
Synthesis and Phase Behavior of a Platform of CO2-Soluble Functional Gradient Copolymers Bearing Metal-Complexing Units

 , ,
, ,  ,
,
Abstract
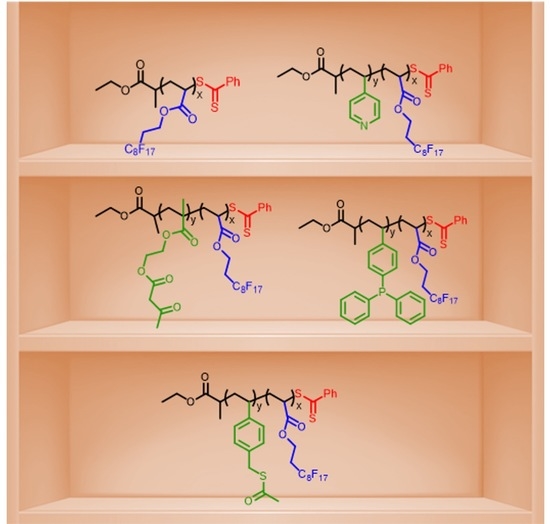
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Monomer and Polymer Syntheses
2.2.1. Synthesis of S-(Vinylbenzyl) Ethanethioate (StySAc) Monomer
2.2.2. Synthesis of Poly(1,1,2,2-tetrahydroperfluorodecylacrylate) (P(FDA)) Homopolymer
2.2.3. Synthesis of Poly(1,1,2,2-tetrahydroperfluorodecylacrylate-co-4-vinylpyridine) (P(4VP-co-FDA)) Copolymer
2.2.4. Synthesis of Poly(1,1,2,2-tetrahydroperfluorodecylacrylate-co-acetoacetoxyethyl Methacrylate) (P(AAEM-co-FDA)) Copolymer
2.2.5. Synthesis of poly(1,1,2,2-tetrahydroperfluorodecylacrylate-co-4-(diphenylphosphino)styrene) (P(DPPS-co-FDA)) Copolymer
2.2.6. Synthesis of Poly(1,1,2,2-tetrahydroperfluorodecylacrylate-co-S-(Vinylbenzyl) Ethanethioate) (P(StySAc-co-FDA)) Copolymer
2.3. Aminolysis of the Polymers
2.4. Characterizations
2.4.1. Cloud Point Measurements
2.4.2. Nuclear Magnetic Resonance Spectroscopy (NMR)
2.4.3. Thermogravimetric Analysis (TGA)
2.4.4. Differential Scanning Calorimetry (DSC)
2.4.5. MALDI-TOF-MS Mass Spectrometry
3. Results and Discussion
3.1. Synthesis and Characterization of the (Co)Polymers before Aminolysis
3.2. Aminolysis of the (Co)Polymers
3.3. Thermal Polymers Characterization Pre- and Post-Aminolysis
3.4. Polymers Phase Behavior in Dense CO2
3.5. Ability of Polymers to Complex Metals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Seehra, M.S.; Bristow, A.D. Introductory Chapter: Overview of the Properties and Applications of Noble and Precious Metals. In Noble and Precious Metals—Properties, Nanoscale Effects and Applications; InTech: London, UK, 2018. [Google Scholar]
- Jowitt, S.M.; Werner, T.T.; Weng, Z.; Mudd, G.M. Recycling of the Rare Earth Elements. Curr. Opin. Green Sustain. Chem. 2018, 13, 1–7. [Google Scholar] [CrossRef]
- Ciriminna, R.; Pagliaro, M.; Luque, R. Heterogeneous Catalysis under Flow for the 21st Century Fine Chemical Industry. Green Energy Environ. 2021, 6, 161–166. [Google Scholar] [CrossRef]
- Taek Rim, K.; Ho Koo, K.; Sun Park, J. Toxicological Evaluations of Rare Earths and Their Health Impacts to Workers: A Literature Review. Saf. Health Work 2013, 4, 12–26. [Google Scholar] [CrossRef]
- Sunarso, J.; Ismadji, S. Decontamination of Hazardous Substances from Solid Matrices and Liquids Using Supercritical Fluids Extraction: A Review. J. Hazard. Mater. 2009, 161, 1–20. [Google Scholar] [CrossRef]
- Zhang, K.; Li, X.; Song, Z.; Yan, J.; Chen, M.; Yin, J. Human Health Risk Distribution and Safety Threshold of Cadmium in Soil of Coal Chemical Industry Area. Minerals 2021, 11, 678. [Google Scholar] [CrossRef]
- Reck, B.K.; Graedel, T.E. Challenges in Metal Recycling. Science 2012, 337, 690–695. [Google Scholar] [CrossRef]
- Lahiani, A.; Mefteh-Wali, S.; Vasbieva, D.G. The Safe-Haven Property of Precious Metal Commodities in the COVID-19 Era. Resour. Policy 2021, 74, 102340. [Google Scholar] [CrossRef]
- Kumar Jha, M.; Kumari, A.; Kumari Jha, A.; Kumar, V.; Hait, J.; Dhar Pandey, B. Recovery of Lithium and Cobalt from Waste Lithium Ion Batteries of Mobile Phone. Waste Manag. 2013, 33, 1890–1897. [Google Scholar] [CrossRef]
- Jiang, D.; Tan, M.; Guo, Q.; Yan, S. Transfer of Heavy Metal along Food Chain: A Mini-Review on Insect Susceptibility to Entomopathogenic Microorganisms under Heavy Metal Stress. Pest Manag. Sci. 2021, 77, 1115–1120. [Google Scholar] [CrossRef]
- Song, P.; Xu, D.; Yue, J.; Ma, Y.; Dong, S.; Feng, J. Recent Advances in Soil Remediation Technology for Heavy Metal Contaminated Sites: A Critical Review. Sci. Total Environ. 2022, 838, 156417. [Google Scholar] [CrossRef]
- Long, Z.; Huang, Y.; Zhang, W.; Shi, Z.; Yu, D.; Chen, Y.; Liu, C.; Wang, R. Effect of Different Industrial Activities on Soil Heavy Metal Pollution, Ecological Risk, and Health Risk. Environ. Monit. Assess. 2021, 193, 20. [Google Scholar] [CrossRef]
- Deboer, M.A.; Lammertsma, K. Scarcity of Rare Earth Elements. ChemSusChem 2013, 6, 2045–2055. [Google Scholar] [CrossRef]
- Işıldar, A.; Rene, E.R.; van Hullebusch, E.D.; Lens, P.N.L. Electronic Waste as a Secondary Source of Critical Metals: Management and Recovery Technologies. Resour. Conserv. Recycl. 2018, 135, 296–312. [Google Scholar] [CrossRef]
- Tunsu, C.; Petranikova, M.; Gergorić, M.; Ekberg, C.; Retegan, T. Reclaiming Rare Earth Elements from End-of-Life Products: A Review of the Perspectives for Urban Mining Using Hydrometallurgical Unit Operations. Hydrometallurgy 2015, 156, 239–258. [Google Scholar] [CrossRef]
- Pereira, E.B.; Suliman, A.L.; Tanabe, E.H.; Bertuol, D.A. Recovery of Indium from Liquid Crystal Displays of Discarded Mobile Phones Using Solvent Extraction. Miner. Eng. 2018, 119, 67–72. [Google Scholar] [CrossRef]
- Chen, Y.; Qiao, Q.; Cao, J.; Li, H.; Bian, Z. Precious Metal Recovery. Joule 2021, 5, 3097–3115. [Google Scholar] [CrossRef]
- Wan Ngah, W.S.; Hanafiah, M.A.K.M. Removal of Heavy Metal Ions from Wastewater by Chemically Modified Plant Wastes as Adsorbents: A Review. Bioresour. Technol. 2008, 99, 3935–3948. [Google Scholar] [CrossRef]
- Ko, D.; Lee, J.S.; Patel, H.A.; Jakobsen, M.H.; Hwang, Y.; Yavuz, C.T.; Hansen, H.C.B.; Andersen, H.R. Selective Removal of Heavy Metal Ions by Disulfide Linked Polymer Networks. J. Hazard. Mater. 2017, 332, 140–148. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, Z.; Zhang, F.S. Direct Extraction of Palladium and Silver from Waste Printed Circuit Boards Powder by Supercritical Fluids Oxidation-Extraction Process. J. Hazard. Mater. 2016, 318, 216–223. [Google Scholar] [CrossRef]
- Barakat, M.A. New Trends in Removing Heavy Metals from Industrial Wastewater. Arab. J. Chem. 2011, 4, 361–377. [Google Scholar] [CrossRef]
- He, J.; Kappler, A. Recovery of Precious Metals from Waste Streams. Microb. Biotechnol. 2017, 10, 1194–1198. [Google Scholar] [CrossRef]
- Taghvaie Nakhjiri, A.; Sanaeepur, H.; Ebadi Amooghin, A.; Shirazi, M.M.A. Recovery of Precious Metals from Industrial Wastewater towards Resource Recovery and Environmental Sustainability: A Critical Review. Desalination 2022, 527, 115510. [Google Scholar] [CrossRef]
- Islam, A.; Swaraz, A.M.; Teo, S.H.; Taufiq-Yap, Y.H.; Vo, D.-V.N.; Ibrahim, M.L.; Abdulkreem-Alsultan, G.; Rashid, U.; Awual, M.R. Advances in Physiochemical and Biotechnological Approaches for Sustainable Metal Recovery from E-Waste: A Critical Review. J. Clean. Prod. 2021, 323, 129015. [Google Scholar] [CrossRef]
- Krishnan, S.; Zulkapli, N.S.; Kamyab, H.; Taib, S.M.; Din, M.F.B.M.; Majid, Z.A.; Chaiprapat, S.; Kenzo, I.; Ichikawa, Y.; Nasrullah, M.; et al. Current Technologies for Recovery of Metals from Industrial Wastes: An Overview. Environ. Technol. Innov. 2021, 22, 101525. [Google Scholar] [CrossRef]
- Yaashikaa, P.R.; Priyanka, B.; Senthil Kumar, P.; Karishma, S.; Jeevanantham, S.; Indraganti, S. A Review on Recent Advancements in Recovery of Valuable and Toxic Metals from E-Waste Using Bioleaching Approach. Chemosphere 2022, 287, 132230. [Google Scholar] [CrossRef]
- Xi, J.; Ji, G.; Liao, Y.; Wu, Y.; Liu, Q.; Li, M. Research on Separation and Extraction of Valuable Metals from Complex Non-Ferrous Metals Resources by High Pressure Oxygen Leaching Methodology: A Review. J. Sustain. Metall. 2022, 8, 51–63. [Google Scholar] [CrossRef]
- Abbott, A.P.; Frisch, G.; Hartley, J.; Ryder, K.S. Processing of Metals and Metal Oxides Using Ionic Liquids. Green Chem. 2011, 13, 471–481. [Google Scholar] [CrossRef]
- AliAkbari, R.; Marfavi, Y.; Kowsari, E.; Ramakrishna, S. Recent Studies on Ionic Liquids in Metal Recovery from E-Waste and Secondary Sources by Liquid-Liquid Extraction and Electrodeposition: A Review. Mater. Circ. Econ. 2020, 2, 10. [Google Scholar] [CrossRef]
- Cevasco, G.; Chiappe, C. Are Ionic Liquids a Proper Solution to Current Environmental Challenges? Green Chem. 2014, 16, 2375–2385. [Google Scholar] [CrossRef]
- Makanyire, T.; Sanchez-Segado, S.; Jha, A. Separation and Recovery of Critical Metal Ions Using Ionic Liquids. Adv. Manuf. 2016, 4, 33–46. [Google Scholar] [CrossRef]
- Al Hamouz, O.C.S.; Ali, S.A. Removal of Heavy Metal Ions Using a Novel Cross-Linked Polyzwitterionic Phosphonate. Sep. Purif. Technol. 2012, 98, 94–101. [Google Scholar] [CrossRef]
- Gomes, C.P.; Almeida, M.F.; Loureiro, J.M. Gold Recovery with Ion Exchange Used Resins. Sep. Purif. Technol. 2001, 24, 35–57. [Google Scholar] [CrossRef]
- Lebron, Y.A.R.; Moreira, V.R.; Amaral, M.C.S. Metallic Ions Recovery from Membrane Separation Processes Concentrate: A Special Look onto Ion Exchange Resins. Chem. Eng. J. 2021, 425, 131812. [Google Scholar] [CrossRef]
- Strauss, M.L.; Diaz, L.A.; McNally, J.; Klaehn, J.; Lister, T.E. Separation of Cobalt, Nickel, and Manganese in Leach Solutions of Waste Lithium-Ion Batteries Using Dowex M4195 Ion Exchange Resin. Hydrometallurgy 2021, 206, 105757. [Google Scholar] [CrossRef]
- Riley, A.L.; Porter, C.P.; Ogden, M.D. Selective Recovery of Copper from a Synthetic Metalliferous Waste Stream Using the Thiourea-Functionalized Ion Exchange Resin Puromet MTS9140. Eng 2021, 2, 512–530. [Google Scholar] [CrossRef]
- Elfeghe, S.; Sheng, Q.; Zhang, Y. Separation of Lead and Copper Ions in Acidic Media Using an Ion-Exchange Resin with a Thiourea Functional Group. ACS Omega 2022, 7, 13042–13049. [Google Scholar] [CrossRef]
- Nan, J.; Han, D.; Zuo, X. Recovery of Metal Values from Spent Lithium-Ion Batteries with Chemical Deposition and Solvent Extraction. J. Power Sources 2005, 152, 278–284. [Google Scholar] [CrossRef]
- Alvial-Hein, G.; Mahandra, H.; Ghahreman, A. Separation and Recovery of Cobalt and Nickel from End of Life Products via Solvent Extraction Technique: A Review. J. Clean. Prod. 2021, 297, 126592. [Google Scholar] [CrossRef]
- Rao, M.D.; Singh, K.K.; Morrison, C.A.; Love, J.B. Recycling Copper and Gold from E-Waste by a Two-Stage Leaching and Solvent Extraction Process. Sep. Purif. Technol. 2021, 263, 118400. [Google Scholar] [CrossRef]
- Punt, T.; Akdogan, G.; Bradshaw, S.; van Wyk, P. Development of a Novel Solvent Extraction Process Using Citric Acid for Lithium-Ion Battery Recycling. Miner. Eng. 2021, 173, 107204. [Google Scholar] [CrossRef]
- Lei, S.; Sun, W.; Yang, Y. Solvent Extraction for Recycling of Spent Lithium-Ion Batteries. J. Hazard. Mater. 2022, 424, 127654. [Google Scholar] [CrossRef]
- Wang, J.; Xu, W.; Liu, H.; Yu, F.; Wang, H. Extractant Structures and Their Performance for Palladium Extraction and Separation from Chloride Media: A Review. Miner. Eng. 2021, 163, 106798. [Google Scholar] [CrossRef]
- Wai, C.M.; Wang, S.; Yu, J.-J. Solubility Parameters and Solubilities of Metal Dithiocarbamates in Supercritical Carbon Dioxide. Anal. Chem. 1996, 68, 3516–3519. [Google Scholar] [CrossRef]
- Özel, M.Z.; Burford, M.D.; Clifford, A.A.; Bartle, K.D.; Shadrin, A.; Smart, N.G.; Tinker, N.D. Supercritical Fluid Extraction of Cobalt with Fluorinated and Non-Fluorinated β-Diketones. Anal. Chim. Acta 1997, 346, 73–80. [Google Scholar] [CrossRef]
- Halili, J.; Mele, A.; Arbneshi, T.; Mazreku, I. Supercritical CO2 Extraction of Heavy Metals Cu, Zn and Cd from Aqueous Solution Using D Ithizone as Chelating Agent. Am. J. Appl. Sci. 2015, 12, 284–289. [Google Scholar] [CrossRef]
- Mochizuki, S.; Wada, N.; Smith, R.L., Jr.; Inomata, H. Supercritical Fluid Extraction of Alkali Metal Ions Using Crown Ethers with Perfluorocarboxylic Acid from Aqueous Solution. Anal. Commun. 1999, 36, 51–52. [Google Scholar] [CrossRef]
- Fayaz, S.M.; Abdoli, M.A.; Baghdadi, M.; Karbasi, A. Ag Removal from E-Waste Using Supercritical Fluid: Improving Efficiency and Selectivity. Int. J. Environ. Stud. 2021, 78, 459–473. [Google Scholar] [CrossRef]
- Hsu, E.; Durning, C.J.; West, A.C.; Park, A.-H.A. Enhanced Extraction of Copper from Electronic Waste via Induced Morphological Changes Using Supercritical CO2. Resour. Conserv. Recycl. 2021, 168, 105296. [Google Scholar] [CrossRef]
- Fayaz, S.M.; Abdoli, M.A.; Baghdadi, M.; Karbassi, A.R. Extraction of Silver from Computer Printed Circuit Boards Wastes by Supercritical Fluids: Pretreatment Study. Int. J. Environ. Sci. Technol. 2022, 19, 4883–4890. [Google Scholar] [CrossRef]
- Van Dyk, L.D.; Mawire, G.; Potgieter, J.H.; Dworzanowski, M. Selection of a Suitable Ligand for the Supercritical Extraction of Gold from a Low-Grade Refractory Tailing. J. Supercrit. Fluids 2022, 179, 105415. [Google Scholar] [CrossRef]
- Chirat, M.; Ribaut, T.; Clerc, S.; Charton, F.; Fournel, B.; Lacroix-Desmazes, P. Extraction of Cobalt Ion from Textile Using a Complexing Macromolecular Surfactant in Supercritical Carbon Dioxide. Ind. Eng. Chem. Res. 2012, 52, 538–542. [Google Scholar] [CrossRef]
- Li, W.S.J.; Gasc, F.; Pinot, J.; Causse, J.; Poirot, H.; Pinaud, J.; Bouilhac, C.; Simonaire, H.; Barth, D.; Lacroix-Desmazes, P. Extraction of Palladium from Alumina-Supported Catalyst in Supercritical CO2 Using Functional Fluorinated Polymers. J. Supercrit. Fluids 2018, 138, 207–214. [Google Scholar] [CrossRef]
- Ruiu, A.; Bauer-Siebenlist, B.; Senila, M.; Jänisch, T.; Foix, D.; Seaudeau-Pirouley, K.; Lacroix-Desmazes, P. Promising Polymer-Assisted Extraction of Palladium from Supported Catalysts in Supercritical Carbon Dioxide. J. CO2 Util. 2020, 41, 101232. [Google Scholar] [CrossRef]
- Ruiu, A.; Bauer-Siebenlist, B.; Senila, M.; Li, W.S.J.; Seaudeau-Pirouley, K.; Lacroix-Desmazes, P.; Jänisch, T. Supercritical CO2 Extraction of Palladium Oxide from an Aluminosilicate-Supported Catalyst Enhanced by a Combination of Complexing Polymers and Piperidine. Molecules 2021, 26, 684. [Google Scholar] [CrossRef]
- Stoychev, I.; Peters, F.; Kleiner, M.; Clerc, S.; Ganachaud, F.; Chirat, M.; Fournel, B.; Sadowski, G.; Lacroix-Desmazes, P. Phase Behavior of Poly(Dimethylsiloxane)-Poly(Ethylene Oxide) Amphiphilic Block and Graft Copolymers in Compressed Carbon Dioxide. J. Supercrit. Fluids 2012, 62, 211–218. [Google Scholar] [CrossRef]
- Parzuchowski, P.G.; Gregorowicz, J.; Fras, Z.; Wawrzyńska, E.P.; Brudzyńska, E.; Rokicki, G. Hyperbranched Poly(Ether-Siloxane) Amphiphiles of Surprisingly High Solubility in Supercritical Carbon Dioxide. J. Supercrit. Fluids 2014, 95, 222–227. [Google Scholar] [CrossRef]
- Tapriyal, D.; Wang, Y.; Enick, R.M.; Johnson, J.K.; Crosthwaite, J.; Thies, M.C.; Paik, I.H.; Hamilton, A.D. Poly(Vinyl Acetate), Poly((1-O-(Vinyloxy) Ethyl-2,3,4,6-Tetra-O-Acetyl-β-d-Glucopyranoside) and Amorphous Poly(Lactic Acid) Are the Most CO2-Soluble Oxygenated Hydrocarbon-Based Polymers. J. Supercrit. Fluids 2008, 46, 252–257. [Google Scholar] [CrossRef]
- Zhang, S.; Luo, Y.; Yang, H.; Yang, H.-J.; Tan, B. Functional Oligo(Vinyl Acetate) Bearing Bipyridine Moieties by RAFT Polymerization and Extraction of Metal Ions in Supercritical Carbon Dioxide. Polym. Chem. 2013, 4, 3507–3513. [Google Scholar] [CrossRef]
- Rindfleisch, F.; DiNoia, T.P.; McHugh, M.A. Solubility of Polymers and Copolymers in Supercritical CO2. J. Phys. Chem. 1996, 100, 15581–15587. [Google Scholar] [CrossRef]
- Canelas, D.A.; Betts, D.E.; DeSimone, J.M. Dispersion Polymerization of Styrene in Supercritical Carbon Dioxide: Importance of Effective Surfactants. Macromolecules 1996, 29, 2818–2821. [Google Scholar] [CrossRef]
- Lacroix-Desmazes, P.; Andre, P.; Desimone, J.M.; Ruzette, A.-V.; Boutevin, B. Macromolecular Surfactants for Supercritical Carbon Dioxide Applications: Synthesis and Characterization of Fluorinated Block Copolymers Prepared by Nitroxide-Mediated Radical Polymerization. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 3537–3552. [Google Scholar] [CrossRef]
- André, P.; Lacroix-Desmazes, P.; Taylor, D.K.; Boutevin, B. Solubility of Fluorinated Homopolymer and Block Copolymer in Compressed CO2. J. Supercrit. Fluids 2006, 37, 263–270. [Google Scholar] [CrossRef]
- Renault, B.; Cloutet, E.; Lacroix-Desmazes, P.; Cramail, H. Synthesis of Polyurethane/Poly(1,1,2,2-Tetrahydroperfluorodecyl Acrylate) Particles in Supercritical Carbon Dioxide. Macromol. Chem. Phys. 2008, 209, 535–543. [Google Scholar] [CrossRef]
- Ribaut, T.; Oberdisse, J.; Annighofer, B.; Fournel, B.; Sarrade, S.; Haller, H.; Lacroix-Desmazes, P. Solubility and Self-Assembly of Amphiphilic Gradient and Block Copolymers in Supercritical CO2. J. Phys. Chem. B 2011, 115, 836–843. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process—A Third Update. Aust. J. Chem. 2012, 65, 985. [Google Scholar] [CrossRef]
- Stephenson, T.A.; Wilkinson, G. New Complexes of Ruthenium (II) and (III) with Triphenylphosphine, Triphenylarsine, Trichlorostannate, Pyridine and Other Ligands. J. Inorg. Nucl. Chem. 1966, 28, 945–956. [Google Scholar] [CrossRef]
- Anwar, A.; Minhaz, A.; Khan, N.A.; Kalantari, K.; Afifi, A.B.M.; Shah, M.R. Synthesis of Gold Nanoparticles Stabilized by a Pyrazinium Thioacetate Ligand: A New Colorimetric Nanosensor for Detection of Heavy Metal Pd(II). Sens. Actuators B Chem. 2018, 257, 875–881. [Google Scholar] [CrossRef]
- Hancock, R.D.; Martell, A.E. Ligand Design for Selective Complexation of Metal Ions in Aqueous Solution. Chem. Rev. 1989, 89, 1875–1914. [Google Scholar] [CrossRef]
- Matlock, M.M.; Howerton, B.S.; Henke, K.R.; Atwood, D.A. A Pyridine-Thiol Ligand with Multiple Bonding Sites for Heavy Metal Precipitation. J. Hazard. Mater. 2001, 82, 55–63. [Google Scholar] [CrossRef]
- Fukuda, Y.; Nakao, A.; Hayashi, K. Syntheses and Specific Structures of Higher-Order Mixed Chelate Lanthanide Complexes Containing Terpyridine, Acetylacetonate, and Nitrate Ligands. J. Chem. Soc. Dalt. Trans. 2002, 527–533. [Google Scholar] [CrossRef]
- Woehrle, G.H.; Brown, L.O.; Hutchison, J.E. Thiol-Functionalized, 1.5-Nm Gold Nanoparticles through Ligand Exchange Reactions: Scope and Mechanism of Ligand Exchange. J. Am. Chem. Soc. 2005, 127, 2172–2183. [Google Scholar] [CrossRef]
- Severac, R.; Lacroix-Desmazes, P.; Boutevin, B. Reversible Addition-Fragmentation Chain-Transfer (RAFT) Copolymerization of Vinylidene Chloride and Methyl Acrylate. Polym. Int. 2002, 51, 1117–1122. [Google Scholar] [CrossRef]
- Guyot, B.; Boutevin, B. Détermination Des Coefficients d’Alfrey et Price de Monomères Acryliques Fluorés et Du Méthacrylate de Morpholinoéthyle. Eur. Polym. J. 1996, 32, 751–756. [Google Scholar] [CrossRef]
- Odian, G. Chain Polymerization, Princ. Polym, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004; pp. 500–502. [Google Scholar]
- Eastman Chemical Company. Acetoacetoxyethyl Methacrylate (AAEM) Acetoacetyl Chemistry; Publication N-319C; Eastman Chemical Company: Kingport, TN, USA, 1999. [Google Scholar]
- Kondo, S.; Ohtsuka, T.; Ogura, K.; Tsuda, K. Convenient Synthesis and Free-Radical Copolymerization of p-Chloromethylstyrene. J. Macromol. Sci. Part A-Chem. 1979, 13, 767–775. [Google Scholar] [CrossRef]
- Ribaut, T.; Lacroix-Desmazes, P.; Fournel, B.; Sarrade, S. Synthesis of Gradient Copolymers with Complexing Groups by RAFT Polymerization and Their Solubility in Supercritical CO2. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 5448–5460. [Google Scholar] [CrossRef]
- Nakamura, S.; Fouquet, T.; Sato, H. Molecular Characterization of High Molecular Weight Polyesters by Matrix-Assisted Laser Desorption/Ionization High-Resolution Time-of-Flight Mass Spectrometry Combined with On-Plate Alkaline Degradation and Mass Defect Analysis. J. Am. Soc. Mass Spectrom. 2019, 30, 355–367. [Google Scholar] [CrossRef]
- Xu, J.; He, J.; Fan, D.; Wang, X.; Yang, Y. Aminolysis of Polymers with Thiocarbonylthio Termini Prepared by RAFT Polymerization: The Difference between Polystyrene and Polymethacrylates. Macromolecules 2006, 39, 8616–8624. [Google Scholar] [CrossRef]
- Willcock, H.; O’Reilly, R.K. End Group Removal and Modification of RAFT Polymers. Polym. Chem. 2010, 1, 149–157. [Google Scholar] [CrossRef]
- Mari, G.; De Crescentini, L.; Favi, G.; Santeusanio, S.; Mantellini, F. 1,2-Diaza-1,3-Diene-Based Multicomponent Reactions in Sequential Protocols to Synthesize Arylamino-5-Hydrazonothiophene-3-Carboxylates. Eur. J. Org. Chem. 2018, 2018, 6548–6556. [Google Scholar] [CrossRef]
- Haida, P.; Abetz, V. Acid-Mediated Autocatalysis in Vinylogous Urethane Vitrimers. Macromol. Rapid Commun. 2020, 41, 2000273. [Google Scholar] [CrossRef]
- Flanders, M.J.; Gramlich, W.M. Reversible-Addition Fragmentation Chain Transfer (RAFT) Mediated Depolymerization of Brush Polymers. Polym. Chem. 2018, 9, 2328–2335. [Google Scholar] [CrossRef]
- Wang, H.S.; Truong, N.P.; Pei, Z.; Coote, M.L.; Anastasaki, A. Reversing RAFT Polymerization: Near-Quantitative Monomer Generation Via a Catalyst-Free Depolymerization Approach. J. Am. Chem. Soc. 2022, 144, 4678–4684. [Google Scholar] [CrossRef] [PubMed]
- Legge, T.M.; Slark, A.T.; Perrier, S. Thermal Stability of Reversible Addition-Fragmentation Chain Transfer/Macromolecular Architecture Design by Interchange of Xanthates Chain-Transfer Agents. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 6980–6987. [Google Scholar] [CrossRef]
- Xu, J.; He, J.; Fan, D.; Tang, W.; Yang, Y. Thermal Decomposition of Dithioesters and Its Effect on RAFT Polymerization. Macromolecules 2006, 39, 3753–3759. [Google Scholar] [CrossRef]
- Lack, E.; Seidlitz, H. Commercial Scale Decaffeination of Coffee and Tea Using Supercritical CO2. In Extraction of Natural Products Using Near-Critical Solvents; Springer: Dordrecht, The Netherlands, 1993; pp. 101–139. [Google Scholar]
- Kirby, C.F.; McHugh, M.A. Phase Behavior of Polymers in Supercritical Fluid Solvents. Chem. Rev. 1999, 99, 565–602. [Google Scholar] [CrossRef]
- Kilic, S.; Wang, Y.; Johnson, J.K.; Beckman, E.J.; Enick, R.M. Influence of Tert-Amine Groups on the Solubility of Polymers in CO2. Polymer 2009, 50, 2436–2444. [Google Scholar] [CrossRef][Green Version]
- Kilic, S.; Michalik, S.; Wang, Y.; Johnson, J.K.; Enick, R.M.; Beckman, E.J. Phase Behavior of Oxygen-Containing Polymers in CO2. Macromolecules 2007, 40, 1332–1341. [Google Scholar] [CrossRef]
- Gasc, F.; Clerc, S.; Gayon, E.; Campagne, J.-M.; Lacroix-Desmazes, P. Supercritical CO2-Mediated Design of Pd Supported Catalysts Using an Amphiphilic Functional Copolymer. J. Supercrit. Fluids 2015, 105, 136–145. [Google Scholar] [CrossRef]
- Chirat, M.; Clerc, S.; Ribaut, T.; Charton, F.; Fournel, B.; Gayon, E.; Campagne, J.-M.; Lacroix-Desmazes, P. Supercritical CO2-soluble functional amphiphilic fluorinated copolymers synthsized by RAFT polymerization and their palladium and cobalt complexes. Polym. Prepr. 2011, 52, 709–710. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Co)polymer a | Mn,targeted c (g/mol) | DPcomplexing mono d | DPFDA d | Mn,NMR d (g/mol) | fcomplexing mono e (wt%) | fFDA f (wt%) | Yield g (%) |
|---|---|---|---|---|---|---|---|
| P(FDA) | 5020 | 0 | 10.8 | 5850 | 0 | 100 | 78 |
| P(4VP-co-FDA) b | 10,250 | 19.8 | 18.3 | 11,820 | 18.0 | 82.0 | 59 |
| P(AAEM-co-FDA) | 10,090 | 18.9 | 17.7 | 13,480 | 30.6 | 69.4 | 43 |
| P(DPPS-co-FDA) | 10,060 | 7.5 | 17.8 | 11,640 | 19.0 | 81.0 | 61 |
| P(StySAc-co-FDA) | 10,060 | 9.7 | 14.8 | 9790 | 19.6 | 80.4 | 55 |
| (m/z)expt | n | m | (m/z)calc a | ∆ = |(m/z)expt − (m/z)calc| |
|---|---|---|---|---|
| 2563.12 | 1 | 4 | 2563.19 | 0.069 |
| 2777.20 | 2 | 4 | 2777.27 | 0.070 |
| 2991.25 | 3 | 4 | 2991.35 | 0.100 |
| 3081.10 | 1 | 5 | 3081.20 | 0.100 |
| 3295.15 | 2 | 5 | 3295.29 | 0.140 |
| 3509.25 | 3 | 5 | 3509.37 | 0.120 |
| 3599.10 | 1 | 6 | 3599.22 | 0.120 |
| (Co)Polymer | TGA | DSC | ||
|---|---|---|---|---|
| T2wt%loss (°C) | Residual mass at 570 °C(%) | Tg a (°C) | Tm b (°C) | |
| P(FDA) | 263 | 0.3 | - | 63 |
| P(FDA)SH | 197 | 1.3 | - | 56 |
| P(4VP-co-FDA) | 249 | 9.3 | 61 | 85 |
| P(4VP-co-FDA)SH | 274 | 31.9 | 67 | 89 |
| P(AAEM-co-FDA) | 239 | 10.0 | 8 | 41 |
| P(AAEM-co-FDA)SH | 280 | 31.4 | 15 | 42 |
| P(DPPS-co-FDA) | 215 | 13.7 | 67 | - |
| P(DPPS-co-FDA)SH | 262 | 16.7 | 68 | - |
| P(StySAc-co-FDA) | 160 | 1.7 | 24 | 51 |
| P(StySH-co-FDA)SH | 298 | 6.3 | 44 | 65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiu, A.; Bouilhac, C.; Gimello, O.; Seaudeau-Pirouley, K.; Senila, M.; Jänisch, T.; Lacroix-Desmazes, P. Synthesis and Phase Behavior of a Platform of CO2-Soluble Functional Gradient Copolymers Bearing Metal-Complexing Units. Polymers 2022, 14, 2698. https://doi.org/10.3390/polym14132698
Ruiu A, Bouilhac C, Gimello O, Seaudeau-Pirouley K, Senila M, Jänisch T, Lacroix-Desmazes P. Synthesis and Phase Behavior of a Platform of CO2-Soluble Functional Gradient Copolymers Bearing Metal-Complexing Units. Polymers. 2022; 14(13):2698. https://doi.org/10.3390/polym14132698
Chicago/Turabian StyleRuiu, Andrea, Cécile Bouilhac, Olinda Gimello, Karine Seaudeau-Pirouley, Marin Senila, Thorsten Jänisch, and Patrick Lacroix-Desmazes. 2022. "Synthesis and Phase Behavior of a Platform of CO2-Soluble Functional Gradient Copolymers Bearing Metal-Complexing Units" Polymers 14, no. 13: 2698. https://doi.org/10.3390/polym14132698
APA StyleRuiu, A., Bouilhac, C., Gimello, O., Seaudeau-Pirouley, K., Senila, M., Jänisch, T., & Lacroix-Desmazes, P. (2022). Synthesis and Phase Behavior of a Platform of CO2-Soluble Functional Gradient Copolymers Bearing Metal-Complexing Units. Polymers, 14(13), 2698. https://doi.org/10.3390/polym14132698