neutron Beam on the TOSCA Spectrometer. Nucl. Instrum. Methods Phys. Res. A 2017, 870, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Pinna, R.S.; Rudić, S.; Parker, S.F.; Armstrong, J.; Zanetti, M.; Škoro, G.; Waller, S.P.; Zacek, D.; Smith, C.A.; Capstick, M.J.; et al. The Neutron Guide Upgrade of the TOSCA Spectrometer. Nucl. Instrum. Methods Phys. Res. A 2018, 896, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Pinna, R.S.; Zanetti, M.; Rudić, S.; Parker, S.; Armstrong, J.; Waller, S.; Zacek, D.; Smith, C.; Harrison, S.; Gorini, G.; et al. The TOSCA Spectrometer at ISIS: The Guide Upgrade and Beyond. J. Phys. Conf. Ser. 2018, 1021, 012029. [Google Scholar] [CrossRef]
- Zanetti, M.; Bellissima, S.; del Rosso, L.; Masi, F.; Chowdhury, M.; Bonis, A.D.; Fresco, L.D.; Scatigno, C.; Armstrong, J.; Rudić, S.; et al. Neutronic developments on TOSCA and VESPA: Progress to date. Phys. B Condens. Matter. 2019, 562, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Seeger, P.A.; Daemen, L.L.; Larese, J.Z. Resolution of VISION, a Crystal-analyzer Spectrometer. Nucl. Instrum. Methods Phys. Res. 2009, 604, 719–728. [Google Scholar] [CrossRef]
- Ivanov, A.; Jimenéz-Ruiz, M.; Kulda, J. IN1-LAGRANGE – the new ILL Instrument to Explore Vibration Dynamics of Complex Materials. J. Phys. Conf. Ser. 2014, 554, 012001. [Google Scholar] [CrossRef] [Green Version]
- Drużbicki, K.; Pinna, R.S.; Rudić, S.; Jura, M.; Gorini, G.; Fernandez-Alonso, F. Unexpected Cation Dynamics in the Low-Temperature Phase of Methylammonium Lead Iodide: The Need for Improved Models. J. Phys. Chem. Lett. 2016, 7, 4701–4709. [Google Scholar] [CrossRef] [Green Version]
- Demmel, F.; McPhail, D.; Crawford, J.; Maxwell, D.; Pokhilchuk, K.; Garcia-Sakai, V.; Mukhopadyay, S.; Telling, M.; Bermejo, F.; Skipper, N.; et al. Opening the Terahertz Window on the OSIRIS Spectrometer. EPJ Web Conf. 2015, 83, 03003. [Google Scholar] [CrossRef] [Green Version]
- Bernard, G.M.; Wasylishen, R.E.; Ratcliffe, C.I.; Terskikh, V.; Wu, Q.; Buriak, J.M.; Hauger, T. Methylammonium Cation Dynamics in Methylammonium Lead Halide Perovskites: A Solid-State NMR Perspective. J. Phys. Chem. A 2018, 122, 1560–1573. [Google Scholar] [CrossRef] [Green Version]
- Supercritical Water. Chem. Eng. News 1991, 69, 26–39. [CrossRef]
- Bryce, D.L. NMR crystallography: Structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrJ 2017, 4, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Drużbicki, K.; Pajzderska, A.; Chudoba, D.; Jenczyk, J.; Jarek, M.; Mielcarek, J.; Wa̧sicki, J. Elucidating the Structure of Ranitidine Hydrochloride Form II: Insights from Solid-State Spectroscopy and ab initio Simulations. Cryst. Growth Des. 2018, 18, 4671–4681. [Google Scholar] [CrossRef]
- Trzeciak, K.; Kazmierski, S.; Druzbicki, K.; Potrzebowski, M.J. Mapping of Guest Localization in Mesoporous Silica Particles by Solid-state NMR and ab initio Modelling: New Insights into Benzoic Acid and p-Fluorobenzoic Acid Embedded in MCM-41 via Ball Milling. J. Phys. Chem. C 2021, in press. [Google Scholar]
- Souza, B.E.; Rudić, S.; Titov, K.; Babal, A.S.; Taylor, J.D.; Tan, J.C. Guest–host Interactions of Nanoconfined Anti-cancer Drug in Metal–organic Framework Exposed by Terahertz Dynamics. Chem. Commun. 2019, 55, 3868–3871. [Google Scholar] [CrossRef] [PubMed]
- Łuczyńska, K.; Drużbicki, K.; Runka, T.; Pałka, N.; Węsicki, J. Vibrational Response of Felodipine in the THz Domain: Optical and Neutron Spectroscopy Versus Plane-Wave DFT Modeling. J. Infrared Millim. Terahertz Waves 2019, 41, 1301–1336. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Kristallogr. Cryst. Mater. 2005, 220. [Google Scholar] [CrossRef] [Green Version]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational Density-functional Perturbation Theory for Dielectrics and Lattice Dynamics. Phys. Rev. B 2006, 73. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Baseggio, O.; Bonfà, P.; Brunato, D.; Car, R.; Carnimeo, I.; Cavazzoni, C.; de Gironcoli, S.; Delugas, P.; Ruffino, F.F.; et al. Quantum ESPRESSO Toward the Exascale. J. Chem. Phys. 2020, 152, 154105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonze, X.; Jollet, F.; Araujo, F.A.; Adams, D.; Amadon, B.; Applencourt, T.; Audouze, C.; Beuken, J.M.; Bieder, J.; Bokhanchuk, A.; et al. Recent developments in the ABINIT software package. Comput. Phys. Commun. 2016, 205, 106–131. [Google Scholar] [CrossRef] [Green Version]
- Hafner, J.; Kresse, G. The Vienna AB-Initio Simulation Program VASP: An Efficient and Versatile Tool for Studying the Structural, Dynamic, and Electronic Properties of Materials. In Properties of Complex Inorganic Solids; Springer: New York, NY, USA, 1997; pp. 69–82. [Google Scholar] [CrossRef]
- Enkovaara, J.; Rostgaard, C.; Mortensen, J.J.; Chen, J.; Dułak, M.; Ferrighi, L.; Gavnholt, J.; Glinsvad, C.; Haikola, V.; Hansen, H.A.; et al. Electronic Structure Calculations with GPAW: A Real-space Implementation of the Projector Augmented-wave Method. J. Phys. Condens. Matter 2010, 22, 253202. [Google Scholar] [CrossRef]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.; Reuter, K.; Scheffler, M. ab initio Molecular Simulations with Numeric Atom-centered Orbitals. Comput. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar] [CrossRef] [Green Version]
- Shang, H.; Carbogno, C.; Rinke, P.; Scheffler, M. Lattice Dynamics Calculations Based on Density-functional Perturbation Theory in Real Space. Comput. Phys. Commun. 2017, 215, 26–46. [Google Scholar] [CrossRef]
- García, A.; Papior, N.; Akhtar, A.; Artacho, E.; Blum, V.; Bosoni, E.; Brandimarte, P.; Brandbyge, M.; Cerdá, J.I.; Corsetti, F.; et al. Siesta: Recent Developments and Applications. J. Chem. Phys. 2020, 152, 204108. [Google Scholar] [CrossRef] [PubMed]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Balasubramani, S.G.; Chen, G.P.; Coriani, S.; Diedenhofen, M.; Frank, M.S.; Franzke, Y.J.; Furche, F.; Grotjahn, R.; Harding, M.E.; Hättig, C.; et al. TURBOMOLE: Modular Program Suite for ab initio Quantum-chemical and Condensed-matter Simulations. J. Chem. Phys. 2020, 152, 184107. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Ben, M.D.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An Electronic Structure and Molecular Dynamics Software Package—Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef] [PubMed]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.H.; Marks, L.D. WIEN2k: An APWlo Program for Calculating the Properties of Solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, J.K.; Sharma, S.; Nordström, L.; Cricchio, F. Available online: elk.sourceforge.net (accessed on 28 April 2021).
- Parlinski, K.; Li, Z.Q.; Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic ZrO2. Phys. Rev. Lett. 1997, 78, 4063–4066. [Google Scholar] [CrossRef]
- Baroni, S.; Giannozzi, P.; Testa, A. Green’s-function Approach to Linear Response in Solids. Phys. Rev. Lett. 1987, 58, 1861–1864. [Google Scholar] [CrossRef]
- Gonze, X. Perturbation Expansion of Variational Principles at Arbitrary Order. Phys. Rev. A 1995, 52, 1086–1095. [Google Scholar] [CrossRef]
- Gonze, X. Adiabatic Density-Functional Perturbation Theory. Phys. Rev. A 1995, 52, 1096–1114. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Chaput, L.; Togo, A.; Tanaka, I.; Hug, G. Phonon-phonon Interactions in Transition Metals. Phys. Rev. B 2011, 84. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shang, S.L.; Fang, H.; Liu, Z.K.; Chen, L.Q. First-principles Calculations of Lattice Dynamics and Thermal Properties of Polar Solids. NPJ Comput. Mater. 2016, 2. [Google Scholar] [CrossRef]
- Lloyd-Williams, J.H.; Monserrat, B. Lattice Dynamics and Electron-phonon Coupling Calculations Using Nondiagonal Supercells. Phys. Rev. B 2015, 92. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Cuesta, A. aCLIMAX 4.0.1, The new version of the software for analyzing and interpreting INS spectra. Comput. Phys. Commun. 2004, 157, 226–238. [Google Scholar] [CrossRef]
- Dymkowski, K.; Parker, S.F.; Fernandez-Alonso, F.; Mukhopadhyay, S. AbINS: The Modern Software for INS Interpretation. Phys. B Condens. Matter 2018, 551, 443–448. [Google Scholar] [CrossRef]
- Cheng, Y.Q.; Daemen, L.L.; Kolesnikov, A.I.; Ramirez-Cuesta, A.J. Simulation of Inelastic Neutron Scattering Spectra Using OCLIMAX. J. Chem. Theory Comput. 2019, 15, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Farhi, E. iFit: A Simple Library to Analyze Data. Available online: http://ifit.mccode.org/Models_Phonons.html (accessed on 28 April 2021).
- Euphonic. Available online: https://euphonic.readthedocs.io/en/latest/ (accessed on 28 April 2021).
- Parlinski, K. PhononA. Available online: http://www.computingformaterials.com/ (accessed on 28 April 2021).
- Kieslich, G.; Skelton, J.M.; Armstrong, J.; Wu, Y.; Wei, F.; Svane, K.L.; Walsh, A.; Butler, K.T. Hydrogen Bonding versus Entropy: Revealing the Underlying Thermodynamics of the Hybrid Organic–Inorganic Perovskite [CH3NH3]PbBr3. Chem. Mater. 2018, 30, 8782–8788. [Google Scholar] [CrossRef] [Green Version]
- Brehm, M.; Thomas, M.; Gehrke, S.; Kirchner, B. TRAVIS—A Free Analyzer for Trajectories from Molecular Simulation. J. Chem. Phys. 2020, 152, 164105. [Google Scholar] [CrossRef] [Green Version]
- Goret, G.; Aoun, B.; Pellegrini, E. MDANSE: An Interactive Analysis Environment for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 1–5. [Google Scholar] [CrossRef]
- Cheng, Y.Q.; Kolesnikov, A.I.; Ramirez-Cuesta, A.J. Simulation of Inelastic Neutron Scattering Spectra Directly from Molecular Dynamics Trajectories. J. Chem. Theory Comput. 2020, 16, 7702–7708. [Google Scholar] [CrossRef] [PubMed]
- Fransson, E.; Slabanja, M.; Erhart, P.; Wahnström, G. dynasor—A Tool for Extracting Dynamical Structure Factors and Current Correlation Functions from Molecular Dynamics Simulations. Adv. Theory Simul. 2021, 4, 2000240. [Google Scholar] [CrossRef]
- Carreras, A.; Togo, A.; Tanaka, I. DynaPhoPy: A code for extracting phonon quasiparticles from molecular dynamics simulations. Comput. Phys. Commun. 2017, 221, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, D.B.; Sun, T.; Wentzcovitch, R. phq: A Fortran code to compute phonon quasiparticle properties and dispersions. Comput. Phys. Commun. 2019, 243, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.F.; Leich, V.; Hönig, J.; Albers, P.W. Investigation of Commercial Graphenes. ChemistryOpen 2020, 9, 1060–1064. [Google Scholar] [CrossRef]
- Pontiroli, D.; Aramini, M.; Gaboardi, M.; Mazzani, M.; Sanna, S.; Caracciolo, F.; Carretta, P.; Cavallari, C.; Rols, S.; Tatti, R.; et al. Tracking the Hydrogen Motion in Defective Graphene. J. Phys. Chem. C 2014, 118, 7110–7116. [Google Scholar] [CrossRef]
- Natkaniec, I.; Sheka, E.F.; Drużbicki, K.; Hołderna-Natkaniec, K.; Gubin, S.P.; Buslaeva, E.Y.; Tkachev, S.V. Computationally Supported Neutron Scattering Study of Parent and Chemically Reduced Graphene Oxide. J. Phys. Chem. C 2015, 119, 18650–18662. [Google Scholar] [CrossRef]
- Cavallari, C.; Pontiroli, D.; Jiménez-Ruiz, M.; Johnson, M.; Aramini, M.; Gaboardi, M.; Parker, S.F.; Riccó, M.; Rols, S. Hydrogen Motions in Defective Graphene: The Role of Surface Defects. Phys. Chem. Chem. Phys. 2016, 18, 24820–24824. [Google Scholar] [CrossRef]
- Cavallari, C.; Rols, S.; Fischer, H.E.; Brunelli, M.; Gaboardi, M.; Magnani, G.; Riccò, M.; Pontiroli, D. Neutron Scattering Study of Nickel Decorated Thermally Exfoliated Graphite Oxide. Int. J. Hydrogen Energy 2019, 44, 30999–31007. [Google Scholar] [CrossRef]
- Vottero, E.; Carosso, M.; Jiménez-Ruiz, M.; Pellegrini, R.; Groppo, E.; Piovano, A. How do the Graphenic Domains Terminate in Activated Carbons and Carbon-supported Metal Catalysts? Carbon 2020, 169, 357–369. [Google Scholar] [CrossRef]
- Blackburn, J.L.; Engtrakul, C.; Bult, J.B.; Hurst, K.; Zhao, Y.; Xu, Q.; Parilla, P.A.; Simpson, L.J.; Rocha, J.d.R.; Hudson, M.R.; et al. Spectroscopic Identification of Hydrogen Spillover Species in Ruthenium-Modified High Surface Area Carbons by Diffuse Reflectance Infrared Fourier Transform Spectroscopy. J. Phys. Chem. C 2012, 116, 26744–26755. [Google Scholar] [CrossRef]
- Tsao, C.S.; Liu, Y.; Chuang, H.Y.; Tseng, H.H.; Chen, T.Y.; Chen, C.H.; Yu, M.S.; Li, Q.; Lueking, A.; Chen, S.H. Hydrogen Spillover Effect of Pt-Doped Activated Carbon Studied by Inelastic Neutron Scattering. J. Phys. Chem. Lett. 2011, 2322–2325. [Google Scholar] [CrossRef]
- Cavallari, C.; Pontiroli, D.; Jiménez-Ruiz, M.; Ivanov, A.; Mazzani, M.; Gaboardi, M.; Aramini, M.; Brunelli, M.; Riccò, M.; Rols, S. Hydrogen on Graphene Investigated by Inelastic Neutron Scattering. J. Phys. Conf. Ser. 2014, 554, 012009. [Google Scholar] [CrossRef] [Green Version]
- Drużbicki, K.; Natkaniec, I. Vibrational Properties of Water Retained in Graphene Oxide. Chem. Phys. Lett. 2014, 600, 106–111. [Google Scholar] [CrossRef]
- Romanelli, G.; Senesi, R.; Zhang, X.; Loh, K.P.; Andreani, C. Probing the Effects of 2D Confinement on Hydrogen Dynamics in Water and Ice Adsorbed in Graphene Oxide Sponges. Phys. Chem. Chem. Phys. 2015, 17, 31680–31684. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, G.; Liscio, A.; Senesi, R.; Zamboni, R.; Treossi, E.; Liscio, F.; Giambastiani, G.; Palermo, V.; Fernandez-Alonso, F.; Andreani, C. Soft Confinement of Water in Graphene-oxide Membranes. Carbon 2016, 108, 199–203. [Google Scholar] [CrossRef]
- Osti, N.C.; Naguib, M.; Ganeshan, K.; Shin, Y.K.; Ostadhossein, A.; van Duin, A.C.T.; Cheng, Y.; Daemen, L.L.; Gogotsi, Y.; Mamontov, E.; et al. Influence of Metal Ions Intercalation on the Vibrational Dynamics of Water Confined Between MXene Layers. Phys. Rev. Mater. 2017, 1. [Google Scholar] [CrossRef]
- Barroso-Bujans, F.; Fernandez-Alonso, F.; Colmenero, J. Neutron Spectroscopy as a Probe of Macromolecular Structure and Dynamics under Extreme Spatial Confinement. J. Phys. Conf. Ser. 2014, 549, 012009. [Google Scholar] [CrossRef]
- Barroso-Bujans, F.; Fernandez-Alonso, F.; Cerveny, S.; Parker, S.F.; Alegría, A.; Colmenero, J. Polymers Under Extreme Two-dimensional Confinement: Poly(ethylene oxide) in Graphite Oxide. Soft Matter 2011, 7, 7173. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Bujans, F.; Fernandez-Alonso, F.; Cerveny, S.; Arrese-Igor, S.; Alegría, A.; Colmenero, J. Two-Dimensional Subnanometer Confinement of Ethylene Glycol and Poly(ethylene oxide) by Neutron Spectroscopy: Molecular Size Effects. Macromolecules 2012, 45, 3137–3144. [Google Scholar] [CrossRef]
- Barroso-Bujans, F.; Fernandez-Alonso, F.; Pomposo, J.A.; Cerveny, S.; Alegría, A.; Colmenero, J. Macromolecular Structure and Vibrational Dynamics of Confined Poly(ethylene oxide): From Subnanometer 2D-Intercalation into Graphite Oxide to Surface Adsorption onto Graphene Sheets. ACS Macro Lett. 2012, 1, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Barroso-Bujans, F.; Palomino, P.; Cerveny, S.; Fernandez-Alonso, F.; Rudić, S.; Alegría, A.; Colmenero, J.; Enciso, E. Confinement of Poly(ethylene oxide) in the Nanometer-scale Pores of Resins and Carbon Nanoparticles. Soft Matter 2013, 9, 10960. [Google Scholar] [CrossRef]
- Vilela, C.; Freire, C.S.R.; Araújo, C.; Rudić, S.; Silvestre, A.J.D.; Vaz, P.D.; Ribeiro-Claro, P.J.A.; Nolasco, M.M. Understanding the Structure and Dynamics of Nanocellulose-Based Composites with Neutral and Ionic Poly(methacrylate) Derivatives Using Inelastic Neutron Scattering and DFT Calculations. Molecules 2020, 25, 1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilela, C.; Cordeiro, D.M.; Boas, J.V.; Barbosa, P.; Nolasco, M.; Vaz, P.D.; Rudić, S.; Ribeiro-Claro, P.; Silvestre, A.J.; Oliveira, V.B.; et al. Poly(4-styrene Sulfonic Acid)/Bacterial Cellulose Membranes: Electrochemical Performance in a Single-chamber Microbial Fuel Cell. Bioresour. Technol. Rep. 2020, 9, 100376. [Google Scholar] [CrossRef]
- Parker, S.F.; Shah, S. Characterisation of Hydration Water in Nafion Membrane. RSC Adv. 2021, 11, 9381–9385. [Google Scholar] [CrossRef]
- Lovell, A.; Fernandez-Alonso, F.; Skipper, N.T.; Refson, K.; Bennington, S.M.; Parker, S.F. Quantum Delocalization of Molecular Hydrogen in Alkali-Graphite Intercalates. Phys. Rev. Lett. 2008, 101, 126101. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Alonso, F.; Bermejo, F.J.; Cabrillo, C.; Loutfy, R.O.; Leon, V.; Saboungi, M.L. Nature of the Bound States of Molecular Hydrogen in Carbon Nanohorns. Phys. Rev. Lett. 2007, 98, 215503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzystyniak, M.; Romanelli, G.; Fernandez-Alonso, F. Non-destructive Quantitation of Hydrogen via Mass-resolved Neutron Spectroscopy. Analyst 2019, 144, 3936–3941. [Google Scholar] [CrossRef] [PubMed]
- Schur, D.V.; Zaginaichenko, S.Y.; Savenko, A.F.; Bogolepov, V.A.; Anikina, N.S.; Zolotarenko, A.; Matysina, Z.A.; Veziroglu, T.N.; Skryabina, N.E. Experimental Evaluation of Total Hydrogen Capacity for Fullerite C60. Int. J. Hydrogen Energy 2011, 36, 1143–1151. [Google Scholar] [CrossRef]
- Mauron, P.; Remhof, A.; Bliersbach, A.; Borgschulte, A.; Züttel, A.; Sheptyakov, D.; Gaboardi, M.; Choucair, M.; Pontiroli, D.; Aramini, M.; et al. Reversible Hydrogen Absorption in Sodium Intercalated Fullerenes. Int. J. Hydrogen Energy 2012, 37, 14307–14314. [Google Scholar] [CrossRef]
- Gaboardi, M.; Amadé, N.S.; Aramini, M.; Milanese, C.; Magnani, G.; Sanna, S.; Riccò, M.; Pontiroli, D. Extending the Hydrogen Storage Limit in Fullerene. Carbon 2017, 120, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Mauron, P.; Gaboardi, M.; Pontiroli, D.; Remhof, A.; Riccò, M.; Züttel, A. Hydrogen Desorption Kinetics in Metal Intercalated Fullerides. J. Phys. Chem. C 2015, 119, 1714–1719. [Google Scholar] [CrossRef]
- Amadè, N.S.; Pontiroli, D.; Maidich, L.; Riccò, M.; Gaboardi, M.; Magnani, G.; Carretta, P.; Sanna, S. Molecular and Ionic Dynamics in NaxLi6–xC60. J. Phys. Chem. C 2017, 121, 6554–6560. [Google Scholar] [CrossRef]
- Gaboardi, M.; Amadè, N.S.; Riccò, M.; Milanese, C.; Girella, A.; Gioventù, M.; Fernandez-Alonso, F. Synthesis and Characterization of Mixed Sodium and Lithium Fullerides for Hydrogen Storage. Int. J. Hydrogen Energy 2018, 43, 16766–16773. [Google Scholar] [CrossRef] [Green Version]
- Mauron, P.; Gaboardi, M.; Remhof, A.; Bliersbach, A.; Sheptyakov, D.; Aramini, M.; Vlahopoulou, G.; Giglio, F.; Pontiroli, D.; Riccò, M.; et al. Hydrogen Sorption in Li12C60. J. Phys. Chem. C 2013, 117, 22598–22602. [Google Scholar] [CrossRef]
- Gaboardi, M.; Duyker, S.; Milanese, C.; Magnani, G.; Peterson, V.K.; Pontiroli, D.; Sharma, N.; Riccò, M. In Situ Neutron Powder Diffraction of Li6C60 for Hydrogen Storage. J. Phys. Chem. C 2015, 119, 19715–19721. [Google Scholar] [CrossRef]
- Aramini, M.; Gaboardi, M.; Vlahopoulou, G.; Pontiroli, D.; Cavallari, C.; Milanese, C.; Riccò, M. Muon Spin Relaxation Reveals the Hydrogen Storage Mechanism in Light Alkali Metal Fullerides. Carbon 2014, 67, 92–97. [Google Scholar] [CrossRef]
- Giglio, F.; Pontiroli, D.; Gaboardi, M.; Aramini, M.; Cavallari, C.; Brunelli, M.; Galinetto, P.; Milanese, C.; Riccò, M. Li12C60: A Lithium Clusters Intercalated Fulleride. Chem. Phys. Lett. 2014, 609, 155–160. [Google Scholar] [CrossRef]
- Gaboardi, M.; Cavallari, C.; Magnani, G.; Pontiroli, D.; Rols, S.; Riccò, M. Hydrogen Storage Mechanism and Lithium Dynamics in Li12C60 investigated by μSR. Carbon 2015, 90, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Maidich, L.; Pontiroli, D.; Gaboardi, M.; Lenti, S.; Magnani, G.; Riva, G.; Carretta, P.; Milanese, C.; Marini, A.; Riccò, M.; et al. Investigation of Li and H Dynamics in Li6C60 and Li6C60Hy. Carbon 2016, 96, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Sarzi Amadè, N.; Gaboardi, M.; Magnani, G.; Riccò, M.; Pontiroli, D.; Milanese, C.; Girella, A.; Carretta, P.; Sanna, S. H and Li dynamics in Li12C60 and Li12C60Hy. Int. J. Hydrogen Energy 2017, 42, 22544–22550. [Google Scholar] [CrossRef] [Green Version]
- Gaboardi, M.; Milanese, C.; Magnani, G.; Girella, A.; Pontiroli, D.; Cofrancesco, P.; Marini, A.; Riccò, M. Optimal Hydrogen Storage in Sodium Substituted Lithium Fullerides. Phys. Chem. Chem. Phys. 2017, 19, 21980–21986. [Google Scholar] [CrossRef] [Green Version]
- Scaravonati, S.; Magnani, G.; Gaboardi, M.; Allodi, G.; Riccò, M.; Pontiroli, D. Electrochemical Intercalation of Fullerene and Hydrofullerene with Sodium. Carbon 2018, 130, 11–18. [Google Scholar] [CrossRef]
- Aramini, M.; Magnani, G.; Pontiroli, D.; Milanese, C.; Girella, A.; Bertoni, G.; Gaboardi, M.; Zacchini, S.; Marini, A.; Riccò, M. Nickel Addition to Optimize the Hydrogen Storage Performance of Lithium Intercalated Fullerides. Mater. Res. Bull. 2020, 126, 110848. [Google Scholar] [CrossRef]
- Sartori, S.; Guzik, M.N.; Knudsen, K.D.; Sørby, M.H.; Teprovich, J.A.; Zidan, R.; Hauback, B.C. Stability and Phase Formation in the (Li/Na)6C60-H Systems Studied by Neutron Scattering. J. Phys. Chem. C 2018, 122, 18346–18355. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.A.; Teprovich, J.A.; Compton, R.; Schwartz, V.; Veith, G.M.; Zidan, R. Evaluation of the physi- and chemisorption of hydrogen in alkali (Na, Li) doped fullerenes. Int. J. Hydrogen Energy 2015, 40, 2710–2716. [Google Scholar] [CrossRef]
- Yoshida, A.; Okuyama, T.; Terada, T.; Naito, S. Reversible hydrogen storage/release phenomena on lithium fulleride (LinC60) and their mechanistic investigation by solid-state NMR spectroscopy. J. Mater. Chem. 2011, 21, 9480. [Google Scholar] [CrossRef]
- Teprovich, J.A.; Wellons, M.S.; Lascola, R.; Hwang, S.J.; Ward, P.a.; Compton, R.N.; Zidan, R. Synthesis and characterization of a lithium-doped fullerane (Lix-C60-Hy) for reversible hydrogen storage. Nano Lett. 2012, 12, 582–589. [Google Scholar] [CrossRef]
- Teprovich, J.A.; Knight, D.A.; Peters, B.; Zidan, R. Comparative study of reversible hydrogen storage in alkali-doped fulleranes. J. Alloy. Compd. 2013, 580, S364–S367. [Google Scholar] [CrossRef]
- Pontiroli, D.; D’Alessio, D.; Gaboardi, M.; Magnani, G.; Milanese, C.; Duyker, S.G.; Peterson, V.K.; Sharma, N.; Riccò, M. Ammonia-storage in Lithium Intercalated Fullerides. J. Mater. Chem. A 2015, 3, 21099–21105. [Google Scholar] [CrossRef] [Green Version]
- Durand, P.; Dubitsky, Y.; Rosseinsky, M.J.; Zaopo, A. Expanded fullerides and electron localisation—Lithium-rich ammoniated C(60) phases. Dalton Trans. 2004, 3137–3143. [Google Scholar] [CrossRef]
- Fullagar, W.; Reynolds, P.; White, J. Lithium and sodium fullerides prepared in liquid ammonia. Solid State Commun. 1997, 104, 23–27. [Google Scholar] [CrossRef]
- Aramini, M.; Milanese, C.; Pontiroli, D.; Gaboardi, M.; Girella, A.; Bertoni, G.; Riccò, M. Addition of Transition Metals to Lithium Intercalated Fullerides Enhances Hydrogen Storage Properties. Int. J. Hydrogen Energy 2014, 39, 2124–2131. [Google Scholar] [CrossRef]
- Eklund, P.C.; Rao, A.M. (Eds.) Fullerene Polymers and Fullerene Polymer Composites; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar] [CrossRef]
- Rols, S.; Cambedouzou, J.; Bantignies, J.L.; Rachdi, F.; Sauvajol, J.L.; Agafonov, V.; Rakhmanina, A.V.; Davydov, V.A.; Hennion, B.; Kahn, R. Lattice Dynamics of Pressure-polymerized Phases of C60: A Neutron Scattering Investigation. Phys. Rev. B 2004, 70. [Google Scholar] [CrossRef]
- Rols, S.; Bantignies, J.L.; Maurin, D.; Sauvajol, J.L.; Agafonov, V.; Rakhmanina, A.V.; Davydov, V.A. Low-Frequency Phonons in High-Pressure High-Temperature C60 Polymers. Fuller. Nanotub. Carbon Nanostructures 2005, 12, 263–268. [Google Scholar] [CrossRef]
- Cambedouzou, J.; Rols, S.; Almairac, R.; Sauvajol, J.L.; Kataura, H.; Schober, H. Low-frequency Excitations of C60 Chains Inserted Inside Single-walled Carbon Nanotubes. Phys. Rev. B 2005, 71. [Google Scholar] [CrossRef] [Green Version]
- Pontiroli, D.; Aramini, M.; Gaboardi, M.; Mazzani, M.; Gorreri, A.; Riccò, M.; Margiolaki, I.; Sheptyakov, D. Ionic conductivity in the Mg intercalated fullerene polymer Mg2C60. Carbon 2013, 51, 143–147. [Google Scholar] [CrossRef]
- Kubozono, Y.; Takabayashi, Y.; Kambe, T.; Fujiki, S.; Kashino, S.; Emura, S. Structure and physical properties of Na4C60 under ambient and high pressures. Phys. Rev. B 2001, 63, 045418. [Google Scholar] [CrossRef] [Green Version]
- Pekker, S.; Jánossy, A.; Mihaly, L.; Chauvet, O.; Carrard, M.; Forró, L. Single-Crystalline (KC60)n: A Conducting Linear Alkali Fulleride Polymer. Science 1994, 265, 1077–1078. [Google Scholar] [CrossRef]
- Stephens, P.W.; Bortel, G.; Faigel, G.; Tegze, M.; Jánossy, A.; Pekker, S.; Oszlanyi, G.; Forró, L. Polymeric fullerene chains in RbC60 and KC60. Nature 1994, 370, 636–639. [Google Scholar] [CrossRef]
- Chauvet, O.; Oszlànyi, G.; Forro, L.; Stephens, P.; Tegze, M.; Faigel, G.; Jànossy, A. Quasi-one-dimensional electronic structure in orthorhombic RbC60. Phys. Rev. Lett. 1994, 72, 2721–2724. [Google Scholar] [CrossRef] [PubMed]
- Rols, S.; Pontiroli, D.; Cavallari, C.; Gaboardi, M.; Aramini, M.; Richard, D.; Johnson, M.R.; Zanotti, J.M.; Suard, E.; Maccarini, M.; et al. Structure and Dynamics of the Fullerene Polymer Li4C60 Studied with Neutron Scattering. Phys. Rev. B 2015, 92. [Google Scholar] [CrossRef] [Green Version]
- Rols, S.; Pontiroli, D.; Aramini, M.; Gaboardi, M.; Cavallari, C.; Riccò, M.; Suard, E.; Johnson, M.R.; Richard, D. Lattice Dynamics of the Ionic Superconductor Li4C60. Inelastic Neutron Scattering and Powder Averaged Lattice Dynamics (PALD) investigations. Acta Crystallogr. A 2016, 72, s76. [Google Scholar] [CrossRef] [Green Version]
- Riccò, M.; Belli, M.; Mazzani, M.; Pontiroli, D.; Quintavalle, D.; Jánossy, A.; Csányi, G. Superionic Conductivity in the Li4C60 Fulleride Polymer. Phys. Rev. Lett. 2009, 102, 145901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosi, N.L. Hydrogen Storage in Microporous Metal-Organic Frameworks. Science 2003, 300, 1127–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowsell, J.L.C.; Eckert, J.; Yaghi, O.M. Characterization of H2Binding Sites in Prototypical Metal-Organic Frameworks by Inelastic Neutron Scattering. J. Am. Chem. Soc. 2005, 127, 14904–14910. [Google Scholar] [CrossRef]
- Ye, S.; Xu, M.; Bacic, Z.; Lawler, R.; Turro, N.J. Quantum Dynamics of a Hydrogen Molecule Inside an Anisotropic Open-Cage Fullerene: Coupled Translation-Rotation Eigenstates and Comparison with Inelastic Neutron Scattering Spectroscopy. J. Phys. Chem. A 2010, 114, 9936–9947. [Google Scholar] [CrossRef]
- Horsewill, A.J.; Panesar, K.S.; Rols, S.; Johnson, M.R.; Murata, Y.; Komatsu, K.; Mamone, S.; Danquigny, A.; Cuda, F.; Maltsev, S.; et al. Quantum Translator-Rotator: Inelastic Neutron Scattering of Dihydrogen Molecules Trapped inside Anisotropic Fullerene Cages. Phys. Rev. Lett. 2009, 102. [Google Scholar] [CrossRef] [Green Version]
- Horsewill, A.J.; Goh, K.; Rols, S.; Ollivier, J.; Johnson, M.R.; Levitt, M.H.; Carravetta, M.; Mamone, S.; Murata, Y.; Chen, J.Y.C.; et al. Quantum Rotation and Translation of Hydrogen Molecules Encapsulated Inside C60: Temperature Dependence of Inelastic Neutron Scattering Spectra. Philos. Trans. R. Soc. A 2013, 371, 20110627. [Google Scholar] [CrossRef]
- Horsewill, A.J.; Panesar, K.S.; Rols, S.; Ollivier, J.; Johnson, M.R.; Carravetta, M.; Mamone, S.; Levitt, M.H.; Murata, Y.; Komatsu, K.; et al. Inelastic Neutron Scattering Investigations of the Quantum Molecular Dynamics of a H2 Molecule Entrapped Inside a Fullerene Cage. Phys. Rev. B 2012, 85. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Alonso, F.; Cabrillo, C.; Fernández-Perea, R.; Bermejo, F.J.; González, M.A.; Mondelli, C.; Farhi, E. Solid para-Hydrogen as the Paradigmatic Quantum Crystal: Three Observables Probed by Ultrahigh-resolution Neutron Spectroscopy. Phys. Rev. B 2012, 86. [Google Scholar] [CrossRef] [Green Version]
- Cabrillo, C.; Fernández-Alonso, F.; Fernández-Perea, R.; Bermejo, F.J.; González, M.A.; Mondelli, C.; Farhi, E. Crystallization of para-Hydrogen: A Quantum Phase Transition at Finite Temperature? J. Phys. Conf. Ser. 2015, 663, 012006. [Google Scholar] [CrossRef]
- Xu, M.; Jiménez-Ruiz, M.; Johnson, M.R.; Rols, S.; Ye, S.; Carravetta, M.; Denning, M.S.; Lei, X.; Bačić, Z.; Horsewill, A.J. Confirming a Predicted Selection Rule in Inelastic Neutron Scattering Spectroscopy: The Quantum Translator-Rotator H2 Entrapped Inside C60. Phys. Rev. Lett. 2014; 113. [Google Scholar] [CrossRef] [Green Version]
- Easun, T.L.; Moreau, F.; Yan, Y.; Yang, S.; Schröder, M. Structural and Dynamic Studies of Substrate Binding in Porous Metal–organic Frameworks. Chem. Soc. Rev. 2017, 46, 239–274. [Google Scholar] [CrossRef] [Green Version]
- Kibble, M.G.; Ramirez-Cuesta, A.J.; Goodway, C.M.; Evans, B.E.; Kirichek, O. Hydrogen Gas Sample Environment for TOSCA. J. Phys. Conf. Ser. 2014, 554, 012006. [Google Scholar] [CrossRef] [Green Version]
- Redfern, L.R.; Farha, O.K. Mechanical properties of metal–organic frameworks. Chem. Sci. 2019, 10, 10666–10679. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.U.; Boutin, A.; Fuchs, A.H.; Coudert, F.X. Anisotropic Elastic Properties of Flexible Metal-Organic Frameworks: How Soft are Soft Porous Crystals? Phys. Rev. Lett. 2012, 109. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.C.; Civalleri, B.; Lin, C.C.; Valenzano, L.; Galvelis, R.; Chen, P.F.; Bennett, T.D.; Mellot-Draznieks, C.; Zicovich-Wilson, C.M.; Cheetham, A.K. Exceptionally Low Shear Modulus in a Prototypical Imidazole-Based Metal-Organic Framework. Phys. Rev. Lett. 2012, 108. [Google Scholar] [CrossRef] [Green Version]
- Widmer, R.N.; Lampronti, G.I.; Chibani, S.; Wilson, C.W.; Anzellini, S.; Farsang, S.; Kleppe, A.K.; Casati, N.P.M.; MacLeod, S.G.; Redfern, S.A.T.; et al. Rich Polymorphism of a Metal–Organic Framework in Pressure–Temperature Space. J. Am. Chem. Soc. 2019, 141, 9330–9337. [Google Scholar] [CrossRef] [Green Version]
- Ryder, M.; Civalleri, B.; Bennett, T.; Henke, S.; Rudić, S.; Cinque, G.; Fernandez-Alonso, F.; Tan, J.C. Identifying the Role of Terahertz Vibrations in Metal-Organic Frameworks: From Gate-Opening Phenomenon to Shear-Driven Structural Destabilization. Phys. Rev. Lett. 2014, 113. [Google Scholar] [CrossRef] [Green Version]
- Fairen-Jimenez, D.; Moggach, S.A.; Wharmby, M.T.; Wright, P.A.; Parsons, S.; D’uren, T. Opening the Gate: Framework Flexibility in ZIF-8 Explored by Experiments and Simulations. J. Am. Chem. Soc. 2011, 133, 8900–8902. [Google Scholar] [CrossRef] [Green Version]
- Casco, M.E.; Cheng, Y.Q.; Daemen, L.L.; Fairen-Jimenez, D.; Ramos-Fernández, E.V.; Ramirez-Cuesta, A.J.; Silvestre-Albero, J. Gate-opening Effect in ZIF-8: The First Experimental Proof using Inelastic Neutron Scattering. Chem. Commun. 2016, 52, 3639–3642. [Google Scholar] [CrossRef] [Green Version]
- Weinrauch, I.; Savchenko, I.; Denysenko, D.; Souliou, S.M.; Kim, H.H.; Tacon, M.L.; Daemen, L.L.; Cheng, Y.; Mavrandonakis, A.; Ramirez-Cuesta, A.J.; et al. Capture of Heavy Hydrogen Isotopes in a Metal-organic Framework with Active Cu(I) Sites. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, M.; da Silva, I.; Johnson, M.; Carter, J.H.; Newby, R.; Suyetin, M.; Besley, E.; Manuel, P.; Rudić, S.; Fitch, A.N.; et al. Observation of Binding and Rotation of Methane and Hydrogen within a Functional Metal–Organic Framework. J. Am. Chem. Soc. 2016, 138, 9119–9127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, M.; Cheng, Y.; Easun, T.L.; Eyley, J.E.; Argent, S.P.; Warren, M.R.; Lewis, W.; Murray, C.; Tang, C.C.; Frogley, M.D.; et al. Selective Adsorption of Sulfur Dioxide in a Robust Metal-Organic Framework Material. Adv. Mater. 2016, 28, 8705–8711. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Forrest, K.A.; Mostrom, M.; Hunt, J.R.; Furukawa, H.; Eckert, J.; Space, B. The Rotational Dynamics of H2 Adsorbed in Covalent Organic Frameworks. Phys. Chem. Chem. Phys. 2017, 19, 13075–13082. [Google Scholar] [CrossRef]
- Cuadrado-Collados, C.; Fernández-Català, J.; Fauth, F.; Cheng, Y.Q.; Daemen, L.L.; Ramirez-Cuesta, A.J.; Silvestre-Albero, J. Understanding the Breathing Phenomena in Nano-ZIF-7 upon Gas Adsorption. J. Mater. Chem. A 2017, 5, 20938–20946. [Google Scholar] [CrossRef] [Green Version]
- Casco, M.E.; Fernández-Catalá, J.; Cheng, Y.; Daemen, L.; Ramirez-Cuesta, A.J.; Cuadrado-Collados, C.; Silvestre-Albero, J.; Ramos-Fernandez, E.V. Understanding ZIF-8 Performance upon Gas Adsorption by Means of Inelastic Neutron Scattering. ChemistrySelect 2017, 2, 2750–2753. [Google Scholar] [CrossRef]
- Zhang, X.; da Silva, I.; Godfrey, H.G.W.; Callear, S.K.; Sapchenko, S.A.; Cheng, Y.; Vitórica-Yrezábal, I.; Frogley, M.D.; Cinque, G.; Tang, C.C.; et al. Confinement of Iodine Molecules into Triple-Helical Chains within Robust Metal–Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 16289–16296. [Google Scholar] [CrossRef]
- Lu, Z.; Godfrey, H.G.W.; da Silva, I.; Cheng, Y.; Savage, M.; Tuna, F.; McInnes, E.J.L.; Teat, S.J.; Gagnon, K.J.; Frogley, M.D.; et al. Modulating Supramolecular Binding of Carbon Dioxide in a Redox-active Porous Metal-organic Framework. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Ryder, M.; de Voorde, B.V.; Civalleri, B.; Bennett, T.D.; Mukhopadhyay, S.; Cinque, G.; Fernandez-Alonso, F.; Vos, D.D.; Rudić, S.; Tan, J.C. Detecting Molecular Rotational Dynamics Complementing the Low-Frequency Terahertz Vibrations in a Zirconium-Based Metal-Organic Framework. Phys. Rev. Lett. 2017, 118. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; da Silva, I.; Kolokolov, D.I.; Han, X.; Li, J.; Smith, G.; Cheng, Y.; Daemen, L.L.; Morris, C.G.; Godfrey, H.G.W.; et al. Post-synthetic Modulation of the Charge Distribution in a Metal–organic Framework for Optimal Binding of Carbon Dioxide and Sulfur Dioxide. Chem. Sci. 2019, 10, 1472–1482. [Google Scholar] [CrossRef]
- Duong, T.D.; Sapchenko, S.A.; da Silva, I.; Godfrey, H.G.W.; Cheng, Y.; Daemen, L.L.; Manuel, P.; Ramirez-Cuesta, A.J.; Yang, S.; Schröder, M. Optimal Binding of Acetylene to a Nitro-Decorated Metal–Organic Framework. J. Am. Chem. Soc. 2018, 140, 16006–16009. [Google Scholar] [CrossRef] [Green Version]
- Duong, T.D.; Sapchenko, S.A.; da Silva, I.; Godfrey, H.G.W.; Cheng, Y.; Daemen, L.L.; Manuel, P.; Frogley, M.D.; Cinque, G.; Ramirez-Cuesta, A.J.; et al. Observation of Binding of Carbon Dioxide to Nitro-decorated Metal–organic Frameworks. Chem. Sci. 2020, 11, 5339–5346. [Google Scholar] [CrossRef]
- Gandara-Loe, J.; Missyul, A.; Fauth, F.; Daemen, L.L.; Cheng, Y.Q.; Ramirez-Cuesta, A.J.; Ravikovitch, P.I.; Silvestre-Albero, J. New Insights into the Breathing Phenomenon in ZIF-4. J. Mater. Chem. A 2019, 7, 14552–14558. [Google Scholar] [CrossRef]
- Doan, H.; Cheng, F.; Dyirakumunda, T.; Elsegood, M.; Chin, J.; Rowe, O.; Redshaw, C.; Ting, V. Using Supercritical CO2 in the Preparation of Metal-Organic Frameworks: Investigating Effects on Crystallisation. Crystals 2019, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Butler, K.T.; Vervoorts, P.; Ehrenreich, M.G.; Armstrong, J.; Skelton, J.M.; Kieslich, G. Experimental Evidence for Vibrational Entropy as Driving Parameter of Flexibility in the Metal–Organic Framework ZIF-4(Zn). Chem. Mater. 2019, 31, 8366–8372. [Google Scholar] [CrossRef]
- Zhao, P.; Fang, H.; Mukhopadhyay, S.; Li, A.; Rudić, S.; McPherson, I.J.; Tang, C.C.; Fairen-Jimenez, D.; Tsang, S.C.E.; Redfern, S.A.T. Structural Dynamics of a Metal–organic Framework Induced by CO2 Migration in its Non-uniform Porous Structure. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Humby, J.D.; Benson, O.; Smith, G.L.; Argent, S.P.; da Silva, I.; Cheng, Y.; Rudić, S.; Manuel, P.; Frogley, M.D.; Cinque, G.; et al. Host–guest Selectivity in a Series of Isoreticular Metal–organic Frameworks: Observation of Acetylene-to-alkyne and Carbon Dioxide-to-amide Interactions. Chem. Sci. 2019, 10, 1098–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, X.; Lyu, K.; Li, L.; Li, J.; Kimberley, L.; Wang, B.; Liu, L.; Cheng, Y.; Frogley, M.D.; Rudić, S.; et al. Integration of Mesopores and Crystal Defects in Metal-organic Frameworks via Templated Electrosynthesis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Titov, K.; Eremin, D.B.; Kashin, A.S.; Boada, R.; Souza, B.E.; Kelley, C.S.; Frogley, M.D.; Cinque, G.; Gianolio, D.; Cibin, G.; et al. OX-1 Metal–Organic Framework Nanosheets as Robust Hosts for Highly Active Catalytic Palladium Species. ACS Sustain. Chem. Eng. 2019, 7, 5875–5885. [Google Scholar] [CrossRef]
- Smith, G.L.; Eyley, J.E.; Han, X.; Zhang, X.; Li, J.; Jacques, N.M.; Godfrey, H.G.W.; Argent, S.P.; McPherson, L.J.M.; Teat, S.J.; et al. Reversible Coordinative Binding and Separation of Sulfur Dioxide in a Robust Metal–organic Framework with Open Copper Sites. Nat. Mater. 2019, 18, 1358–1365. [Google Scholar] [CrossRef]
- Cuadrado-Collados, C.; Mouchaham, G.; Daemen, L.; Cheng, Y.; Ramirez-Cuesta, A.; Aggarwal, H.; Missyul, A.; Eddaoudi, M.; Belmabkhout, Y.; Silvestre-Albero, J. Quest for an Optimal Methane Hydrate Formation in the Pores of Hydrolytically Stable Metal–Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 13391–13397. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.E.; Möslein, A.F.; Titov, K.; Taylor, J.D.; Rudić, S.; Tan, J.C. Green Reconstruction of MIL-100 (Fe) in Water for High Crystallinity and Enhanced Guest Encapsulation. ACS Sustain. Chem. Eng. 2020, 8, 8247–8255. [Google Scholar] [CrossRef]
- Li, J.; Zhou, Z.; Han, X.; Zhang, X.; Yan, Y.; Li, W.; Smith, G.L.; Cheng, Y.; MPherson, L.J.M.; Teat, S.J.; et al. Guest-Controlled Incommensurate Modulation in a Meta-Rigid Metal–Organic Framework Material. J. Am. Chem. Soc. 2020, 142, 19189–19197. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Lu, W.; Chen, Y.; da Silva, I.; Li, J.; Lin, L.; Li, W.; Sheveleva, A.M.; Godfrey, H.G.W.; Lu, Z.; et al. High Ammonia Adsorption in MFM-300 Materials: Dynamics and Charge Transfer in Host–Guest Binding. J. Am. Chem. Soc. 2021, 143, 3153–3161. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yildirim, T. Lattice Dynamics of Metal-organic Frameworks: Neutron Inelastic Scattering and First-principles Calculations. Phys. Rev. B 2006, 74. [Google Scholar] [CrossRef] [Green Version]
- Forster, P.M.; Eckert, J.; Heiken, B.D.; Parise, J.B.; Yoon, J.W.; Jhung, S.H.; Chang, J.S.; Cheetham, A.K. Adsorption of Molecular Hydrogen on Coordinatively Unsaturated Ni(II) Sites in a Nanoporous Hybrid Material. J. Am. Chem. Soc. 2006, 128, 16846–16850. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Brown, C.; Neumann, D.; Peterson, V.; Kepert, C. Inelastic Neutron Scattering of H2 adsorbed in HKUST-1. J. Alloys Compd. 2007, 446–447, 385–388. [Google Scholar] [CrossRef]
- Liu, Y.; Eubank, J.; Cairns, A.; Eckert, J.; Kravtsov, V.; Luebke, R.; Eddaoudi, M. Assembly of Metal–Organic Frameworks (MOFs) Based on Indium-Trimer Building Blocks: A Porous MOF with soc Topology and High Hydrogen Storage. Angew. Chem. Int. Ed. 2007, 46, 3278–3283. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Her, J.H.; Dailly, A.; Ramirez-Cuesta, A.J.; Neumann, D.A.; Brown, C.M. Reversible Structural Transition in MIL-53 with Large Temperature Hysteresis. J. Am. Chem. Soc. 2008, 130, 11813–11818. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kabbour, H.; Brown, C.M.; Neumann, D.A.; Ahn, C.C. Increasing the Density of Adsorbed Hydrogen with Coordinatively Unsaturated Metal Centers in Metal-Organic Frameworks. Langmuir 2008, 24, 4772–4777. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.S.; Ma, S.; Forster, P.; Yuan, D.; Eckert, J.; López, J.; Murphy, B.; Parise, J.; Zhou, H.C. Enhancing H2Uptake by “Close-Packing” Alignment of Open Copper Sites in Metal-Organic Frameworks. Angew. Chem. Int. Ed. 2008, 47, 7263–7266. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Eckert, J.; Forster, P.M.; Yoon, J.W.; Hwang, Y.K.; Chang, J.S.; Collier, C.D.; Parise, J.B.; Zhou, H.C. Further Investigation of the Effect of Framework Catenation on Hydrogen Uptake in Metal-Organic Frameworks. J. Am. Chem. Soc. 2008, 130, 15896–15902. [Google Scholar] [CrossRef]
- Brown, C.M.; Liu, Y.; Yildirim, T.; Peterson, V.K.; Kepert, C.J. Hydrogen Adsorption in HKUST-1: A Combined Inelastic Neutron Scattering and First-principles Study. Nanotechnology 2009, 20, 204025. [Google Scholar] [CrossRef] [PubMed]
- Nouar, F.; Eckert, J.; Eubank, J.F.; Forster, P.; Eddaoudi, M. Zeolite-likeMetal-Organic Frameworks (ZMOFs) as Hydrogen Storage Platform: Lithium and Magnesium Ion-Exchange and H2-(rho-ZMOF) Interaction Studies. J. Am. Chem. Soc. 2009, 131, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, P.D.C.; Georgiev, P.A.; Eckert, J.; Blom, R.; Strässle, T.; Unruh, T. Interaction of Hydrogen with Accessible Metal Sites in the Metal–organic frameworks M2(dhtp) (CPO-27-M;M = Ni, Co, Mg). Chem. Commun. 2010, 46, 4962. [Google Scholar] [CrossRef]
- Sumida, K.; Her, J.H.; Dincă, M.; Murray, L.J.; Schloss, J.M.; Pierce, C.J.; Thompson, B.A.; FitzGerald, S.A.; Brown, C.M.; Long, J.R. Neutron Scattering and Spectroscopic Studies of Hydrogen Adsorption in Cr3(BTC)2—A Metal-Organic Framework with Exposed Cr2 Sites. J. Phys. Chem. C 2011, 115, 8414–8421. [Google Scholar] [CrossRef]
- Sumida, K.; Brown, C.M.; Herm, Z.R.; Chavan, S.; Bordiga, S.; Long, J.R. Hydrogen Storage Properties and Neutron Scattering Studies of Mg2(dobdc)—A Metal–organic Framework with Open Mg2Adsorption Sites. Chem. Commun. 2011, 47, 1157–1159. [Google Scholar] [CrossRef]
- Tranchemontagne, D.J.; Park, K.S.; Furukawa, H.; Eckert, J.; Knobler, C.B.; Yaghi, O.M. Hydrogen Storage in New Metal–Organic Frameworks. J. Phys. Chem. C 2012, 116, 13143–13151. [Google Scholar] [CrossRef]
- Eubank, J.F.; Nouar, F.; Luebke, R.; Cairns, A.J.; Wojtas, L.; Alkordi, M.; Bousquet, T.; Hight, M.R.; Eckert, J.; Embs, J.P.; et al. On Demand: The Singular rht Net, an Ideal Blueprint for the Construction of a Metal-Organic Framework (MOF) Platform. Angew. Chem. Int. Ed. 2012, 51, 10099–10103. [Google Scholar] [CrossRef]
- Queen, W.L.; Bloch, E.D.; Brown, C.M.; Hudson, M.R.; Mason, J.A.; Murray, L.J.; Ramirez-Cuesta, A.J.; Peterson, V.K.; Long, J.R. Hydrogen Adsorption in the Metal–organic Frameworks Fe2(dobdc) and Fe2(O2)(dobdc). Dalton Trans. 2012, 41, 4180. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, M.B.; Getman, R.B.; Lee, J.Y.; Roberts, J.M.; Sarjeant, A.A.; Scheidt, K.A.; Georgiev, P.A.; Embs, J.P.; Eckert, J.; Farha, O.K.; et al. A Zwitterionic Metal–organic Framework with Free Carboxylic Acid Sites that Exhibits Enhanced Hydrogen Adsorption Energies. CrystEngComm 2013, 15, 9408. [Google Scholar] [CrossRef]
- Callear, S.K.; Ramirez-Cuesta, A.J.; David, W.I.; Millange, F.; Walton, R.I. High-resolution Inelastic Neutron Scattering and Neutron Powder Diffraction Study of the Adsorption of Dihydrogen by the Cu(II) Metal–organic Framework Material HKUST-1. Chem. Phys. 2013, 427, 9–17. [Google Scholar] [CrossRef]
- Munn, A.S.; Ramirez-Cuesta, A.J.; Millange, F.; Walton, R.I. Interaction of Methanol with the Flexible Metal-organic Framework MIL-53(Fe) Observed by Inelastic Neutron Scattering. Chem. Phys. 2013, 427, 30–37. [Google Scholar] [CrossRef]
- Pham, T.; Forrest, K.A.; Eckert, J.; Georgiev, P.A.; Mullen, A.; Luebke, R.; Cairns, A.J.; Belmabkhout, Y.; Eubank, J.F.; McLaughlin, K.; et al. Investigating the Gas Sorption Mechanism in an rht-Metal–Organic Framework through Computational Studies. J. Phys. Chem. C 2013, 118, 439–456. [Google Scholar] [CrossRef]
- Rosnes, M.H.; Opitz, M.; Frontzek, M.; Lohstroh, W.; Embs, J.P.; Georgiev, P.A.; Dietzel, P.D.C. Intriguing Differences in Hydrogen Adsorption in CPO-27 Materials Induced by Metal Substitution. J. Mater. Chem. A 2015, 3, 4827–4839. [Google Scholar] [CrossRef]
- Nugent, P.; Pham, T.; McLaughlin, K.; Georgiev, P.A.; Lohstroh, W.; Embs, J.P.; Zaworotko, M.J.; Space, B.; Eckert, J. Dramatic Effect of Pore Size Reduction on the Dynamics of Hydrogen Adsorbed in Metal–organic Materials. J. Mater. Chem. A 2014, 2, 13884. [Google Scholar] [CrossRef]
- Pham, T.; Forrest, K.A.; Georgiev, P.A.; Lohstroh, W.; Xue, D.X.; Hogan, A.; Eddaoudi, M.; Space, B.; Eckert, J. A High Rotational Barrier for Physisorbed Hydrogen in an fcu-metal–organic Framework. Chem. Commun. 2014, 50, 14109–14112. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Ramirez-Cuesta, A.J.; Schröder, M. Inelastic Neutron Scattering Study of Binding of para-Hydrogen in an Ultra-microporous Metal–organic Framework. Chem. Phys. 2014, 428, 111–116. [Google Scholar] [CrossRef]
- Pham, T.; Forrest, K.A.; Falcão, E.H.L.; Eckert, J.; Space, B. Exceptional H2 Sorption Characteristics in a Mg2-based Metal–organic Framework With Small Pores: Insights from Experimental and Theoretical Studies. Phys. Chem. Chem. Phys. 2016, 18, 1786–1796. [Google Scholar] [CrossRef]
- Forrest, K.A.; Pham, T.; Georgiev, P.A.; Pinzan, F.; Cioce, C.R.; Unruh, T.; Eckert, J.; Space, B. Investigating H2 Sorption in a Fluorinated Metal–Organic Framework with Small Pores Through Molecular Simulation and Inelastic Neutron Scattering. Langmuir 2015, 31, 7328–7336. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Forrest, K.A.; Banerjee, R.; Orcajo, G.; Eckert, J.; Space, B. Understanding the H2Sorption Trends in the M-MOF-74 Series (M = Mg, Ni, Co, Zn). J. Phys. Chem. C 2014, 119, 1078–1090. [Google Scholar] [CrossRef]
- Cairns, A.J.; Eckert, J.; Wojtas, L.; Thommes, M.; Wallacher, D.; Georgiev, P.A.; Forster, P.M.; Belmabkhout, Y.; Ollivier, J.; Eddaoudi, M. Gaining Insights on the H2–Sorbent Interactions: Robust soc-MOF Platform as a Case Study. Chem. Mater. 2016, 28, 7353–7361. [Google Scholar] [CrossRef]
- Rosen, P.F.; Calvin, J.J.; Dickson, M.S.; Katsenis, A.D.; Friščić, T.; Navrotsky, A.; Ross, N.L.; Kolesnikov, A.I.; Woodfield, B.F. Heat Capacity and Thermodynamic Functions of Crystalline Forms of the Metal-organic Framework Zinc 2-Methylimidazolate, Zn(MeIm)2. J. Chem. Thermodyn. 2019, 136, 160–169. [Google Scholar] [CrossRef]
- Vanpoucke, D.E.P.; Lejaeghere, K.; Speybroeck, V.V.; Waroquier, M.; Ghysels, A. Mechanical Properties from Periodic Plane Wave Quantum Mechanical Codes: The Challenge of the Flexible Nanoporous MIL-47(V) Framework. J. Phys. Chem. C 2015, 119, 23752–23766. [Google Scholar] [CrossRef] [Green Version]
- Salzmann, C.G.; Radaelli, P.G.; Mayer, E.; Finney, J.L. Ice XV: A New Thermodynamically Stable Phase of Ice. Phys. Rev. Lett. 2009, 103. [Google Scholar] [CrossRef] [Green Version]
- Gasser, T.M.; Thoeny, A.V.; Fortes, A.D.; Loerting, T. Structural Characterization of Ice XIX as the Second Polymorph Related to Ice VI. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef]
- Salzmann, C.G. Advances in the Experimental Exploration of Water’s Phase Diagram. J. Chem. Phys. 2019, 150, 060901. [Google Scholar] [CrossRef] [PubMed]
- Wollan, E.O.; Davidson, W.L.; Shull, C.G. Neutron Diffraction Study of the Structure of Ice. Phys. Rev. 1949, 75, 1348–1352. [Google Scholar] [CrossRef]
- Rahman, A.; Stillinger, F.H. Molecular Dynamics Study of Liquid Water. J. Chem. Phys. 1971, 55, 3336–3359. [Google Scholar] [CrossRef]
- Soper, A. The Radial Distribution Functions of Water and Ice from 220 to 673 K and at Pressures up to 400 MPa. Chem. Phys. 2000, 258, 121–137. [Google Scholar] [CrossRef]
- Soper, A.; Phillips, M. A New Determination of the Structure of Water at 25 °C. Chem. Phys. 1986, 107, 47–60. [Google Scholar] [CrossRef]
- Rosu-Finsen, A.; Amon, A.; Armstrong, J.; Fernandez-Alonso, F.; Salzmann, C.G. Deep-Glassy Ice VI Revealed with a Combination of Neutron Spectroscopy and Diffraction. J. Phys. Chem. Lett. 2020, 11, 1106–1111. [Google Scholar] [CrossRef] [Green Version]
- Li, J. Inelastic Neutron Scattering Studies of Hydrogen Bonding in Ices. J. Chem. Phys. 1996, 105, 6733–6755. [Google Scholar] [CrossRef]
- Li, J.; Kolesnikov, A. Neutron Spectroscopic Investigation of Dynamics of Water Ice. J. Mol. Liq. 2002, 100, 1–39. [Google Scholar] [CrossRef]
- Klotz, S.; Strässle, T.; Salzmann, C.G.; Philippe, J.; Parker, S.F. Incoherent Inelastic Neutron Scattering Measurements on Ice VII: Are There Two Kinds of Hydrogen Bonds in Ice? EPL 2005, 72, 576–582. [Google Scholar] [CrossRef]
- Andreani, C.; Romanelli, G.; Parmentier, A.; Senesi, R.; Kolesnikov, A.I.; Ko, H.Y.; Andrade, M.F.C.; Car, R. Hydrogen Dynamics in Supercritical Water Probed by Neutron Scattering and Computer Simulations. J. Phys. Chem. Lett. 2020, 11, 9461–9467. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, A.I.; Reiter, G.F.; Choudhury, N.; Prisk, T.R.; Mamontov, E.; Podlesnyak, A.; Ehlers, G.; Seel, A.G.; Wesolowski, D.J.; Anovitz, L.M. Quantum Tunneling of Water in Beryl: A New State of the Water Molecule. Phys. Rev. Lett. 2016, 116. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, A.I.; Reiter, G.F.; Prisk, T.R.; Krzystyniak, M.; Romanelli, G.; Wesolowski, D.J.; Anovitz, L.M. Inelastic and Deep Inelastic Neutron Spectroscopy of Water Molecules under Ultra-confinement. J. Phys. Conf. Ser. 2018, 1055, 012002. [Google Scholar] [CrossRef]
- Komatsu, K.; Noritake, F.; Machida, S.; Sano-Furukawa, A.; Hattori, T.; Yamane, R.; Kagi, H. Partially Ordered State of Ice XV. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Loerting, T. Order and Disorder—A Story about Ice. Available online: https://chemistrycommunity.nature.com/posts/order-and-disorder-a-story-about-ice (accessed on 28 April 2021).
- Gasser, T.M.; Thoeny, A.V.; Plaga, L.J.; Köster, K.W.; Etter, M.; Böhmer, R.; Loerting, T. Experiments Indicating a Second Hydrogen Ordered Phase of Ice VI. Chem. Sci. 2018, 9, 4224–4234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosu-Finsen, A.; Salzmann, C.G. Origin of the Low-temperature Endotherm of Acid-doped Ice VI: New Hydrogen-ordered Phase of Ice or Deep Glassy States? Chem. Sci. 2019, 10, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, G.; Pan, D. A Closer Look at Supercritical Water. Proc. Natl. Acad. Sci. USA 2013, 110, 6250–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakaboylu, O.; Harinck, J.; Smit, K.; de Jong, W. Supercritical Water Gasification of Biomass: A Literature and Technology Overview. Energies 2015, 8, 859–894. [Google Scholar] [CrossRef]
- Oka, Y.; Koshizuka, S. Supercritical-pressure, Once-through Cycle Light Water Cooled Reactor Concept. J. Nucl. Sci. Technol. 2001, 38, 1081–1089. [Google Scholar] [CrossRef]
- Deguchi, S.; Tsujii, K. Supercritical Water: A Fascinating Medium for Soft Matter. Soft Matter 2007, 3, 797. [Google Scholar] [CrossRef]
- Boero, M.; Terakura, K.; Ikeshoji, T.; Liew, C.C.; Parrinello, M. Water at Supercritical Conditions: A First Principles Study. J. Chem. Phys. 2001, 115, 2219–2227. [Google Scholar] [CrossRef]
- Mayers, J.; Andreani, C.; Baciocco, G. Initial State Effects in Deep Inelastic Neutron Scattering. Phys. Rev. B 1989, 39, 2022–2028. [Google Scholar] [CrossRef]
- Andreani, C.; Colognesi, D.; Degiorgi, E.; Ricci, M.A. Proton Dynamics in Supercritical Water. J. Chem. Phys. 2001, 115, 11243–11248. [Google Scholar] [CrossRef] [Green Version]
- Blostein, J.; Dawidowski, J.; Granada, J. On the Analysis of Deep Inelastic Neutron Scattering Experiments for Light Nuclei. Phys. B Condens. Matter 2001, 304, 357–367. [Google Scholar] [CrossRef]
- Andreani, C.; Colognesi, D.; Mayers, J.; Reiter, G.F.; Senesi, R. Measurement of Momentum Distribution of Light Atoms and Molecules in Condensed Matter Systems Using Inelastic Neutron Scattering. Adv. Phys. 2005, 54, 377–469. [Google Scholar] [CrossRef]
- Pantalei, C.; Pietropaolo, A.; Senesi, R.; Imberti, S.; Andreani, C.; Mayers, J.; Burnham, C.; Reiter, G. Proton Momentum Distribution of Liquid Water from Room Temperature to the Supercritical Phase. Phys. Rev. Lett. 2008, 100. [Google Scholar] [CrossRef] [Green Version]
- Andreani, C.; Senesi, R.; Krzystyniak, M.; Romanelli, G.; Fernandez-Alonso, F. Atomic Quantum Dynamics in Materials Research. In Neutron Scattering—Applications in Biology, Chemistry, and Materials Science; Elsevier: Amsterdam, The Netherlands, 2017; pp. 403–457. [Google Scholar] [CrossRef]
- Stern, J.; Loerting, T. Crystallisation of the Amorphous Ices in the Intermediate Pressure Regime. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Han, J.; Wang, H.; Car, R.; E, W. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics. Phys. Rev. Lett. 2018, 120. [Google Scholar] [CrossRef] [Green Version]
- Behler, J.; Parrinello, M. Generalized Neural-Network Representation of High-Dimensional Potential-Energy Surfaces. Phys. Rev. Lett. 2007, 98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, M.; Wu, X.; Wang, H.; Weinan, E.; Car, R. Deep Neural Network for the Dielectric Response of Insulators. Phys. Rev. B 2020, 102. [Google Scholar] [CrossRef]
- Sommers, G.M.; Andrade, M.F.C.; Zhang, L.; Wang, H.; Car, R. Raman Spectrum and Polarizability of Liquid Water from Deep Neural Networks. Phys. Chem. Chem. Phys. 2020, 22, 10592–10602. [Google Scholar] [CrossRef]
- Schütt, K.T.; Gastegger, M.; Tkatchenko, A.; Müller, K.R.; Maurer, R.J. Unifying Machine Learning and Quantum Chemistry with a Deep Neural Network for Molecular Wavefunctions. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Schran, C.; Behler, J.; Marx, D. Automated Fitting of Neural Network Potentials at Coupled Cluster Accuracy: Protonated Water Clusters as Testing Ground. J. Chem. Theory Comput. 2019, 16, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmiela, S.; Sauceda, H.E.; Müller, K.R.; Tkatchenko, A. Towards Exact Molecular Dynamics Simulations with Machine-learned Force Fields. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Remsing, R.C.; Zhang, Y.; Sun, Z.; Ruzsinszky, A.; Peng, H.; Yang, Z.; Paul, A.; Waghmare, U.; Wu, X.; et al. Accurate First-principles Structures and Energies of Diversely Bonded Systems from an Efficient Density Functional. Nat. Chem. 2016, 8, 831–836. [Google Scholar] [CrossRef]
- Car, R. Fixing Jacob’s Ladder. Nat. Chem. 2016, 8, 820–821. [Google Scholar] [CrossRef]
- Gill, P.M.W. Obituary: Density Functional Theory (1927–1993). Aust. J. Chem. 2001, 54, 661. [Google Scholar] [CrossRef]
- Chen, M.; Ko, H.Y.; Remsing, R.C.; Andrade, M.F.C.; Santra, B.; Sun, Z.; Selloni, A.; Car, R.; Klein, M.L.; Perdew, J.P.; et al. Ab initio Theory and Modeling of Water. Proc. Natl. Acad. Sci. USA 2017, 114, 10846–10851. [Google Scholar] [CrossRef] [Green Version]
- LaCount, M.D.; Gygi, F. Ensemble First-principles Molecular Dynamics Simulations of Water Using the SCAN meta-GGA Density Functional. J. Chem. Phys. 2019, 151, 164101. [Google Scholar] [CrossRef] [PubMed]
- Pestana, L.R.; Mardirossian, N.; Head-Gordon, M.; Head-Gordon, T. ab initio Molecular Dynamics Simulations of Liquid Water using High Quality meta-GGA Functionals. Chem. Sci. 2017, 8, 3554–3565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.; Ciardi, G.; Sidler, D.; Hamm, P.; Shalit, A. Impact of Nuclear Quantum Effects on the Structural Inhomogeneity of Liquid Water. Proc. Natl. Acad. Sci. USA 2019, 116, 2458–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceriotti, M.; Fang, W.; Kusalik, P.G.; McKenzie, R.H.; Michaelides, A.; Morales, M.A.; Markland, T.E. Nuclear Quantum Effects in Water and Aqueous Systems: Experiment, Theory, and Current Challenges. Chem. Rev. 2016, 116, 7529–7550. [Google Scholar] [CrossRef] [PubMed]
- Novikov, V.N.; Sokolov, A.P. Quantum Effects in Dynamics of Water and Other Liquids of Light Molecules. Eur. Phys. J. E 2017, 40. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, D.M.; Manolopoulos, D.E.; Pipolo, S.; Laage, D.; Hynes, J.T. Nuclear Quantum Effects in Water Reorientation and Hydrogen-Bond Dynamics. J. Phys. Chem. Lett. 2017, 8, 2602–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauceda, H.E.; Vassilev-Galindo, V.; Chmiela, S.; Müller, K.R.; Tkatchenko, A. Dynamical Strengthening of Covalent and Non-covalent Molecular Interactions by Nuclear Quantum Effects at Finite Temperature. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Markland, T.E.; Ceriotti, M. Nuclear Quantum Effects Enter the Mainstream. Nat. Rev. Chem. 2018, 2. [Google Scholar] [CrossRef] [Green Version]
- Balan, E.; Lazzeri, M.; Delattre, S.; Méheut, M.; Refson, K.; Winkler, B. Anharmonicity of Inner-OH Stretching Modes in Hydrous Phyllosilicates: Assessment from First-principles Frozen-phonon Calculations. Phys. Chem. Miner. 2007, 34, 621–625. [Google Scholar] [CrossRef]
- Lin, C.Y.; George, M.W.; Gill, P.M.W. EDF2: A Density Functional for Predicting Molecular Vibrational Frequencies. Aust. J. Chem. 2004, 57, 365. [Google Scholar] [CrossRef] [Green Version]
- Zhong, K.; Yu, C.C.; Dodia, M.; Bonn, M.; Nagata, Y.; Ohto, T. Vibrational Mode Frequency Correction of Liquid Water in Density Functional Theory Molecular Dynamics Simulations With van der Waals Correction. Phys. Chem. Chem. Phys. 2020, 22, 12785–12793. [Google Scholar] [CrossRef]
- Pestana, L.R.; Marsalek, O.; Markland, T.E.; Head-Gordon, T. The Quest for Accurate Liquid Water Properties from First Principles. J. Phys. Chem. Lett. 2018, 9, 5009–5016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsalek, O.; Markland, T.E. Quantum Dynamics and Spectroscopy of ab initio Liquid Water: The Interplay of Nuclear and Electronic Quantum Effects. J. Phys. Chem. Lett. 2017, 8, 1545–1551. [Google Scholar] [CrossRef] [Green Version]
- Elton, D.C.; Fernández-Serra, M. The Hydrogen-bond Network of Water Supports Propagating Optical Phonon-like Modes. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Rekik, N.; Suleiman, J.; Blaise, P.; Wojcik, M.J.; Flakus, H.T.; Nakajima, T. Electrical Anharmonicity in Hydrogen Bonded Systems: Complete Interpretation of the IR spectra of the Cl–H Stretching Band in the Gaseous (CH3)2OHCl complex. Phys. Chem. Chem. Phys. 2017, 19, 5917–5931. [Google Scholar] [CrossRef]
- Ivanov, S.D.; Witt, A.; Shiga, M.; Marx, D. Communications: On Artificial Frequency Shifts in Infrared Spectra Obtained from Centroid Molecular Dynamics: Quantum Liquid Water. J. Chem. Phys. 2010, 132, 031101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenins, G.; Willatt, M.J.; Althorpe, S.C. Path-integral Dynamics of Water Using Curvilinear Centroids. J. Chem. Phys. 2019, 151, 054109. [Google Scholar] [CrossRef] [Green Version]
- Habershon, S.; Manolopoulos, D.E. Zero Point Energy Leakage in Condensed Phase Dynamics: An Assessment of Quantum Simulation Methods for Liquid Water. J. Chem. Phys. 2009, 131, 244518. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Ceriotti, M.; Manolopoulos, D.E. How to Remove the Spurious Resonances from Ring Polymer Molecular Dynamics. J. Chem. Phys. 2014, 140, 234116. [Google Scholar] [CrossRef] [Green Version]
- Benson, R.L.; Trenins, G.; Althorpe, S.C. Which Quantum Statistics–classical Dynamics Method is Best for Water? Faraday Discuss. 2020, 221, 350–366. [Google Scholar] [CrossRef]
- Lin, L.; Morrone, J.A.; Car, R.; Parrinello, M. Displaced Path Integral Formulation for the Momentum Distribution of Quantum Particles. Phys. Rev. Lett. 2010, 105. [Google Scholar] [CrossRef] [Green Version]
- Morrone, J.A.; Srinivasan, V.; Sebastiani, D.; Car, R. Proton Momentum Distribution in Water: An Open Path Integral Molecular Dynamics Study. J. Chem. Phys. 2007, 126, 234504. [Google Scholar] [CrossRef]
- Poltavsky, I.; Tkatchenko, A. Modeling Quantum Nuclei with Perturbed Path Integral Molecular Dynamics. Chem. Sci. 2016, 7, 1368–1372. [Google Scholar] [CrossRef] [Green Version]
- Poltavsky, I.; Kapil, V.; Ceriotti, M.; Kim, K.S.; Tkatchenko, A. Accurate Description of Nuclear Quantum Effects with High-Order Perturbed Path Integrals (HOPPI). J. Chem. Theory Comput. 2020, 16, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Leal, L.; Kolesnikov, A.I. Analysis of the Time-of-Flight Neutron Scattering Cross-section Data for Light Water Measured at the SEQUOIA Spectrometer, Spallation Neutron Source (SNS). EPJ Web Conf. 2020, 239, 14007. [Google Scholar] [CrossRef]
- Li, X.Z.; Walker, B.; Michaelides, A. Quantum Nature of the Hydrogen Bond. Proc. Natl. Acad. Sci. USA 2011, 108, 6369–6373. [Google Scholar] [CrossRef] [Green Version]
- Senesi, R.; Flammini, D.; Kolesnikov, A.I.; Murray, É.D.; Galli, G.; Andreani, C. The Quantum Nature of the OH Stretching Mode in Ice and Water Probed by Neutron Scattering Experiments. J. Chem. Phys. 2013, 139, 074504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmentier, A.; Shephard, J.J.; Romanelli, G.; Senesi, R.; Salzmann, C.G.; Andreani, C. Evolution of Hydrogen Dynamics in Amorphous Ice with Density. J. Phys. Chem. Lett. 2015, 6, 2038–2042. [Google Scholar] [CrossRef]
- Kapil, V.; Cuzzocrea, A.; Ceriotti, M. Anisotropy of the Proton Momentum Distribution in Water. J. Phys. Chem. B 2018, 122, 6048–6054. [Google Scholar] [CrossRef] [Green Version]
- Ceriotti, M.; Parrinello, M.; Markland, T.E.; Manolopoulos, D.E. Efficient Stochastic Thermostatting of Path Integral Molecular Dynamics. J. Chem. Phys. 2010, 133, 124104. [Google Scholar] [CrossRef] [Green Version]
- Ceriotti, M.; Bussi, G.; Parrinello, M. Colored-Noise Thermostats à la Carte. J. Chem. Theory Comput. 2010, 6, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Ceriotti, M.; Manolopoulos, D.E.; Parrinello, M. Accelerating the Convergence of Path Integral Dynamics with a Generalized Langevin Equation. J. Chem. Phys. 2011, 134, 084104. [Google Scholar] [CrossRef]
- Marsalek, O.; Markland, T.E. ab initio Molecular Dynamics with Nuclear Quantum Effects at Classical Cost: Ring Polymer Contraction for Density Functional Theory. J. Chem. Phys. 2016, 144, 054112. [Google Scholar] [CrossRef] [Green Version]
- Kapil, V.; Rossi, M.; Marsalek, O.; Petraglia, R.; Litman, Y.; Spura, T.; Cheng, B.; Cuzzocrea, A.; Meißner, R.H.; Wilkins, D.M.; et al. i-PI 2.0: A universal force engine for advanced molecular simulations. Comput. Phys. Commun. 2019, 236, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Ceriotti, M.; More, J.; Manolopoulos, D.E. i-PI: A Python Interface for ab initio Path Integral Molecular Dynamics Simulations. Comput. Phys. Commun. 2014, 185, 1019–1026. [Google Scholar] [CrossRef] [Green Version]
- Romanelli, G.; Ceriotti, M.; Manolopoulos, D.E.; Pantalei, C.; Senesi, R.; Andreani, C. Direct Measurement of Competing Quantum Effects on the Kinetic Energy of Heavy Water upon Melting. J. Phys. Chem. Lett. 2013, 4, 3251–3256. [Google Scholar] [CrossRef] [Green Version]
- Morrone, J.A.; Lin, L.; Car, R. Tunneling and Delocalization Effects in Hydrogen Bonded Systems: A Study in Position and Momentum space. J. Chem. Phys. 2009, 130, 204511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammini, D.; Pietropaolo, A.; Senesi, R.; Andreani, C.; McBride, F.; Hodgson, A.; Adams, M.A.; Lin, L.; Car, R. Spherical Momentum Distribution of the Protons in Hexagonal Ice from Modeling of Inelastic Neutron Scattering Data. J. Chem. Phys. 2012, 136, 024504. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Ceriotti, M.; Manolopoulos, D.E. Nuclear Quantum Effects in H and OH– Diffusion along Confined Water Wires. J. Phys. Chem. Lett. 2016, 7, 3001–3007. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Behler, J.; Ceriotti, M. Nuclear Quantum Effects in Water at the Triple Point: Using Theory as a Link Between Experiments. J. Phys. Chem. Lett. 2016, 7, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Gaiduk, A.P.; Gygi, F.; Galli, G. Density and Compressibility of Liquid Water and Ice from First-Principles Simulations with Hybrid Functionals. J. Phys. Chem. Lett. 2015, 6, 2902–2908. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Engel, E.A.; Behler, J.; Dellago, C.; Ceriotti, M. ab initio Thermodynamics of Liquid and Solid Water. Proc. Natl. Acad. Sci. USA 2019, 116, 1110–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, H.; Car, R.; Weinan, E. The Phase Diagram of a Deep Potential Water Model. arXiv 2020, arXiv:2102.04804. [Google Scholar]
- Reinhardt, A.; Cheng, B. Quantum-mechanical Exploration of the Phase Diagram of Water. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef]
- Bokdam, M.; Lahnsteiner, J.; Ramberger, B.; Schäfer, T.; Kresse, G. Assessing Density Functionals Using Many Body Theory for Hybrid Perovskites. Phys. Rev. Lett. 2017, 119. [Google Scholar] [CrossRef] [PubMed]
- Lahnsteiner, J.; Kresse, G.; Heinen, J.; Bokdam, M. Finite-temperature Structure of the MAPbI3 perovskite: Comparing Density Functional Approximations and Force Fields to Experiment. Phys. Rev. Mater. 2018, 2. [Google Scholar] [CrossRef] [Green Version]
- Jinnouchi, R.; Lahnsteiner, J.; Karsai, F.; Kresse, G.; Bokdam, M. Phase Transitions of Hybrid Perovskites Simulated by Machine-Learning Force Fields Trained on the Fly with Bayesian Inference. Phys. Rev. Lett. 2019, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapil, V.; Wieme, J.; Vandenbrande, S.; Lamaire, A.; Speybroeck, V.V.; Ceriotti, M. Modeling the Structural and Thermal Properties of Loaded Metal–Organic Frameworks. An Interplay of Quantum and Anharmonic Fluctuations. J. Chem. Theory Comput. 2019, 15, 3237–3249. [Google Scholar] [CrossRef] [Green Version]
- Melillo, J.H.; Cerveny, S. Isotope Effect on the Dynamics of Hydrophilic Solutions at Supercooled Temperatures. In ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2021; pp. 263–281. [Google Scholar] [CrossRef]
- Markland, T.E.; Morrone, J.A.; Berne, B.J.; Miyazaki, K.; Rabani, E.; Reichman, D.R. Quantum fluctuations can promote or inhibit glass formation. Nat. Phys. 2011, 7, 134–137. [Google Scholar] [CrossRef] [Green Version]
- Gainaru, C.; Agapov, A.L.; Fuentes-Landete, V.; Amann-Winkel, K.; Nelson, H.; Köster, K.W.; Kolesnikov, A.I.; Novikov, V.N.; Richert, R.; Böhmer, R.; et al. Anomalously Large Isotope Effect in the Glass Transition of Water. Proc. Natl. Acad. Sci. USA 2014, 111, 17402–17407. [Google Scholar] [CrossRef] [Green Version]
- Shulumba, N.; Hellman, O.; Minnich, A.J. Lattice Thermal Conductivity of Polyethylene Molecular Crystals from First-Principles Including Nuclear Quantum Effects. Phys. Rev. Lett. 2017, 119. [Google Scholar] [CrossRef] [Green Version]
- Jakowski, J.; Huang, J.; Garashchuk, S.; Luo, Y.; Hong, K.; Keum, J.; Sumpter, B.G. Deuteration as a Means to Tune Crystallinity of Conducting Polymers. J. Phys. Chem. Lett. 2017, 8, 4333–4340. [Google Scholar] [CrossRef]
- Chang, D.; Li, T.; Li, L.; Jakowski, J.; Huang, J.; Keum, J.K.; Lee, B.; Bonnesen, P.V.; Zhou, M.; Garashchuk, S.; et al. Selectively Deuterated Poly(ϵ-caprolactone)s: Synthesis and Isotope Effects on the Crystal Structures and Properties. Macromolecules 2018, 51, 9393–9404. [Google Scholar] [CrossRef]
- Cai, W.; Dunuwille, M.; He, J.; Taylor, T.V.; Hinton, J.K.; MacLean, M.C.; Molaison, J.J.; dos Santos, A.M.; Sinogeikin, S.; Deemyad, S. Deuterium Isotope Effects in Polymerization of Benzene under Pressure. J. Phys. Chem. Lett. 2017, 8, 1856–1864. [Google Scholar] [CrossRef]
Figure 1.
Regions of Q and E simultaneously accessible to neutrons, and qualitative comparison with other experimental probes. For ease of comparison, associated length and time scales are given by the bottom and right axes, respectively. Neutron spectroscopic techniques include the use of epithermal (hot neutrons), thermal (time-of-flight and crystal spectroscopy), and cold/ultracold wavelengths (backscattering and NSE). Blue text entries around the main figure illustrate areas of scientific and technological application. Reprinted with permission from Ref. [63].
Figure 1.
Regions of Q and E simultaneously accessible to neutrons, and qualitative comparison with other experimental probes. For ease of comparison, associated length and time scales are given by the bottom and right axes, respectively. Neutron spectroscopic techniques include the use of epithermal (hot neutrons), thermal (time-of-flight and crystal spectroscopy), and cold/ultracold wavelengths (backscattering and NSE). Blue text entries around the main figure illustrate areas of scientific and technological application. Reprinted with permission from Ref. [63].

Figure 2.
Color map of the elastic properties of several classes of materials around ambient conditions, including those discussed in the present and subsequent sections of this work. Please note the logarithmic scales of both vertical and horizontal axes. Adapted from Ref. [74] with permission from The Royal Society of Chemistry.
Figure 2.
Color map of the elastic properties of several classes of materials around ambient conditions, including those discussed in the present and subsequent sections of this work. Please note the logarithmic scales of both vertical and horizontal axes. Adapted from Ref. [74] with permission from The Royal Society of Chemistry.

Figure 3.
Phase diagram of solid solutions over the temperature range 10–365 K at ambient pressure. Pure MAPbI(FAPbI) corrresponds to the left (right) edges of the diagram. For further details, see the main text. Adapted with permission from Refs. [75,81]. Copyright (2021) American Chemical Society.
Figure 3.
Phase diagram of solid solutions over the temperature range 10–365 K at ambient pressure. Pure MAPbI(FAPbI) corrresponds to the left (right) edges of the diagram. For further details, see the main text. Adapted with permission from Refs. [75,81]. Copyright (2021) American Chemical Society.

Figure 4.
Phonon dispersion relations in the cubic phase of MAPbI3, measured on both perdeuterated and hydrogenous single-crystals using IXS (cyan circles [123]) and INS-TAS (orange squares [109] and green circles [113], respectively). Wave vectors on the abscissa are given in reduced lattice units. Longitudinal and Transverse Acoustic (LA and TA, respectively) branches are presented along with the lowest-energy Transverse Optical (TO) mode. Adapted with permission from Ref. [109].
Figure 4.
Phonon dispersion relations in the cubic phase of MAPbI3, measured on both perdeuterated and hydrogenous single-crystals using IXS (cyan circles [123]) and INS-TAS (orange squares [109] and green circles [113], respectively). Wave vectors on the abscissa are given in reduced lattice units. Longitudinal and Transverse Acoustic (LA and TA, respectively) branches are presented along with the lowest-energy Transverse Optical (TO) mode. Adapted with permission from Ref. [109].

Figure 5.
INS spectra of hydrogenous MAPbI3 in the orthorhombic phase at low temperatures (T = 5–20 K). These spectra were recorded on five different instruments: 1T (TAS; single-crystal [116]); AMATERAS (TOF; Ei = 54 meV; single-crystal [121]; TOSCA [144], VISION [109], TOSCA+ [75] (indirect-geometry; powders). Adapted with permission from Refs. [75,109,116,121,144]. Copyright (2021) American Chemical Society.
Figure 5.
INS spectra of hydrogenous MAPbI3 in the orthorhombic phase at low temperatures (T = 5–20 K). These spectra were recorded on five different instruments: 1T (TAS; single-crystal [116]); AMATERAS (TOF; Ei = 54 meV; single-crystal [121]; TOSCA [144], VISION [109], TOSCA+ [75] (indirect-geometry; powders). Adapted with permission from Refs. [75,109,116,121,144]. Copyright (2021) American Chemical Society.
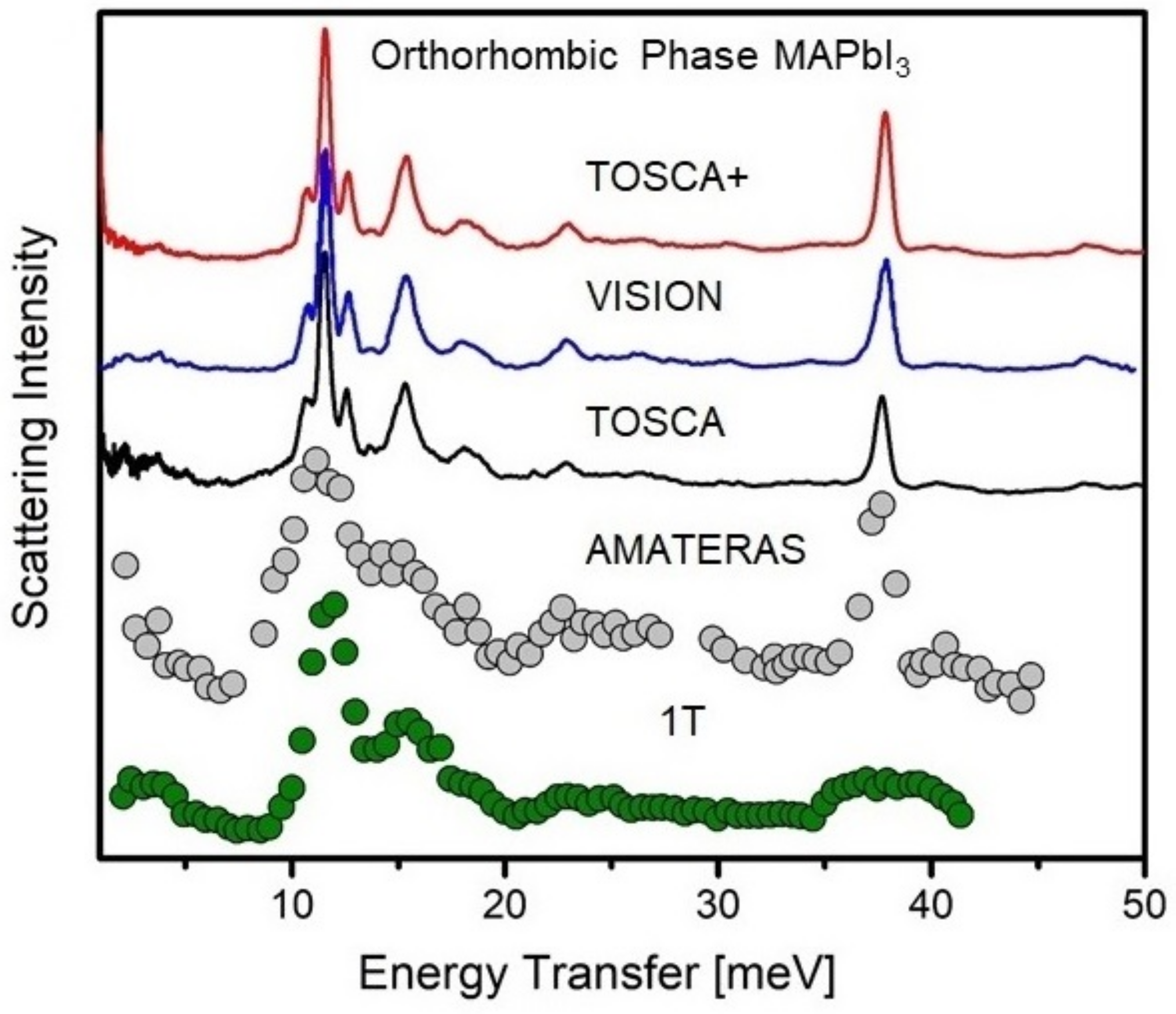
Figure 6.
(a) The lower two panels show experimental INS spectra of the MA0.4FA0.6PbI solid-solution recorded on TOSCA+ at T = 10 K, along with the difference spectrum obtained by substracting the signal from FAPbI, highlighting contributions from the MA+ cations [75]. These data are compared with the results of AIMD simulations in the upper two panels of (a). The theoretical INS spectrum of the MA0.4FA0.6 mixed-cation composition was constructed by stoichiometric weighting of the partial hydrogen-projected VDOS. MA+ cations were selected based on the analysis of the individual VDOSs for each of the twelve distinct types used in the structural model (see the bold curves in the top panel). The molecular model in (a) illustrates the lowest-energy internal mode of MA+. (b) Structural model used to describe the mixture, with FA+ omitted to ease visualization. Distinct MA+ cations are labeled with Roman numerals. (c) Distribution of closest nitrogen-iodine distances for each of the twelve MA+ cations. Two extreme cases are highlighted as bold curves (labeled as II and XII). Their time-evolved structures over a 30-ps interval are presented as cumulative models, as shown in the insets in (c). The same color coding has been used in all panels. [Unpublished data].
Figure 6.
(a) The lower two panels show experimental INS spectra of the MA0.4FA0.6PbI solid-solution recorded on TOSCA+ at T = 10 K, along with the difference spectrum obtained by substracting the signal from FAPbI, highlighting contributions from the MA+ cations [75]. These data are compared with the results of AIMD simulations in the upper two panels of (a). The theoretical INS spectrum of the MA0.4FA0.6 mixed-cation composition was constructed by stoichiometric weighting of the partial hydrogen-projected VDOS. MA+ cations were selected based on the analysis of the individual VDOSs for each of the twelve distinct types used in the structural model (see the bold curves in the top panel). The molecular model in (a) illustrates the lowest-energy internal mode of MA+. (b) Structural model used to describe the mixture, with FA+ omitted to ease visualization. Distinct MA+ cations are labeled with Roman numerals. (c) Distribution of closest nitrogen-iodine distances for each of the twelve MA+ cations. Two extreme cases are highlighted as bold curves (labeled as II and XII). Their time-evolved structures over a 30-ps interval are presented as cumulative models, as shown in the insets in (c). The same color coding has been used in all panels. [Unpublished data].

Figure 7.
INS spectra of bulk (red) and confined (green) PEO. The black trace corresponds to the INS response of the graphite oxide substrate. Adapted from Ref. [202] with permission from The Royal Society of Chemistry.
Figure 7.
INS spectra of bulk (red) and confined (green) PEO. The black trace corresponds to the INS response of the graphite oxide substrate. Adapted from Ref. [202] with permission from The Royal Society of Chemistry.

Figure 8.
(a) Structure of Li4C60 obtained from DFT (VASP) geometry optimization within space group I2/m. Gray sticks are carbon bonds; magenta and brown spheres are carbon atoms involved in covalent intermolecular bonds, including [2 + 2] bridges and single bonds. Red and yellow spheres represent two different types of intercalated ions, LiT and LiO, respectively. (b) View along the c-axis of one polymeric plane (the Li ions have been omitted for clarity). (c) (Top) Generalized Density of States (GDOS) derived from INS data collected between 300 K (polymer phase) and 700 K (monomer phase): red, 300 K; yellow, 610 K; brown, 630 K; gray, 640 K; and black, 700 K). (Bottom) GDOS extracted from the MD simulations at 800 K (see text) in the monomer phase (black solid lines, total GDOS; gray solid line, carbon GDOS; red area, lithium GDOS). The INS spectra were collected on Mibemol (LLB, Saclay, FR). Reproduced from Ref. [246] with permission from APS (https://doi.org/10.1103/PhysRevLett.113.215502) (accessed on 28 April 2021).
Figure 8.
(a) Structure of Li4C60 obtained from DFT (VASP) geometry optimization within space group I2/m. Gray sticks are carbon bonds; magenta and brown spheres are carbon atoms involved in covalent intermolecular bonds, including [2 + 2] bridges and single bonds. Red and yellow spheres represent two different types of intercalated ions, LiT and LiO, respectively. (b) View along the c-axis of one polymeric plane (the Li ions have been omitted for clarity). (c) (Top) Generalized Density of States (GDOS) derived from INS data collected between 300 K (polymer phase) and 700 K (monomer phase): red, 300 K; yellow, 610 K; brown, 630 K; gray, 640 K; and black, 700 K). (Bottom) GDOS extracted from the MD simulations at 800 K (see text) in the monomer phase (black solid lines, total GDOS; gray solid line, carbon GDOS; red area, lithium GDOS). The INS spectra were collected on Mibemol (LLB, Saclay, FR). Reproduced from Ref. [246] with permission from APS (https://doi.org/10.1103/PhysRevLett.113.215502) (accessed on 28 April 2021).

Figure 9.
(a) Nanoporous hybrid framework structures of ZIF-4, ZIF-7, and ZIF-8. The inorganic building blocks are represented by purple ZnN4 tetrahedra. (b) Experimental (TOSCA T = 10 K) and theoretical (DFT; CRYSTAL14) INS spectra for these compounds. Adapted from Ref. [264] with permission from APS (https://doi.org/10.1103/PhysRevLett.113.215502) (accessed on 28 April 2021).
Figure 9.
(a) Nanoporous hybrid framework structures of ZIF-4, ZIF-7, and ZIF-8. The inorganic building blocks are represented by purple ZnN4 tetrahedra. (b) Experimental (TOSCA T = 10 K) and theoretical (DFT; CRYSTAL14) INS spectra for these compounds. Adapted from Ref. [264] with permission from APS (https://doi.org/10.1103/PhysRevLett.113.215502) (accessed on 28 April 2021).
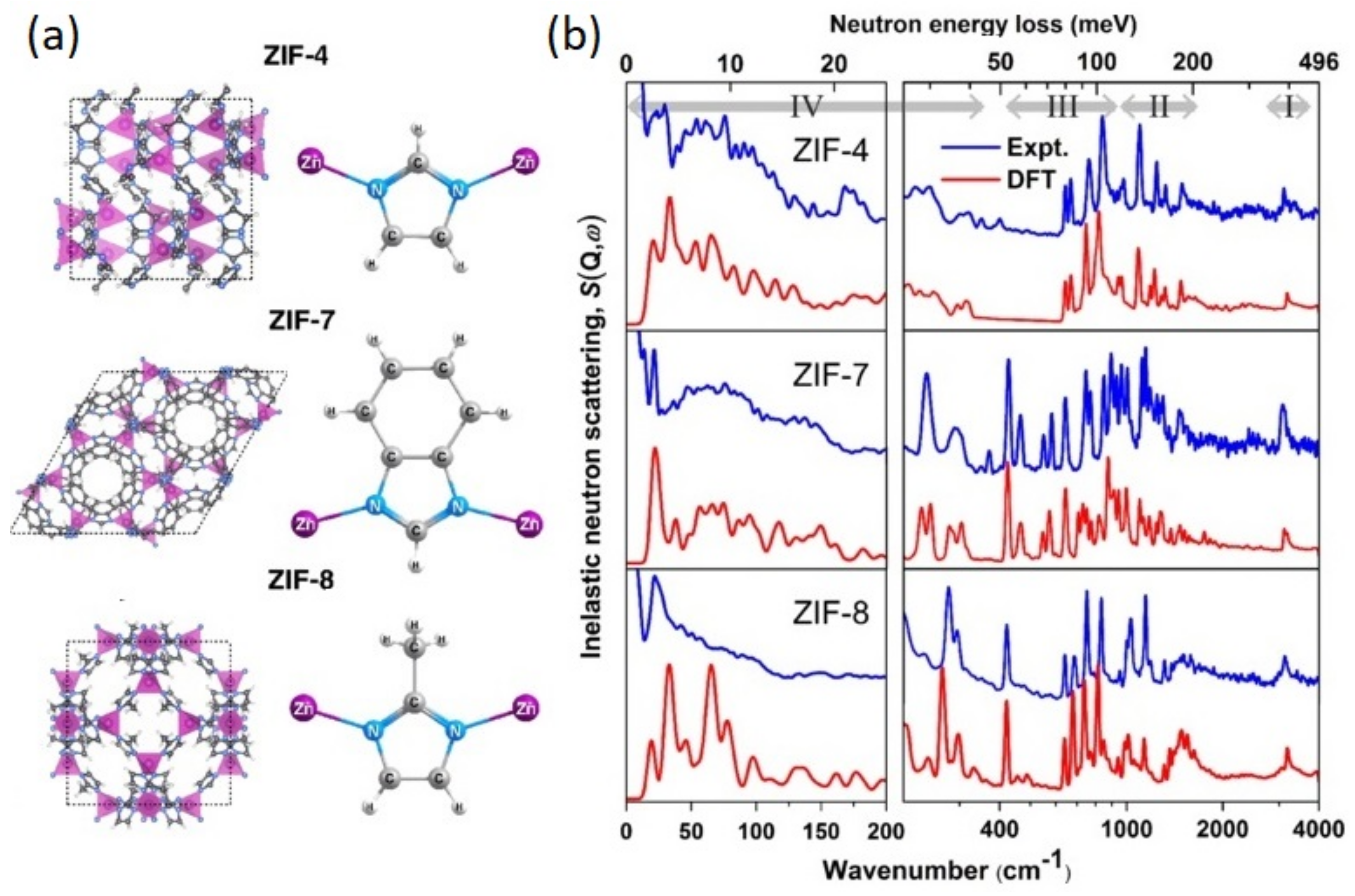
Figure 10.
(a) Experimental (TOSCA+) and simulated (DFT; VASP) INS spectra of closed- (cp) and open-pore (op) ZIF-4(Zn) structures. (b) Schematic diagram of the low-temperature structure of ZIF-4(Zn)-cp and the high-temperature structure ZIF-4(Zn)-op, looking down the b-axis in both phases. A volume contraction of approximately 24% associated with the cp-to-op phase transition is related to a rotation of the imidazolate linkers. Four of such rotations are highlighted by green stars. Reprinted with permission from Ref. [281]. Copyright (2021) American Chemical Society.
Figure 10.
(a) Experimental (TOSCA+) and simulated (DFT; VASP) INS spectra of closed- (cp) and open-pore (op) ZIF-4(Zn) structures. (b) Schematic diagram of the low-temperature structure of ZIF-4(Zn)-cp and the high-temperature structure ZIF-4(Zn)-op, looking down the b-axis in both phases. A volume contraction of approximately 24% associated with the cp-to-op phase transition is related to a rotation of the imidazolate linkers. Four of such rotations are highlighted by green stars. Reprinted with permission from Ref. [281]. Copyright (2021) American Chemical Society.

Figure 14.
Bottom: experimental INS spectrum of liquid H2O measured on SEQUOIA at ambient conditions. Two different incident energies have been used, as indicated in the figure [388]. Middle: experimental IR spectrum of H2O at ambient conditions, along with the results of AIMD predictions using vdW-corrected hybrid-DFT (revPBE0-D3 ML-FF) and different computational schemes—classical MD and approximate path-integral simulations (TRPMD and PACMD) [376]. Top: cartoon illustrating competing quantum effects in the H-bonding between two water molecules. There are two qualitatively different contributions to the vibrational ZPE: (i) the one arising from the two bending vibrational modes, in the plane of the water molecule and perpendicular to it (not shown); (ii) a second one associated with the O–H stretch. As the O–O distance R decreases, the contribution of the stretch decreases, and that of the bend increases. Consequently, the two contributions weaken and strengthen the intermolecular H-bond, respectively. [367]. Adapted with permission from Refs. [367,376,388]. Copyright (2021) American Chemical Society.
Figure 14.
Bottom: experimental INS spectrum of liquid H2O measured on SEQUOIA at ambient conditions. Two different incident energies have been used, as indicated in the figure [388]. Middle: experimental IR spectrum of H2O at ambient conditions, along with the results of AIMD predictions using vdW-corrected hybrid-DFT (revPBE0-D3 ML-FF) and different computational schemes—classical MD and approximate path-integral simulations (TRPMD and PACMD) [376]. Top: cartoon illustrating competing quantum effects in the H-bonding between two water molecules. There are two qualitatively different contributions to the vibrational ZPE: (i) the one arising from the two bending vibrational modes, in the plane of the water molecule and perpendicular to it (not shown); (ii) a second one associated with the O–H stretch. As the O–O distance R decreases, the contribution of the stretch decreases, and that of the bend increases. Consequently, the two contributions weaken and strengthen the intermolecular H-bond, respectively. [367]. Adapted with permission from Refs. [367,376,388]. Copyright (2021) American Chemical Society.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Drużbicki, K.; Gaboardi, M.; Fernandez-Alonso, F. Dynamics & Spectroscopy with Neutrons—Recent Developments & Emerging Opportunities. Polymers 2021, 13, 1440. https://doi.org/10.3390/polym13091440
AMA Style
Drużbicki K, Gaboardi M, Fernandez-Alonso F. Dynamics & Spectroscopy with Neutrons—Recent Developments & Emerging Opportunities. Polymers. 2021; 13(9):1440. https://doi.org/10.3390/polym13091440
Chicago/Turabian StyleDrużbicki, Kacper, Mattia Gaboardi, and Felix Fernandez-Alonso. 2021. "Dynamics & Spectroscopy with Neutrons—Recent Developments & Emerging Opportunities" Polymers 13, no. 9: 1440. https://doi.org/10.3390/polym13091440
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.