
Ring-Opening Polymerization (ROP) and Catalytic Rearrangement as a Way to Obtain Siloxane Mono- and Telechelics, as Well as Well-Organized Branching Centers: History and Prospects



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
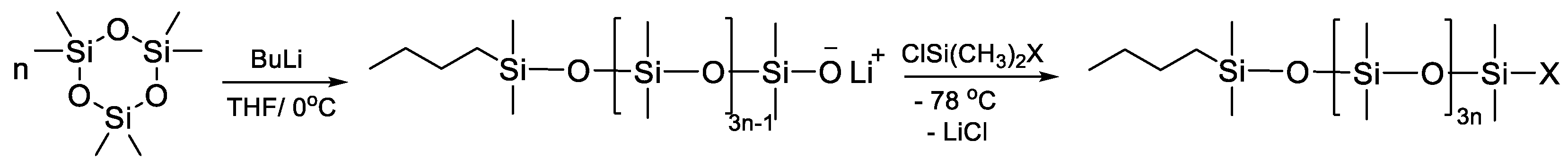{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
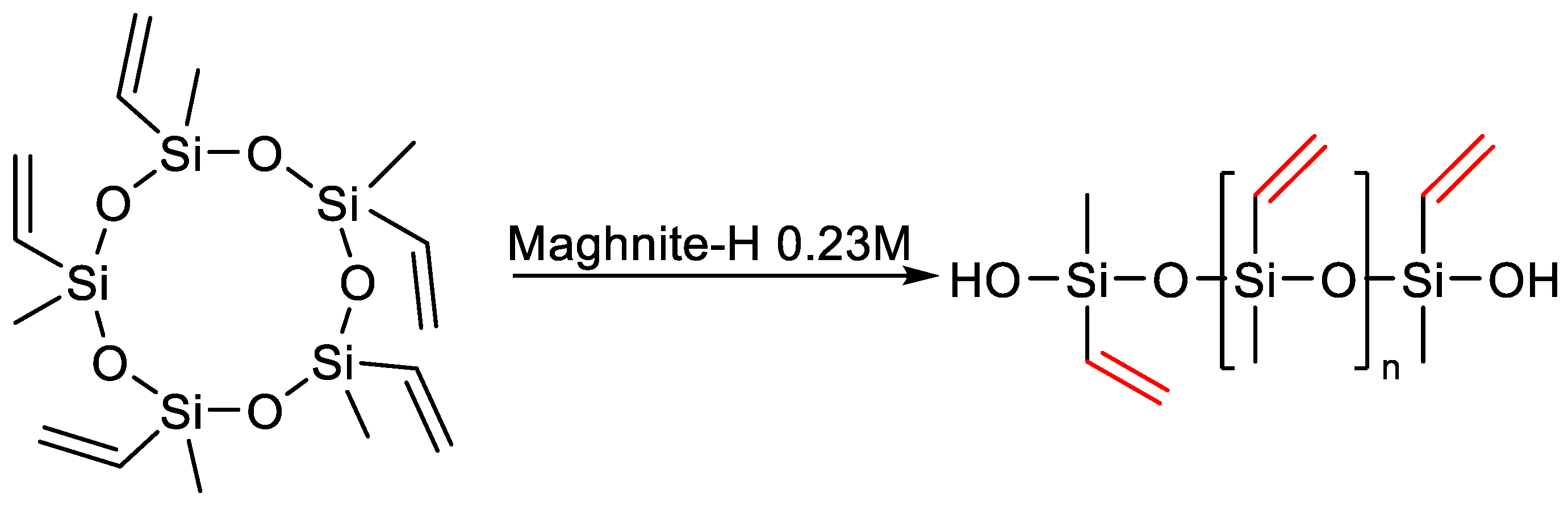{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
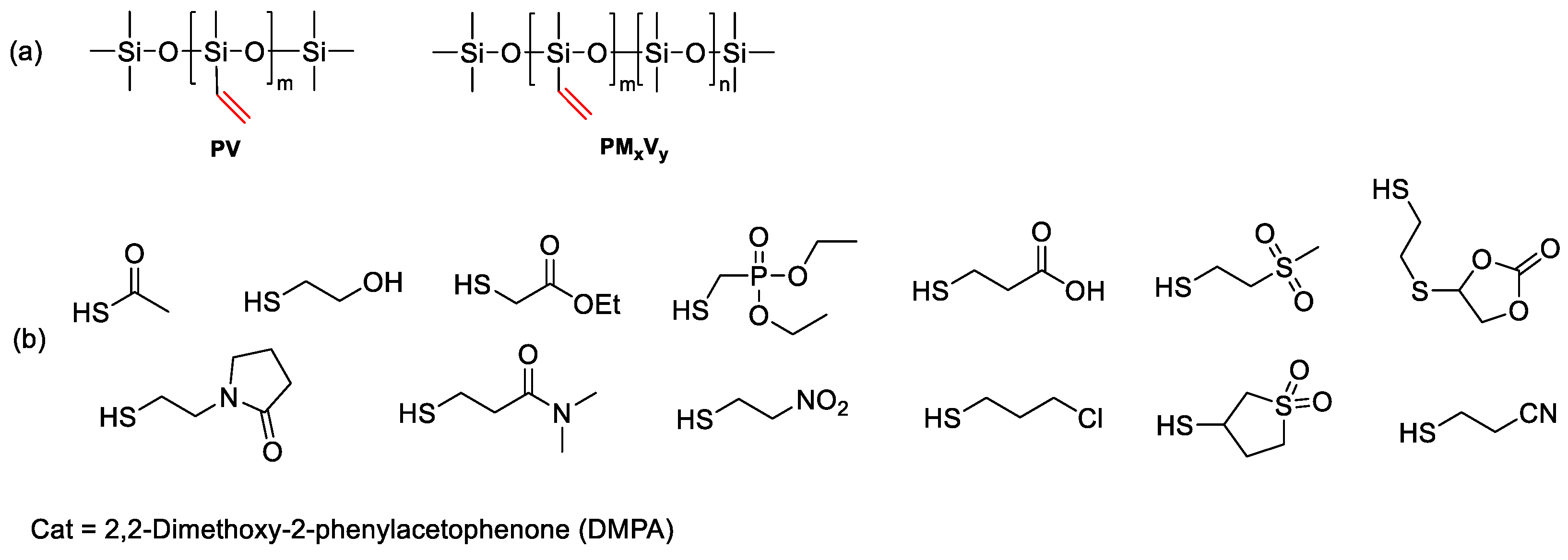{kind=link}
{kind=link}
{kind=link}
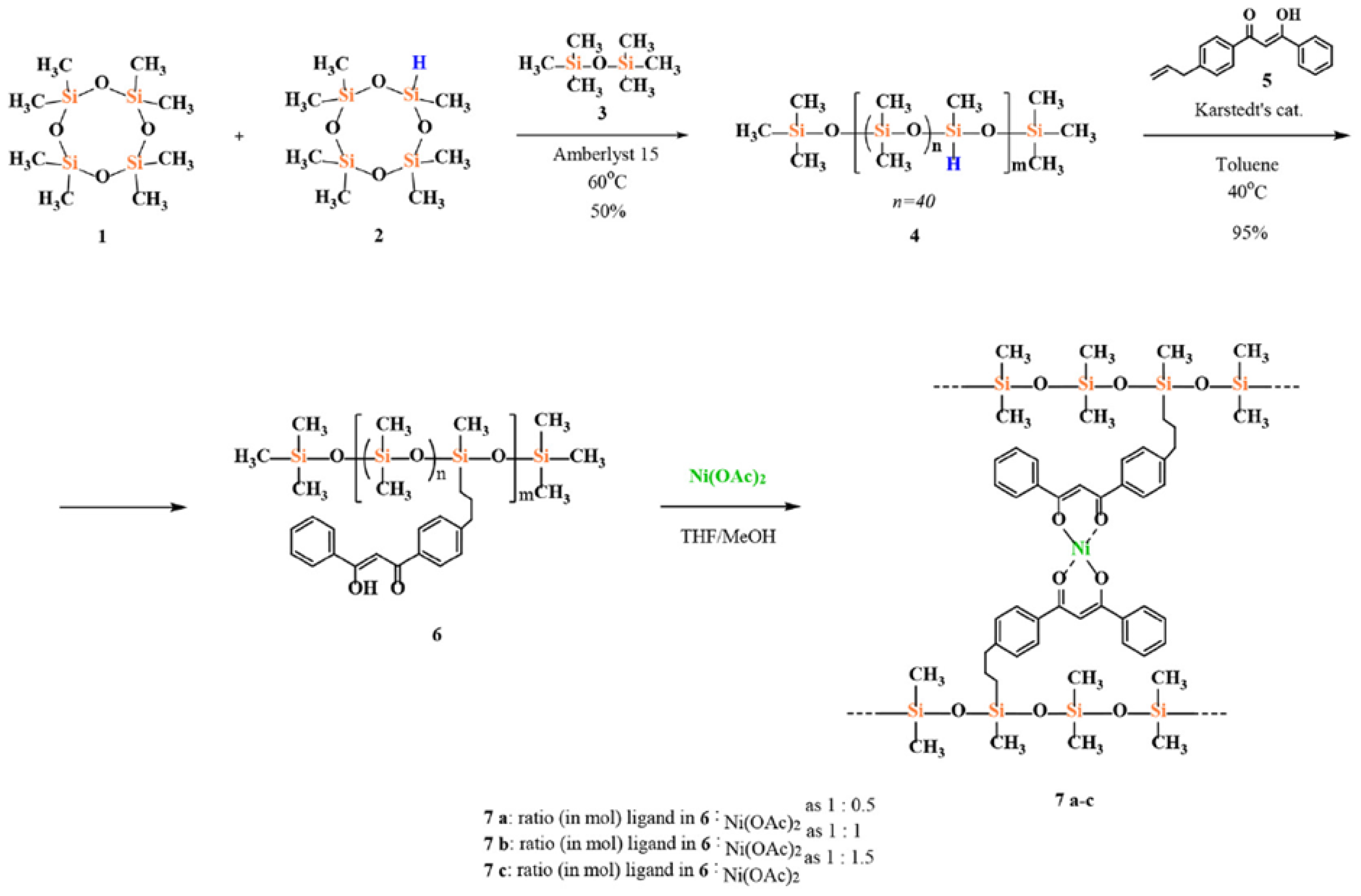{kind=link}
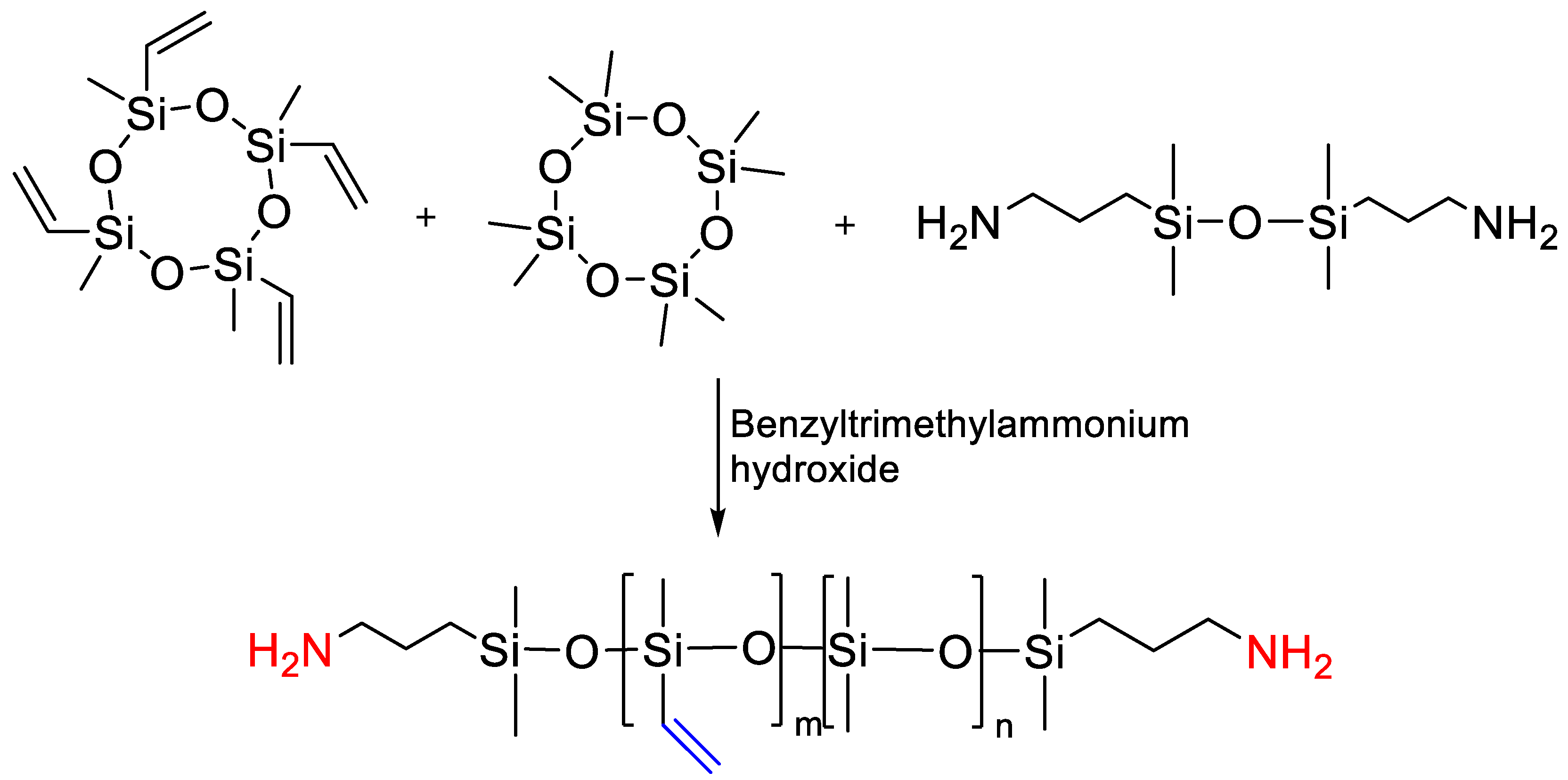{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Preparation of Diorganosiloxane Telechelics by the ROP Method
2.1. Preparation of Organosiloxane Telechelics by Anionic ROP
2.1.1. AROP Initiators
2.1.2. Influence of the Structure of the Initial Organocyclosiloxane on the AROP Process
2.1.3. Preparation of Monochelical PDMS by AROP
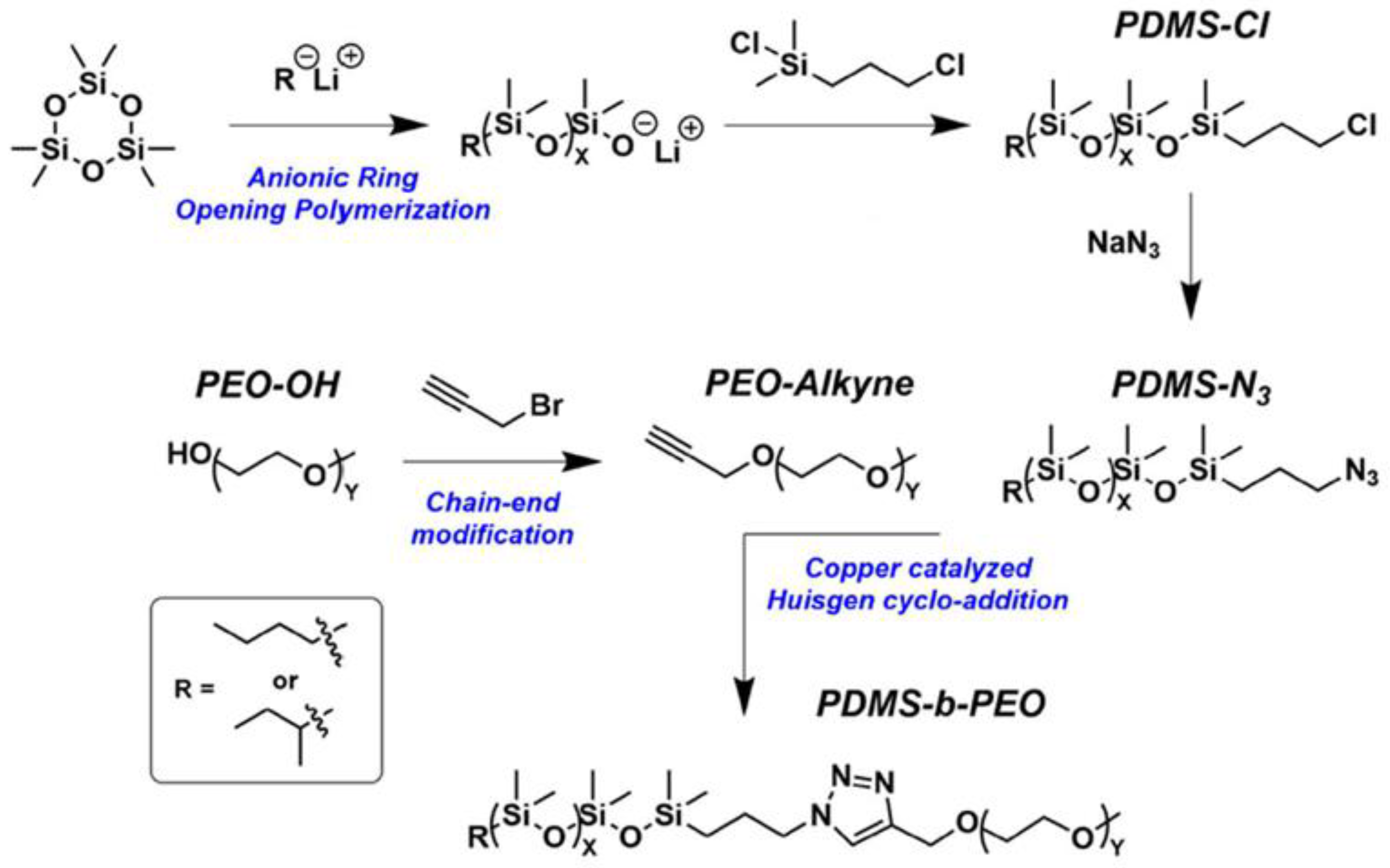
2.1.4. Obtaining Functional Telechelics by AROP
2.2. Preparation of Organosiloxane Telechelics by Cationic ROP
2.2.1. CROP Initiators
2.2.2. Obtaining Siloxane Telechelics by CROP
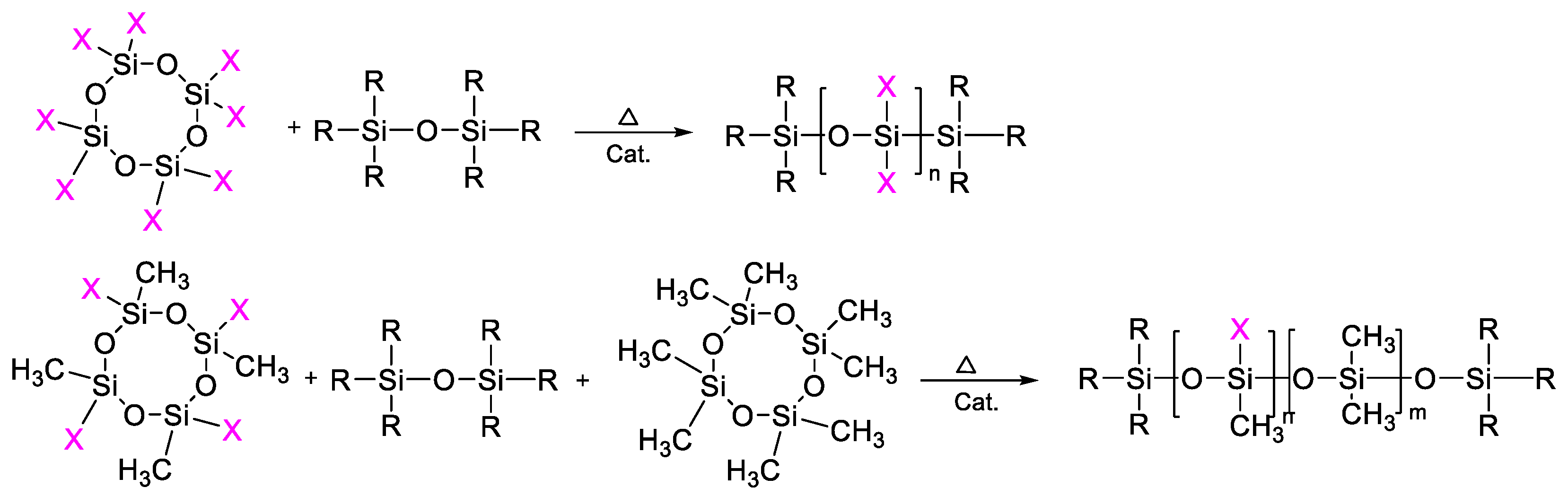
2.3. Catalytic Rearrangement Reactions for the Preparation of PDMS Copolymers
3. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Köhler, T.; Gutacker, A.; Mejiá, E. Industrial Synthesis of Reactive Silicones: Reaction Mechanisms and Processes. Org. Chem. Front. 2020, 7, 4108–4120. [Google Scholar] [CrossRef]
- McDonald, J.C.; Whitesides, G.M. Poly(Dimethylsiloxane) as a Material for Fabricating Microfluidic Devices. Acc. Chem. Res. 2002, 35, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Nahrup, J.S.; Mark, J.E.; Sakr, A. Poly(Dimethylsiloxane) Coatings for Controlled Drug Release. III. Drug Release Profiles and Swelling Properties of the Free-Standing Films. J. Appl. Polym. Sci. 2005, 96, 494–501. [Google Scholar] [CrossRef]
- Berthier, E.; Young, E.W.K.; Beebe, D. Engineers Are from PDMS-Land, Biologists Are from Polystyrenia. Lab Chip 2012, 12, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.K.; Grandisson, C.; Golemanov, K.; Ahuja, R.; Berret, J.F. Silicone Incorporation into an Esterquat Based Fabric Softener in Presence of Guar Polymers. Colloids Surf. A Physicochem. Eng. Asp. 2021, 615, 126175. [Google Scholar] [CrossRef]
- Chandrasekhar, V. Inorganic and Organometallic Polymers; Springer: Berlin, Germany, 2005. [Google Scholar]
- Voronkov, M.G.; Zelchan, G.I.; Lukevits, E.Y. Silicon and Life: Biochemistry, Toxicology and Pharmacology of Silicon Compounds. Zinat. Riga 1978, 9, 7–8. [Google Scholar]
- Zalewski, K.; Chyłek, Z.; Trzciński, W.A. A Review of Polysiloxanes in Terms of Their Application in Explosives. Polymers 2021, 13, 1080. [Google Scholar] [CrossRef]
- Luis, E.; Liu, H.; Song, J.; Yeong, W.Y. A Review of Medical Silicone 3D-Printing Technologies and Clinical Applications. J. Orthop. Res. Ther. 2018, 7, 1104. [Google Scholar] [CrossRef]
- Liu, J.; Yao, Y.; Li, X.; Zhang, Z. Fabrication of Advanced Polydimethylsiloxane-Based Functional Materials: Bulk Modifications and Surface Functionalizations. Chem. Eng. J. 2021, 408, 127262. [Google Scholar] [CrossRef]
- Andriot, M.; DeGroot, J.; Meeks, R.; Gerlach, E.; Jungk, M.; Wolf, A.; Chao, S.; Colas, A.; de Buyl, F.; Dupont, A.; et al. Silicones in Industrial Applications. In Inorganic Polymers; Oxford University Press: Oxford, UK, 2009; pp. 61–161. [Google Scholar]
- Zhang, J.; Xiao, P. 3D Printing of Photopolymers. Polym. Chem. 2018, 9, 1530–1540. [Google Scholar] [CrossRef]
- Liravi, F.; Toyserkani, E. Additive Manufacturing of Silicone Structures: A Review and Prospective. Addit. Manuf. 2018, 24, 232–242. [Google Scholar] [CrossRef]
- Sousa, R.P.C.L.; Ferreira, B.; Azenha, M.; Costa, S.P.G.; Silva, C.J.R.; Figueira, R.B. PDMS Based Hybrid Sol-Gel Materials for Sensing Applications in Alkaline Environments: Synthesis and Characterization. Polymers 2020, 12, 371. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F.; West, R. The Nature of the Silicon-Oxygen Bond. Organometallics 2011, 30, 5815–5824. [Google Scholar] [CrossRef]
- Yllgor, I.; Mcgrath, J.E. Polysiloxane Containing Copolymers: A Survey of Recent Developments. Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1988; pp. 1–86. [Google Scholar]
- Tikhonov, P.A.; Vasilenko, N.G.; Gallyamov, M.O.; Cherkaev, G.V.; Vasil’ev, V.G.; Demchenko, N.V.; Buzin, M.I.; Vasil’ev, S.G.; Muzafarov, A.M. Multiarm Star-Shaped Polydimethylsiloxanes with a Dendritic Branching Center. Molecules 2021, 26, 3280. [Google Scholar] [CrossRef]
- Vasilenko, N.G.; Rebrov, E.A.; Muzafarov, M.; EJwein, B.; Striege, B.; Miillere, M. Preparation of Multi-Arm Star Polymers with Polylithiated Carbosilane Dendrimers. Macromol. Chem. Phys. 1998, 199, 889–895. [Google Scholar] [CrossRef]
- Pakula, T.; Zhang, Y.; Matyjaszewski, K.; Lee, H.I.; Boerner, H.; Qin, S.; Berry, G.C. Molecular Brushes as Super-Soft Elastomers. Polymer 2006, 47, 7198–7206. [Google Scholar] [CrossRef]
- Daniel, W.F.M.; Burdyńska, J.; Vatankhah-Varnoosfaderani, M.; Matyjaszewski, K.; Paturej, J.; Rubinstein, M.; Dobrynin, A.V.; Sheiko, S.S. Solvent-Free, Supersoft and Superelastic Bottlebrush Melts and Networks. Nat. Mater. 2016, 15, 183–189. [Google Scholar] [CrossRef]
- Daniels, D.R.; McLeish, T.C.B.; Crosby, B.J.; Young, R.N.; Fernyhough, C.M. Molecular Rheology of Comb Polymer Melts. 1. Linear Viscoelastic Response. Macromolecules 2001, 34, 7025–7033. [Google Scholar] [CrossRef]
- Kapnistos, M.; Vlassopoulos, D.; Roovers, J.; Leal, L.G. Linear Rheology of Architecturally Complex Macromolecules: Comb Polymers with Linear Backbones. Macromolecules 2005, 38, 7852–7862. [Google Scholar] [CrossRef]
- Vatankhah-Varnoosfaderani, M.; Daniel, W.F.M.; Zhushma, A.P.; Li, Q.; Morgan, B.J.; Matyjaszewski, K.; Armstrong, D.P.; Spontak, R.J.; Dobrynin, A.V.; Sheiko, S.S. Bottlebrush Elastomers: A New Platform for Freestanding Electroactuation. Adv. Mater. 2016, 29, 1604209. [Google Scholar] [CrossRef]
- Daniel, W.F.M.; Xie, G.; Vatankhah Varnoosfaderani, M.; Burdyńska, J.; Li, Q.; Nykypanchuk, D.; Gang, O.; Matyjaszewski, K.; Sheiko, S.S. Bottlebrush-Guided Polymer Crystallization Resulting in Supersoft and Reversibly Moldable Physical Networks. Macromolecules 2017, 50, 2103–2111. [Google Scholar] [CrossRef]
- Zhang, J.; Schneiderman, D.K.; Li, T.; Hillmyer, M.A.; Bates, F.S. Design of Graft Block Polymer Thermoplastics. Macromolecules 2016, 49, 9108–9118. [Google Scholar] [CrossRef]
- Xia, Y.; Olsen, B.D.; Kornfield, J.A.; Grubbs, R.H. Efficient Synthesis of Narrowly Dispersed Brush Copolymers and Study of Their Assemblies: The Importance of Side Chain Arrangement. J. Am. Chem. Soc. 2009, 131, 18525–18532. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.; Rzayev, J. Synthesis and Melt Self-Assembly of PS-PMMA-PLA Triblock Bottlebrush Copolymers. Macromolecules 2014, 47, 2864–2874. [Google Scholar] [CrossRef]
- Tezuka, Y. Telechelic polymers. Prog. Polym. Sci. 1992, 17, 471–514. [Google Scholar] [CrossRef]
- Kumar, A.; Gupta, R.K. Fundamentals of Polymer Chemistry; CRC Press: Boca Raton, FL, USA, 1974; p. 614. [Google Scholar]
- Abe, Y.; Gunji, T. Oligo-and polysiloxanes. Prog. Polym. Sci. 2004, 29, 149–182. [Google Scholar] [CrossRef]
- Cerveau, G.; Chappellet, S.; Corriu, R.J.P.; Dabiens, B. Synthesis, Characterization, and Polycondensation of New Stable α,ω-Organo(Bis-Silanediols). Can. J. Chem. 2003, 81, 1213–1222. [Google Scholar] [CrossRef]
- Andrianov, K.A.; Pashintseva, G.I.; Nanush’yan, S.R.; Severnyi, V.V. Thermal and Thermal-Oxidative Stability of Polyorganocarbosiloxanes with a Net-like Structure. Polym. Sci. USSR 1973, 15, 2039–2049. [Google Scholar] [CrossRef]
- Kopylov, V.M.; Khananashvili, L.M.; Shkol’nik, O.V.; Ivanov, A.G. Hydrolytic Polycondensation of Organochlorosilanes. Polym. Sci. 1995, 37, 242–265. [Google Scholar]
- Noll, W. Chemistry and Technology of Silicones; Academic Press: Cambridge, MA, USA, 1968. [Google Scholar]
- Rochow, E.G. Silicon and Silicones; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1987. [Google Scholar]
- Jones, R.G.; Ando, W.; Chojnowski, J. Silicon-Containing Polymers: The Science and Technology of Their Synthesis and Applications; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Kalinina, A.; Strizhiver, N.; Vasilenko, N.; Perov, N.; Demchenko, N.; Muzafarov, A. Polycondensation of Diethoxydimethylsilane in Active Medium. Silicon 2015, 7, 95–106. [Google Scholar] [CrossRef]
- Voronkov, M.G.; Mileshkevich, V.P.; Yuzhelevsky, Y.A. The Siloxane Bond; Consultants Bureau: New York, NY, USA, 1970. [Google Scholar]
- Kendrick, T.C.; White, J.W. The Silicon-Heteroatom Bond; Patai, S., Rappoport, Z., Eds.; John Wiley: Hoboken, NJ, USA, 1989; Volume 3. [Google Scholar]
- Lee, C.L.; Johannson, O.K. Polymerization of cyclosiloxanes. I. Kinetic studies of living polymer-octamethylcyclotetrasiloxane systems. J. Polym. Sci. 1966, A4, 3013. [Google Scholar] [CrossRef]
- Yuzhelevsky, Y.A.; Kagan, E.G.; Dmokhovskaya, E.B. Influence of the nature of substituents at the silicon atom on the equilibrium during the catalytic rearrangement of cyclosiloxanes in solution. Chem. Heterocycl. Compd. 1967, 5, 951. [Google Scholar]
- Beervers, M.S.; Semlyen, J.A. Equilibrium ring concentrations and the statistical conformations of polymer chains: Part 5. Stereoisomeric cyclics in poly(phenylmethylsiloxane) equilibrates. Polymer 1971, 12, 373. [Google Scholar] [CrossRef]
- Tasdelen, M.A.; Kahveci, M.U.; Yagci, Y. Telechelic Polymers by Living and Controlled/Living Polymerization Methods. Prog. Polym. Sci. 2011, 36, 455–567. [Google Scholar] [CrossRef]
- Andrianov, K.A. Anionic polymerization of cyclic organosilicon compounds. Review. Polym. Sci. USSR 1978, 20, 275–288. [Google Scholar] [CrossRef]
- Grubb, W.T.; Osthoff, R.C. Kinetics of the Polymerization of a Cyclic Dimethylsiloxane. J. Am. Chem. Soc. 1955, 77, 1405. [Google Scholar] [CrossRef]
- Yuzhelevsky, Y.A.; Kagan, E.G.; Kogan, E.V.; Klebansky, A.L.; Nikiforov, N.N. Kinetics of anionic polymerization of cyclosiloxanes with 3,3,3-trifluoropropyl groups at the silicon atom. Vysokomolek. Soed. 1969, 11, 1539. [Google Scholar] [CrossRef]
- Hurd, D.T.; Osthoff, R.C.; Corrin, M.L. The mechanism of the base-catalyzed rearrangement of organopolysiloxanes. J. Am. Chem. Soc. 1954, 76, 249. [Google Scholar] [CrossRef]
- Gilbert, A.K.; Kantor, S.W. Transient catalysts for the polymerization of organosiloxanes. J. Polym. Sci. 1959, 40, 35. [Google Scholar] [CrossRef]
- Mazurek, M.; Ściborek, M.; Chojnowski, J.; Zavin, B.G.; Zhdanov, A.A. Thermodynamic Enhancement of Oligomers in Dynamic Living Polymer System Involving End-Group Interaction. Eur. Polym. J. 1980, 16, 57. [Google Scholar] [CrossRef]
- Pchelintsev, V.V. About the Association of Alkali Metal Siloxanolates. J. Gen. Chem. 1973, 43, 1200. [Google Scholar]
- Morton, M.; Deisz, M.A.; Bostick, E.E. Base-catalyzed solution polymerization of octamethylcyclotetrasiloxane. J. Polym. Sci. 1964, A2, 513. [Google Scholar] [CrossRef]
- Hölle, H.J.; Lehnen, B.R. Preparation and characterization of polydimethylsiloxanes with narrow molecular weight distribution. Eur. Polym. J. 1975, 11, 663. [Google Scholar] [CrossRef]
- Lee, C.L.; Frye, C.L.; Johannso, O.K. Selective Polymerization of Reactive Cyclosiloxanes to Give Non-Equlibrium Molecular Weight Distributions. Monodisperse Siloxane Polymers. Polymer 1969, 10, 133. [Google Scholar]
- Fessler, W.A.; Juliano, P.C. Reactivity of Solvated Lithium N-Butyldimethylsilanolate with Organosiloxane Substrates. Ind. Eng. Chem. Prod. Res. Dev. 1972, 11, 407–410. [Google Scholar] [CrossRef]
- Frye, C.L.; Salinger, R.M.; Fearon, F.W.G.; Klosowski, J.M.; DeYoung, T. Reactions of organolithium reagents with siloxane substrates. Org. Chem. 1970, 35, 1308. [Google Scholar] [CrossRef]
- Mazurek, M.; Chojnowski, J. Anionic polymerization of siloxanes. 2. Internal multifunctional assistance of siloxane system to the siloxane bond cleavage by alkali metal silanolates. Makromol. Chem. 1977, 178, 1005. [Google Scholar] [CrossRef]
- Suryanarayanan, B.; Peace, B.W.; Mayhan, K.G. Anionic polymerization of five-membered cyclocarbosiloxanes. J. Polym. Sci. 1974, 12, 1089. [Google Scholar] [CrossRef]
- Tugui, C.; Tiron, V.; Dascalu, M.; Sacarescu, L.; Cazacu, M. From Ultra-High Molecular Weight Polydimethylsiloxane to Super-Soft Elastomer. Eur. Polym. J. 2019, 120, 109243. [Google Scholar] [CrossRef]
- Bessmertnykh, A.; Ben, F.; Baceiredo, A.; Mignani, G. Anionic Ring-Opening Polymerization of Cyclic Organosiloxanes Using Phosphorus Ylides as Strong Non-Ionic Bases. J. Organomet. Chem. 2003, 686, 281–285. [Google Scholar] [CrossRef]
- Graiver, D.; Lomas, A.V.; Rasmussen, E.T., Jr.; Wall, K.J. Method for the preparation of potassium silanolates. U.S. Patent 5856546, 5 January 1999. [Google Scholar]
- Molenberg, A.; Möller, M. A fast catalyst system for the ring-opening polymerization of cyclosiloxanes. Macromol. Rapid Commun. 1995, 16, 449–453. [Google Scholar] [CrossRef]
- Eßwein, B.; Molenberg, A.; Möller, M. Use of polyiminophosphazene bases for ring-opening polymerizations. Macromol. Symp. 1996, 107, 331–340. [Google Scholar] [CrossRef]
- Hupfield, P.C.; Taylor, R.G. Ring-Opening Polymerization of Siloxanes Using Phosphazene Base Catalysts. J. Inorg. Organomet. Polym. 1999, 9, 17–34. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, N.; Xia, S.; Liu, S.; Li, Z. Phosphazene Superbase Catalyzed Ring-Opening Polymerization of Cyclotetrasiloxane toward Copolysiloxanes with High Diphenyl Siloxane Content. Polym. Chem. 2019, 10, 2126–2133. [Google Scholar] [CrossRef]
- Rodriguez, M.; Marrot, S.; Kato, T.; Stérin, S.; Fleury, E.; Baceiredo, A. Catalytic Activity of N-Heterocyclic Carbenes in Ring Opening Polymerization of Cyclic Siloxanes. J. Organomet. Chem. 2007, 692, 705–708. [Google Scholar] [CrossRef]
- Yactine, B.; Ratsimihety, A.; Ganachaud, F. Do-It-Yourself Functionalized Silicones Part 2: Synthesis by Ring Opening Polymerization of Commercial Cyclosiloxanes. Polym. Adv. Technol. 2010, 21, 139–149. [Google Scholar] [CrossRef]
- Oka, M.; Takagi, H.; Miyazawa, T.; Waymouth, R.M.; Honda, S. Photocleavable Regenerative Network Materials with Exceptional and Repeatable Viscoelastic Manipulability. Adv. Sci. 2021, 8, 2101143. [Google Scholar] [CrossRef]
- Fuchise, K.; Igarashi, M.; Sato, K.; Shimada, S. Organocatalytic Controlled/Living Ring-Opening Polymerization of Cyclotrisiloxanes Initiated by Water with Strong Organic Base Catalysts. Chem. Sci. 2018, 9, 2879–2891. [Google Scholar] [CrossRef]
- Morton, M.; Bostick, E.E. Bostick Anionic Polymerization of Octamethylcyclotetrasiloxane in Tetrahydrofuran Solution. J. Polym. Sci. Part A Gen. Pap. 1964, 2, 523–538. [Google Scholar] [CrossRef]
- Kantor, S.W.; Grubb, W.T.; Osthoff, R.C. The mechanism of the acid- and base-catalyzed equilibration of siloxanes. J. Am. Chem. Soc. 1954, 176, 5190. [Google Scholar] [CrossRef]
- Suzuki, T.; Okawa, T. Preparation of poly(dimethylsiloxane) macro-monomers by the ‘initiator method’. Polym. Commun. 1988, 29, 225. [Google Scholar] [CrossRef]
- Oulad Hammouch, S.; Beinert, G.J.; Zillox, G.J.; Herz, J.E. Synthesis and characterization of monofunctional polydimethylsiloxanes with a narrow molecular weight distribution. Polymer 1995, 36, 421. [Google Scholar] [CrossRef]
- Sormani, P.M.; Minton, R.J.; McGrath, J.E. Anionic Ring-Opening Polymerization of Octamethylcyclotetrasiloxane in the Presence of 1,3-Bis(Aminopropyl)-1,1,3,3-Tetramethyldisiloxane. In Ring-Opening Polymerization; American Chemical Society: Washington, DC, USA, 1985; pp. 147–160. [Google Scholar]
- Bischoff, R.; Sigwalt, P. Mechanism of octamethylcyclotetrasiloxane polymerization in the presence of siloxanediols. Polym. Int. 1996, 40, 99–109. [Google Scholar] [CrossRef]
- Godovsky, Y.K.; Papkov, V.S. Thermotropic Mesophases in Element-Organic Polymers. Adv. Polym. Sci. 1989, 88, 127. [Google Scholar]
- Lee, M.K.; Meier, D.J. Synthesis and properties of diarylsiloxane and (aryl/methyl) siloxane polymers: 1. Thermal properties. Polymer 1993, 34, 4882–4892. [Google Scholar] [CrossRef]
- Buzin, M.I.; Gerasimov, M.V.; Obolonkova, S.; Papkov, V.S. Solid-State Polymerization of Hexaphenylcyclotrisiloxane. J. Polym. Sci. Part A 1997, 35, 1973–1984. [Google Scholar] [CrossRef]
- Babchinitser, T.M.; Kazaryan, L.G.; Tartakovskaya, L.M.; Vasilenko, N.G.; Zhdanov, A.A.; Korshak, V.V. Crystalline structure of poly (diaryl siloxanes). Polymer 1985, 26, 1527–1530. [Google Scholar] [CrossRef]
- Evmenenko, G.; Yu, C.J.; Kmetko, J.; Dutta, P. Density Anomalies in Thin Liquid Films of Hydride Functional Siloxanes. Langmuir 2002, 18, 5468–5472. [Google Scholar] [CrossRef]
- Morariu, S.; Brunchi, C.E.; Cazacu, M.; Bercea, M. The Behavior of Poly(Dimethylsiloxane-Co-Diphenylsiloxane)s in Good and Theta Solvents. J. Chem. Eng. Data 2011, 56, 1473–1475. [Google Scholar] [CrossRef]
- Shrivastava, A. Polymerization. In Introduction to Plastics Engineering; William Andrew: Norwich, NY, USA, 2018; pp. 17–48. [Google Scholar] [CrossRef]
- Yuzhelevsky, Y.A.; Kagan, E.G.; Timofeeva, N.P.; Doletskaya, T.D.; Klebansky, A.L. Anionic polymerization of methyl (propyl) cyclosiloxanes. Vysokomolek. Soed. 1971, 13, 183. [Google Scholar] [CrossRef]
- Andrianov, K.A.; Yakushkina, S.E.; Terentiev, N.N. Study of the influence of substituents on the reactivity of organocyclosiloxanes in the reaction of anionic polymerization. Vysokomolek. Soed. 1968, 10, 1721. [Google Scholar]
- Borisov, S.N.; Kurlova, T.V.; Yuzhelevsky, Y.A.; Chernyshev, E.A. Anionic polymerization of methylphenylcyclosiloxanes. Vysokomolek. Soed. 1970, 12B, 332. [Google Scholar]
- Andrianov, K.A. On the polymerization of organosilicon cyclic compounds. Vysokomolek. Soed. 1969, 11, 1362. [Google Scholar]
- Bostick, E.E. Novel organosiloxane-silicate copolymers. U.S. Patent 3337496, 22 August 1967. [Google Scholar]
- Eabom, C. Organosilicon Compounds; Butterworths Scientific, 1960. [Google Scholar]
- Kazama, H.; Tezuka, Y.; Imai, K. Syntheses and reactions of uniform size poly (dimethylsiloxane) with various reactive end groups. Polym. J. 1987, 19, 1091–1100. [Google Scholar] [CrossRef][Green Version]
- Tezuka, Y.; Fukushima, A.; Imai, K. Synthesis of poly (vinyl alcohol)/poly (dimethylsiloxane) graft copolymer. Die Makromol. Chem. Macromol. Chem. Phys. 1985, 186, 685–694. [Google Scholar] [CrossRef]
- Cameron, G.G.; Chisholm, M.S. Polymerization of poly (dimethylsiloxane) macromers: 1. Copolymerization with styrene. Polymer 1985, 26, 437–442. [Google Scholar] [CrossRef]
- Peters, M.A.; Belu, A.M.; Linton, R.W.; Dupray, L.; Meyer, T.J.; DeSimone, J.M. Termination of living anionic polymerizations using chlorosilane derivatives: A general synthetic methodology for the synthesis of end-functionalized polymers. J. Am. Chem. Soc. 1995, 117, 3380–3388. [Google Scholar] [CrossRef]
- Vysochinskaya, Y.S.; Anisimov, A.A.; Peregudov, A.S.; Dubovik, A.S.; Orlov, V.N.; Malakhova, Y.N.; Stupnikov, A.A.; Buzin, M.I.; Nikiforova, G.G.; Vasil’ev, V.G.; et al. Star-Shaped Siloxane Polymers with Various Cyclic Cores: Synthesis and Properties. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1233–1246. [Google Scholar] [CrossRef]
- Kawakami, Y.; Miki, Y.; Tsuda, T.; Murthy, R.A.N.; Yamashita, Y. Silicone macromers for graft polymer synthesis. Polym. J. 1982, 14, 913–917. [Google Scholar] [CrossRef]
- Fauquignon, M.; Ibarboure, E.; Carlotti, S.; Brûlet, A.; Schmutz, M.; le Meins, J.F. Large and Giant Unilamellar Vesicle(s) Obtained by Self-Assembly of Poly(Dimethylsiloxane)-b-Poly(Ethylene Oxide) Diblock Copolymers, Membrane Properties and Preliminary Investigation of Their Ability to Form Hybrid Polymer/Lipid Vesicles. Polymers 2019, 11, 2013. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Montarnal, D.; Treat, N.J.; Hustad, P.D.; Christianson, M.D.; Kramer, E.J.; Fredrickson, G.H.; Hawker, C.J. Enhanced Block Copolymer Phase Separation Using Click Chemistry and Ionic Junctions. ACS Macro Lett. 2015, 4, 1332–1336. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Kim, B.; Montarnal, D.; Mester, Z.; Pester, C.W.; McGrath, A.J.; Hill, G.; Kramer, E.J.; Fredrickson, G.H.; Hawker, C.J. Improved Self-Assembly of Poly(Dimethylsiloxane-b-Ethylene Oxide) Using a Hydrogen-Bonding Additive. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2200–2208. [Google Scholar] [CrossRef]
- Elkins, C.L.; Long, T.E. Living Anionic Polymerization of Hexamethylcyclotrisiloxane (D3) Using Functionalized Initiation. Macromolecules 2004, 37, 6657–6659. [Google Scholar] [CrossRef]
- Goff, J.; Sulaiman, S.; Arkles, B. Applications of Hybrid Polymers Generated from Living Anionic Ring Opening Polymerization. Molecules 2021, 26, 2755. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Wang, D.; Yang, Z.; Liang, Y.; Zhang, J.; Feng, S. Facile, Versatile and Efficient Synthesis of Functional Polysiloxanes via Thiol-Ene Chemistry. Eur. Polym. J. 2013, 49, 1050–1056. [Google Scholar] [CrossRef]
- Sivasailam, K.; Cohen, C. Scaling Behavior: Effect of Precursor Concentration and Precursor Molecular Weight on the Modulus and Swelling of Polymeric Networks. J. Rheol. 2000, 44, 897–915. [Google Scholar] [CrossRef]
- O’Malley, J.; Pacansky, T.J.; Stauffer, W. Synthesis and characterization of poly (hexamethylene sebacate)-poly (dimethylsiloxane) block copolymers. Macromolecules 1977, 10, 1197–1199. [Google Scholar] [CrossRef]
- Hoffman, J.J.; Leir, C.M. Tetramethylammonium 3-aminopropyl dimethylsilanolate: A new catalyst for the synthesis of high purity, high molecular weight α, ω-bis (aminopropyl) polydimethylsiloxanes. Polym. Int. 1991, 24, 131–138. [Google Scholar] [CrossRef]
- McGrath, J.E.; Sormani, P.M.; Elsbernd, C.S.; Kilic, S. Kinetics, mechanisms, and synthesis studies of difunctional aminopropyl terminated polydimethylsiloxane oligomers. In Makromolekulare Chemie. Macromolecular Symposia; Hüthig & Wepf Verlag: Basel, Switzerland, 1986; Volume 6, pp. 67–80. [Google Scholar]
- Li, X.; Yu, R.; Zhao, T.; Zhang, Y.; Yang, X.; Zhao, X.; Huang, W. A Self-Healing Polysiloxane Elastomer Based on Siloxane Equilibration Synthesized through Amino-Ene Michael Addition Reaction. Eur. Polym. J. 2018, 108, 399–405. [Google Scholar] [CrossRef]
- Gorodov, V.V.; Bakirov, A.V.; Buzin, M.I.; Vasil’ev, V.G.; Shragin, D.I.; Myakushev, V.D.; Papkov, V.S.; Chvalun, S.N.; Muzafarov, A.M. Synthesis of Telehelic Oligodimethylsiloxanes with 4-Carboxypyrrolidone Fragments: Rheological and Thermal Properties. Russ. Chem. Bull. 2018, 67, 2282–2289. [Google Scholar] [CrossRef]
- Zuo, Y.; Lu, H.; Xue, L.; Wang, X.; Ning, L.; Feng, S. Preparation and Characterization of Luminescent Silicone Elastomer by Thiol-Ene “Click” Chemistry. J. Mater. Chem. C 2014, 2, 2724–2734. [Google Scholar] [CrossRef]
- Drozdov, F.V.; Cherkaev, G.V.; Demchenko, N.V.; Muzafarov, A.M. Synthesis of New Functional Hexamethyltrisiloxanes and Telechelic Polydimethylsiloxanes Based on Them. J. Appl. Polym. Sci. 2018, 135, 46848. [Google Scholar] [CrossRef]
- Fei, H.F.; Xie, W.; Wang, Q.; Gao, X.; Hu, T.; Zhang, Z.; Xie, Z. Controlled Synthesis and Characterization of Poly [Methyl(3,3,3-Trifluoropropyl)Siloxane] with Selective End Groups. RSC Adv. 2014, 4, 56279–56287. [Google Scholar] [CrossRef]
- Sato, K.I.; Ito, S.; Higashihara, T.; Fuchise, K. Precise Synthesis of α,ω-Chain-End Functionalized Poly(Dimethylsiloxane) with Azide Groups Based on Metal-Free Ring-Opening Polymerization and a Quantitative Azidation Reaction. React. Funct. Polym. 2021, 166, 105009. [Google Scholar] [CrossRef]
- Fuchise, K.; Sato, K.; Igarashi, M. Precise Synthesis of Side-Chain-Functionalized Linear Polysiloxanes by Organocatalytic Ring-Opening Polymerization of Monofunctional Cyclotrisiloxanes. Macromolecules 2021, 54, 5204–5217. [Google Scholar] [CrossRef]
- Fuchise, K.; Sato, K.; Igarashi, M. Precise Synthesis of Linear Polysiloxanes End-Functionalized with Alkynylsilyl Groups by Organocatalytic Ring-Opening Polymerization of Cyclotrisiloxanes. Macromolecules 2021, 54, 5765–5773. [Google Scholar] [CrossRef]
- Chojnowski, J.; Mazurek, M.; Šcibiorek, M.; Wilczek, L. Cationic polymerization of siloxanes. Approach to the mechanistic studies. Die Makromol. Chem. Macromol. Chem. Phys. 1974, 175, 3299–3303. [Google Scholar] [CrossRef]
- Wilczek, L.; Chojnowski, J. Acidolytic Ring Opening of Cyclic Siloxane and Acetal Monomers. Role of Hydrogen Bonding in Cationic Polymerization Initiated with Protonic Acids. Macromolecules 1981, 14, 9–17. [Google Scholar] [CrossRef]
- Cypryk, M. Polymerization of Cyclic Siloxanes, Silanes, and Related Monomers; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Toskas, G.; Besztercey, G.; Moreau, M.; Masure, M.; Sigwalt, P. Cationic polymerization of hexamethylcyclotrisiloxane by trifluoromethanesulfonic acid and its derivatives, 2. Reaction involving activated trifluoromethylsulfonates. Macromol. Chem. Phys. 1995, 196, 2715–2735. [Google Scholar] [CrossRef]
- Yashiro, T.; Kricheldorf, H.R.; Schwarz, G. Polymerization of Cyclosiloxanes by Means of Triflic Acid and Metal Triflates. Macromol. Chem. Phys. 2010, 211, 1311–1321. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yamamoto, Y. Method for the preparation of organopolysiloxanes. (To Dow Corning Toray Silicone Co.). U.S. Patent 5241032, 31 August 1993. [Google Scholar]
- Andrianov, K.A.; Shkolnik, M.I.; Kopylow, V.M.; Bravina, N.N. Polymerization of octamethylcyclotetrasiloxane by perchloric-acid. Vysokomol. Soed. B 1974, 16, 893. [Google Scholar]
- Bryk, M.T.; Baglei, N.N.; Kurilenko, O.D. Polymerization of octamethylcyclotetrasiloxane catalysed by the ion exchange resin KU-2. Polym. Sci. USSR 1975, 17, 1187–1195. [Google Scholar] [CrossRef]
- Siciliano, G.R. Continuous process for producing polysiloxane oils utilizing a carbon black catalyst (To General Electric). U.S. Patent 3853933, 10 December 1974. [Google Scholar]
- Kendrick, T.C. The acid-catalysed polymerisation of cyclosiloxanes. Part I. The kinetics of the polymerisation of octamethylcyclotetrasiloxane catalysed by anhydrous ferric chloride–hydrogen chloride. J. Chem. Soc. 1965, 361, 2027–2035. [Google Scholar] [CrossRef]
- Jordan, E.; Lestel, L.; Boileau, S.; Cheradame, H.; Gandini, A. Can lewis acids initiate the polymerization of cyclic siloxanes via direct addition? Die Makromol. Chem. Macromol. Chem. Phys. 1989, 190, 267–276. [Google Scholar] [CrossRef]
- Sigwalt, P.; Gobin, C.; Nicol, P.; Moreau, M.; Masure, M. Inhibiting or cocatalytic effect of water and other additives on cationic polymerization of cyclodimethylsiloxanes. In Makromolekulare Chemie. Macromolecular Symposia; Hüthig & Wepf Verlag: Basel, Switzerland, 1991; Volume 42, pp. 229–240. [Google Scholar]
- Djinović, V.; Antić, V.; Djonlagić, J.; Govedarica, M. Synthesis of α,ω-dicarboxypropyl oligodimethylsiloxanes by ion-exchange resin catalyzed equilibration polymerization. React. Funct. Polym. 2000, 44, 299–306. [Google Scholar] [CrossRef]
- Cancouët, P.; Daudet, E.; Hélary, G.; Moreau, M.; Sauvet, G. Functional Polysiloxanes. I. Microstructure of Poly(Hydrogenmethylsiloxane-Co-Dimethylsiloxane)s Obtained by Cationic Copolymerization. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 826–836. [Google Scholar]
- Ganachaud, F.; Boileau, S. Siloxane-Containing Polymers. In Handbook of Ring-Opening Polymerization; Dubois, P., Coulembier, O., Raquez, J.-M., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 69–95. [Google Scholar]
- Vallejo-Montesinos, J.; Villegas, A.; Jacobo-Azuara, A.; Martínez, J.M.; Ramírez-Oliva, E.; Romero-Izquierdo, A.; Cervantes, J. Synthetic and Natural Silica-Aluminates as Inorganic Acidic Catalysts in Ring Opening Polymerization of Cyclosiloxanes. Appl. Organomet. Chem. 2012, 26, 362–368. [Google Scholar] [CrossRef]
- Kherroub, D.E.; Belbachir, M.; Lamouri, S. Cationic Ring Opening Polymerization of E-Caprolactam by a Montmorillonite Clay Catalyst. Bull. Chem. React. Eng. Catal. 2014, 9, 74–80. [Google Scholar] [CrossRef]
- Amine, H.; Karima, O.; el Amine, B.M.; Belbachir, M.; Meghabar, R. Cationic Ring Opening Polymerization of Glycolide Catalysed by a Montmorillonite Clay Catalyst. J. Polym. Res. 2005, 12, 361–365. [Google Scholar] [CrossRef]
- Kherroub, D.E.; Belbachir, M.; Lamouri, S. Synthesis and Characterization of Polyvinylmethylsiloxanes by Cationic Polymerization Using a Solid Green Catalyst. E-Polymers 2017, 17, 439–448. [Google Scholar] [CrossRef]
- Chakraborty, R.; Soucek, M.D. Synthesis of Amine and Epoxide Telechelic Siloxanes. Macromol. Chem. Phys. 2008, 209, 604–614. [Google Scholar] [CrossRef]
- Antić, V.V.; Antić, M.P.; Govedarica, M.N.; Dvornić, P.R. Kinetics of the Formation of Poly(Methyldecylsiloxane) by Hydrosilylation of Poly(Methylhydrosiloxane) and 1-Decene. Mater. Sci. Forum 2007, 555, 485–490. [Google Scholar] [CrossRef]
- Gorodov, V.V.; Tikhonov, P.A.; Buzin, M.I.; Vasil’ev, V.G.; Milenin, S.A.; Shragin, D.I.; Papkov, V.S.; Muzafarov, A.M. Synthesis and Thermal and Rheological Properties of Polydimethylsiloxanes Modified with Benzoic Acid Fragments. Polym. Sci.-Ser. B 2018, 60, 290–298. [Google Scholar] [CrossRef]
- Klosowsli, J.M.; Severson, S.S. Compositions comprising mercapto-functional organosilicon compounds. U.S. Patent 5994456, 30 November 1999. [Google Scholar]
- Temnikov, M.N.; Kononevich, Y.N.; Meshkov, I.B.; Buzin, M.I.; Vasil’ev, V.G.; Nikiforova, G.G.; Muzafarov, A.M. Simple and Fast Method for Producing Flexible Superhydrophobic Aerogels by Direct Formation of Thiol-Ene Networks in ScCO2. Polymer 2018, 138, 255–266. [Google Scholar] [CrossRef]
- Yilgör, I.; Riffle, J.S.; McGRATH, J.E. Reactive Difunctional Siloxane Oligomers. In Reactive Oligomers; American Chemical Society: Washington, DC, USA, 1985; pp. 161–174. [Google Scholar]
- Riffle, J.S.; Freelin, R.G.; Banthia, A.K.; McGrath, J.E. Interfacial Synthesis Part II: Phase-Transfer Catalyzed Synthesis of Polycarbonate/Polysiloxane Block Copolymers. J. Macromol. Sci. Part A-Chem. 1981, 15, 967–998. [Google Scholar] [CrossRef]
- Kojima, K.; Gore, C.R.; Marvel, C.S. Preparation of polysiloxanes having terminal carboxyl or hydroxyl groups. J. Polym. Sci. Part A—1 Polym. Chem. 1966, 4, 2325–2327. [Google Scholar] [CrossRef]
- Gorodov, V.V.; Demchenko, N.V.; Buzin, M.I.; Shragin, D.I.; Papkov, V.S.; Muzafarov, A.M. Synthesis and thermal and rheological properties of carboxyl-containing polydimethylsiloxanes. Russ. Chem. Bull. 2017, 66, 1290–1299. [Google Scholar] [CrossRef]
- Gorodov, V.V.; Milenin, S.A.; Demchenko, N.V.; Muzafarov, A.M. Carboxyl-Containing Polydimethylsiloxanes: Synthesis and Properties. INEOS Open 2020, 3, 43–54. [Google Scholar] [CrossRef]
- Drozdov, F.V.; Milenin, S.A.; Gorodov, V.V.; Demchenko, N.V.; Buzin, M.I.; Muzafarov, A.M. Crosslinked Polymers Based on Polyborosiloxanes: Synthesis and Properties. J. Organomet. Chem. 2019, 891, 72–77. [Google Scholar] [CrossRef]
- Tasic, A.M.; Pergal, M.V.; Antic, M.P.; Antic, V.V. Synthesis, Structure and Thermogravimetric Analysis of α,ω-Telechelic Polydimethylsiloxanes of Low Molecular Weight. J. Serb. Chem. Soc. 2017, 82, 1395–1416. [Google Scholar] [CrossRef]
- Cheesman, B.T.; Gates, P.J.; Castle, T.C.; Cosgrove, T.; Prescott, S.W. Linear and Star Architecture Methacrylate-Functionalised PDMS. Mater. Today Commun. 2015, 3, 122–129. [Google Scholar] [CrossRef]
- Drozdov, F.V.; Tarasenkov, A.N.; Cherkaev, G.V.; Demchenko, N.V.; Buzin, M.I.; Leites, L.A.; Muzafarov, A.M. Synthesis and Properties of Prepolymers and Their Siloxane Analogues by Thiol-Ene Polyaddition of Limonene with Dithiols. Polym. Int. 2019, 68, 2017–2023. [Google Scholar] [CrossRef]
- Zheng, C.; Qu, L.; Wang, Y.; Xu, T.; Zhang, C. Thermal Insulation and Stability of Polysiloxane Foams Containing Hydroxyl-Terminated Polydimethylsiloxanes. RSC Adv. 2018, 8, 9901–9909. [Google Scholar] [CrossRef] [PubMed]
- Talalaeva, E.V.; Kalinina, A.A.; Chernov, E.V.; Khmelnitskaia, A.G.; Obrezkova, M.A.; Cherkaev, G.V.; Muzafarov, A.M. Synthesis of 1,1,3,3,5,5-Hexamethyl-7, 7-Diorganocyclotetrasiloxanes and Its Copolymers. Polymers 2022, 14, 28. [Google Scholar] [CrossRef]
- Cao, J.; Zuo, Y.; Lu, H.; Yang, Y.; Feng, S. An Unconventional Chromophore in Water-Soluble Polysiloxanes Synthesized via Thiol-Ene Reaction for Metal Ion Detection. J. Photochem. Photobiol. A Chem. 2018, 350, 152–163. [Google Scholar] [CrossRef]
- Mukbaniani, O.; Aneli, J.; Esartia, I.; Tatrishvili, T.; Markarashvili, E.; Jalagonia, N. Siloxane Oligomers with Epoxy Pendant Groups. Macromol. Symp. 2013, 328, 25–37. [Google Scholar] [CrossRef]
- Mukbaniani, O.; Brostow, W.; Aneli, J.; Tatrishvili, T.; Markarashvili, E.; Chigvinadze, M.; Esartia, I. Synthesis and Ionic Conductivity of Siloxane Based Polymer Electrolytes with Pendant Propyl Acetoacetate Groups. Pure Appl. Chem. 2018, 90, 989–999. [Google Scholar] [CrossRef]
- Sheima, Y.; Yuts, Y.; Frauenrath, H.; Opris, D.M. Polysiloxanes Modified with Different Types and Contents of Polar Groups: Synthesis, Structure, and Thermal and Dielectric Properties. Macromolecules 2021, 54, 5737–5749. [Google Scholar] [CrossRef]
- Milenin, S.A.; Khairova, R.R.; Myakushev, V.D.; Buzin, A.I.; Vasiliev, V.G.; Stoikov, I.I.; Muzafarov, A.M. Synthesis of New Organoelement Copolymers Based on Polydimethylsiloxanes and Aminophosphonates. J. Organomet. Chem. 2018, 870, 110–115. [Google Scholar] [CrossRef]
- Perju, E.; Cuervo-Reyes, E.; Shova, S.; Opris, D.M. Synthesis of Novel Cyclosiloxane Monomers Containing Push-Pull Moieties and Their Anionic Ring Opening Polymerization. RSC Adv. 2018, 8, 7569–7578. [Google Scholar] [CrossRef] [PubMed]
- Dünki, S.J.; Nüesch, F.A.; Opris, D.M. Elastomers with Tunable Dielectric and Electromechanical Properties. J. Mater. Chem. C 2016, 4, 10545–10553. [Google Scholar] [CrossRef]
- Kim, E.E.; Kononevich, Y.N.; Anisimov, A.A.; Buzin, M.I.; Vasil’ev, V.G.; Korlyukov, A.A.; Ionov, D.S.; Khanin, D.A.; Shtykova, E.V.; Volkov, V.V.; et al. Cross-Linked Polymer Networks Based on Polysiloxane and Nickel β-Diketonate Precursors. React. Funct. Polym. 2021, 164, 104896. [Google Scholar] [CrossRef]
- Bodkhe, R.B.; Stafslien, S.J.; Cilz, N.; Daniels, J.; Thompson, S.E.M.; Callow, M.E.; Callow, J.A.; Webster, D.C. Polyurethanes with Amphiphilic Surfaces Made Using Telechelic Functional PDMS Having Orthogonal Acid Functional Groups. Prog. Org. Coat. 2012, 75, 38–48. [Google Scholar] [CrossRef]
- Guo, M.; Huang, Y.; Chen, Z.; Zhang, Y.; Zhang, Y.; Zhu, M.; Zhang, J.; Feng, S. Preparation and Properties of Benzylsulfonyl-Containing Silicone Copolymers via Ring-Opening Copolymerization of Macroheterocyclosiloxane and Cyclosiloxane. Chem. A Eur. J. 2021, 27, 7897–7907. [Google Scholar] [CrossRef]
- Fei, H.F.; Han, X.; Liu, B.; Gao, X.; Wang, Q.; Zhang, Z.; Xie, Z. Synthesis of Gradient Copolysiloxanes by Simultaneous Copolymerization of Cyclotrisiloxanes and Mechanism for Kinetics Inverse between Anionic and Cationic Ring-Opening Polymerization. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 835–843. [Google Scholar] [CrossRef]
- Indulekha, K.; Roy, R.E.; Vishnu, A.G.; Rajeev, R.S.; Ninan, K.N.; Gouri, C. Silicone Copolymers Bearing Reactive Vinyl and Hydride Functionalities: Synthesis, Characterisation and Particulate Composite Thereof for Specialty Applications. Mater. Chem. Phys. 2018, 206, 213–223. [Google Scholar] [CrossRef]
- Isaacman, M.J.; Barron, K.A.; Theogarajan, L.S. Clickable Amphiphilic Triblock Copolymers. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2319–2329. [Google Scholar] [CrossRef]
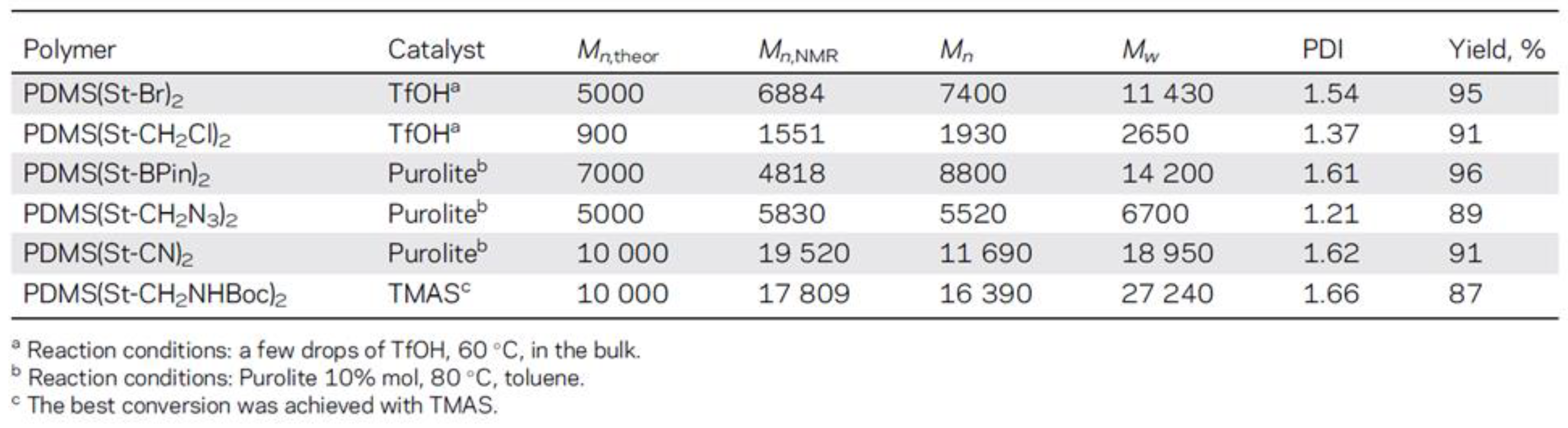
- Milenin, S.A.; Drozdov, F.V.; Bezlepkina, K.A.; Majorov, V.Y.; Muzafarov, A.M. Acid-Catalyzed Rearrangement of Azidopropyl-Siloxane Monomers for the Synthesis of Azidopropyl-Polydimethylsiloxane and Their Carboxylic Acid Derivatives. Macromolecules 2021, 54, 2921–2935. [Google Scholar] [CrossRef]









































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezlepkina, K.A.; Milenin, S.A.; Vasilenko, N.G.; Muzafarov, A.M. Ring-Opening Polymerization (ROP) and Catalytic Rearrangement as a Way to Obtain Siloxane Mono- and Telechelics, as Well as Well-Organized Branching Centers: History and Prospects. Polymers 2022, 14, 2408. https://doi.org/10.3390/polym14122408
Bezlepkina KA, Milenin SA, Vasilenko NG, Muzafarov AM. Ring-Opening Polymerization (ROP) and Catalytic Rearrangement as a Way to Obtain Siloxane Mono- and Telechelics, as Well as Well-Organized Branching Centers: History and Prospects. Polymers. 2022; 14(12):2408. https://doi.org/10.3390/polym14122408
Chicago/Turabian StyleBezlepkina, Kseniya A., Sergey A. Milenin, Natalia G. Vasilenko, and Aziz M. Muzafarov. 2022. "Ring-Opening Polymerization (ROP) and Catalytic Rearrangement as a Way to Obtain Siloxane Mono- and Telechelics, as Well as Well-Organized Branching Centers: History and Prospects" Polymers 14, no. 12: 2408. https://doi.org/10.3390/polym14122408
APA StyleBezlepkina, K. A., Milenin, S. A., Vasilenko, N. G., & Muzafarov, A. M. (2022). Ring-Opening Polymerization (ROP) and Catalytic Rearrangement as a Way to Obtain Siloxane Mono- and Telechelics, as Well as Well-Organized Branching Centers: History and Prospects. Polymers, 14(12), 2408. https://doi.org/10.3390/polym14122408