
In Vivo Degradation Studies of PGA-PLA Block Copolymer and Their Histochemical Analysis for Spinal-Fixing Application


Abstract
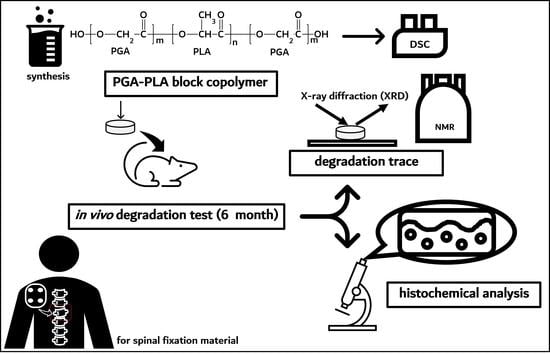
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Method
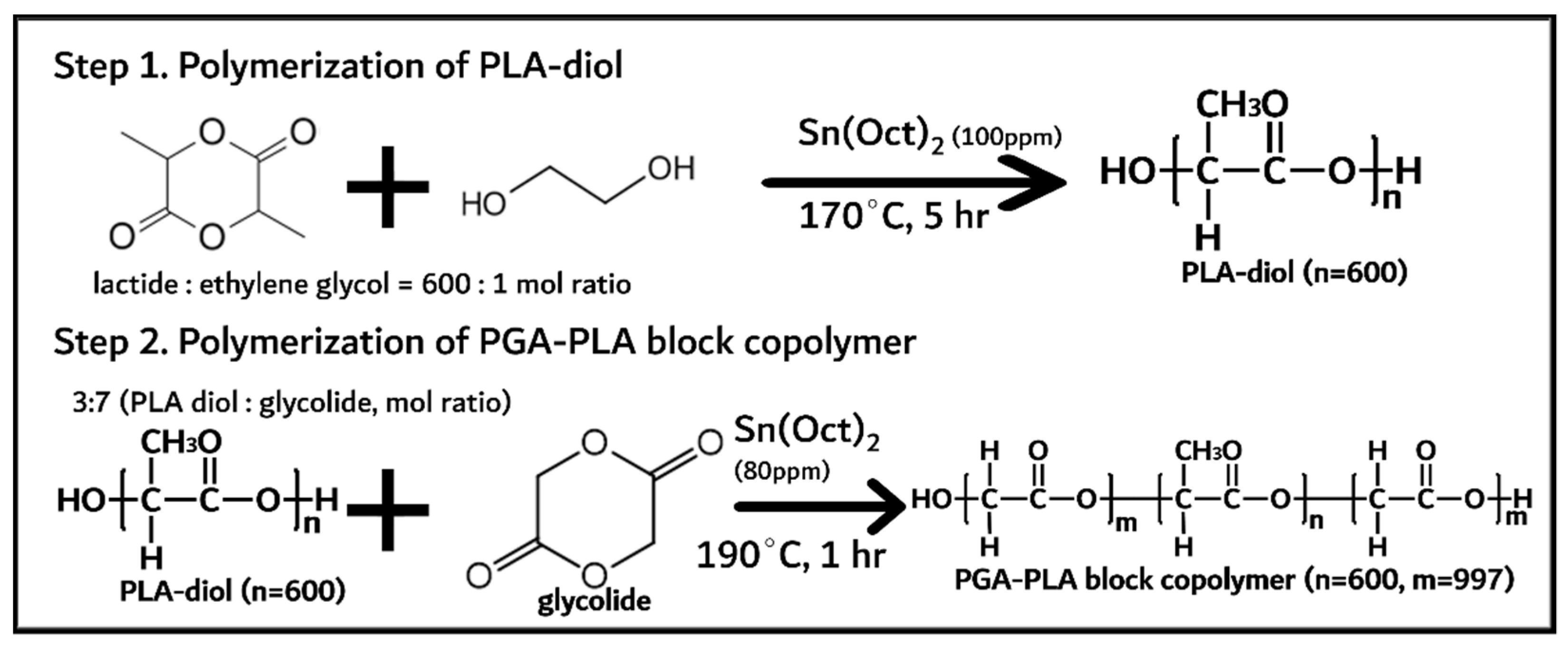
2.2.1. Synthesis PGA–PLA Block Copolymer
2.2.2. In Vivo Animal Degradation Test and Histochemical Analysis
3. Results and Discussion
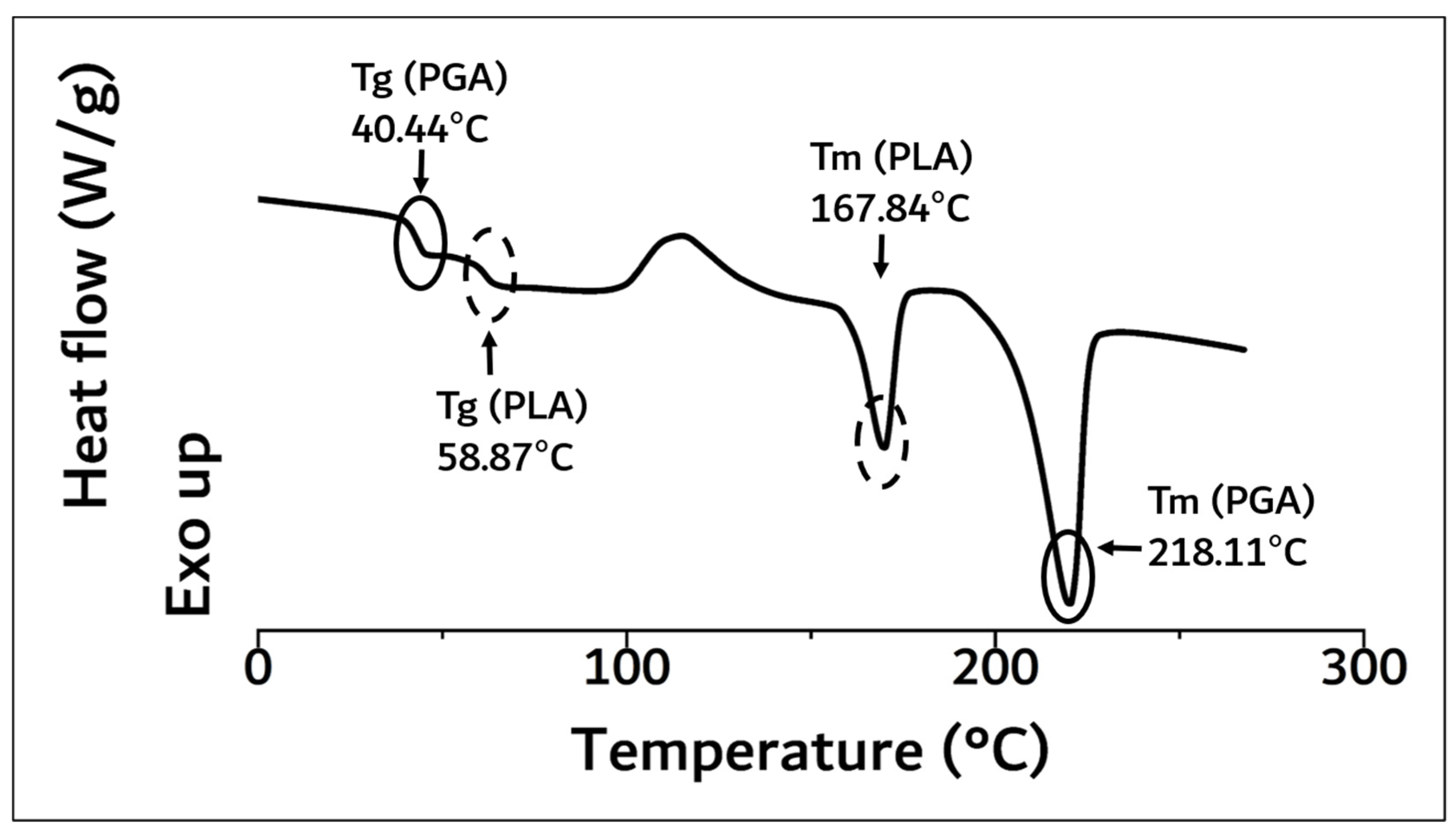
3.1. Synthesis of PGA–PLA Block Copolymer
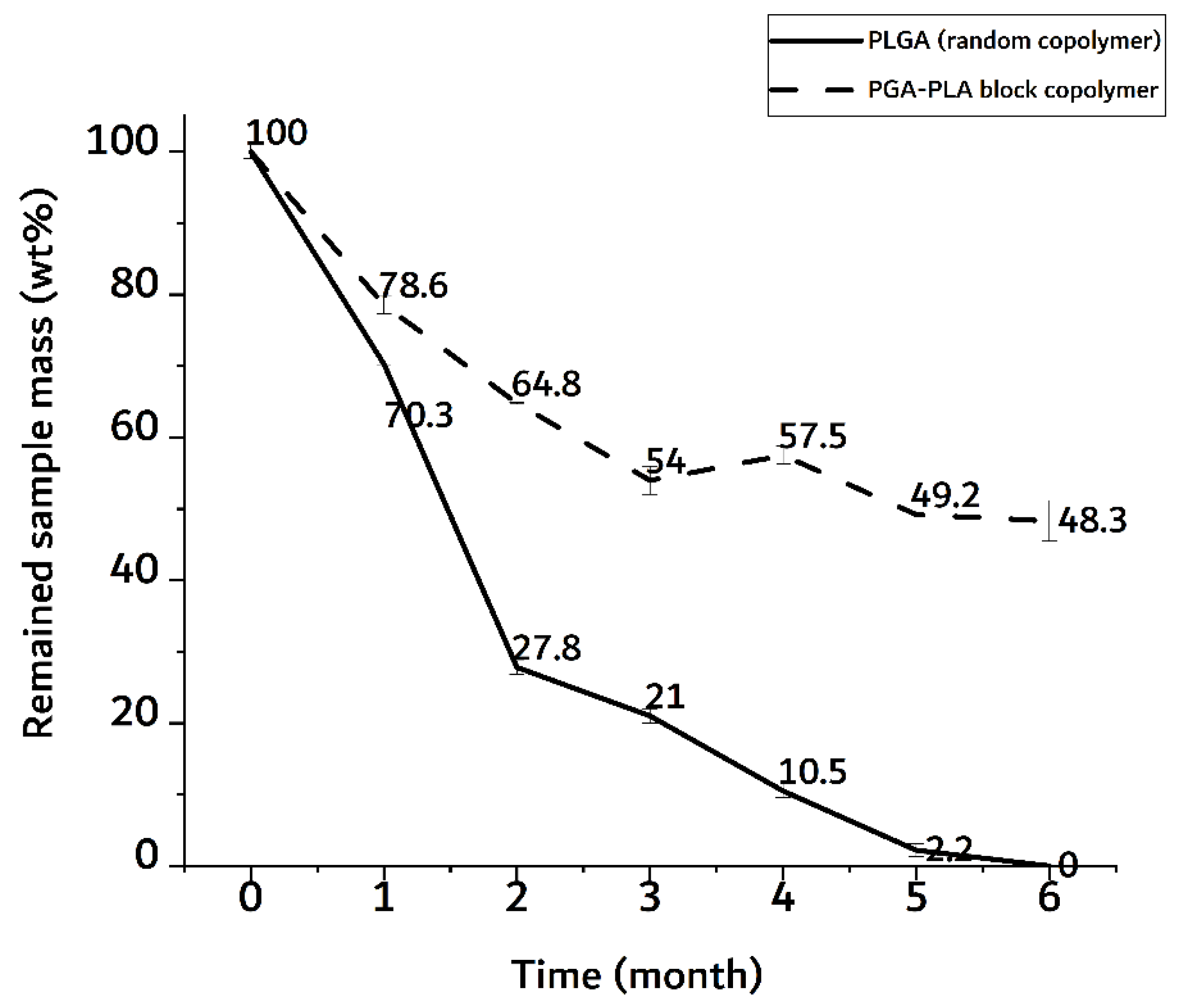
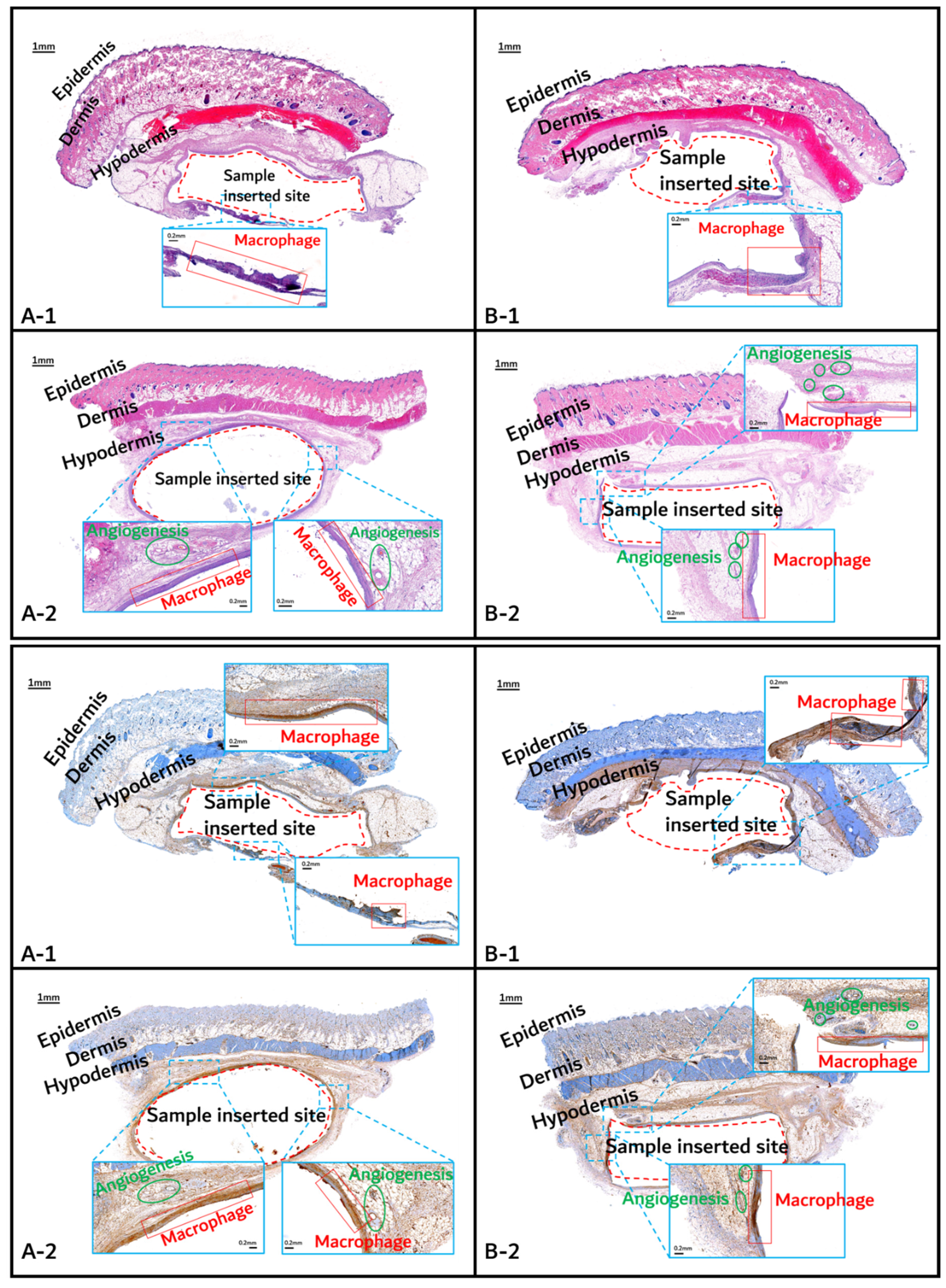
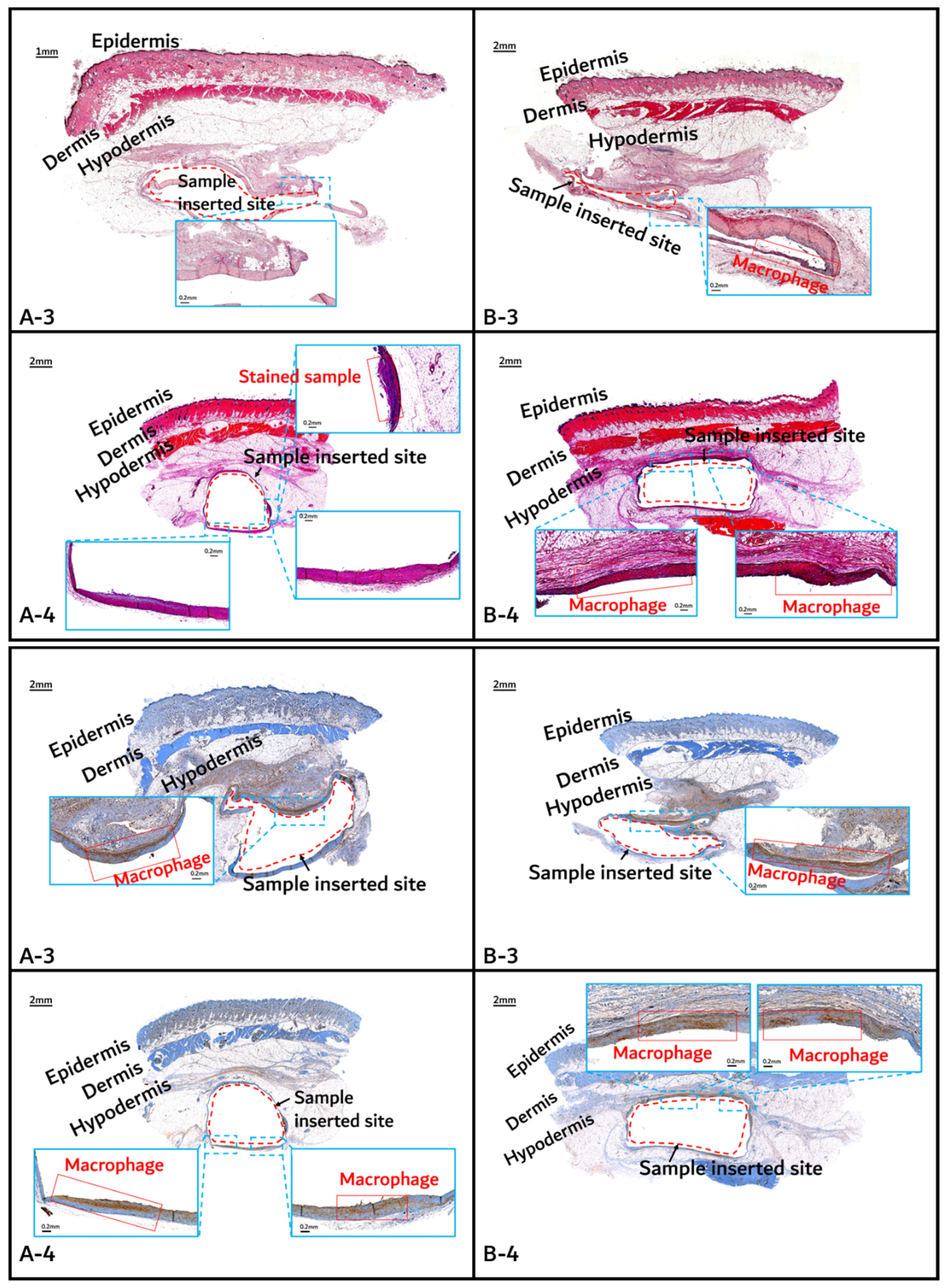
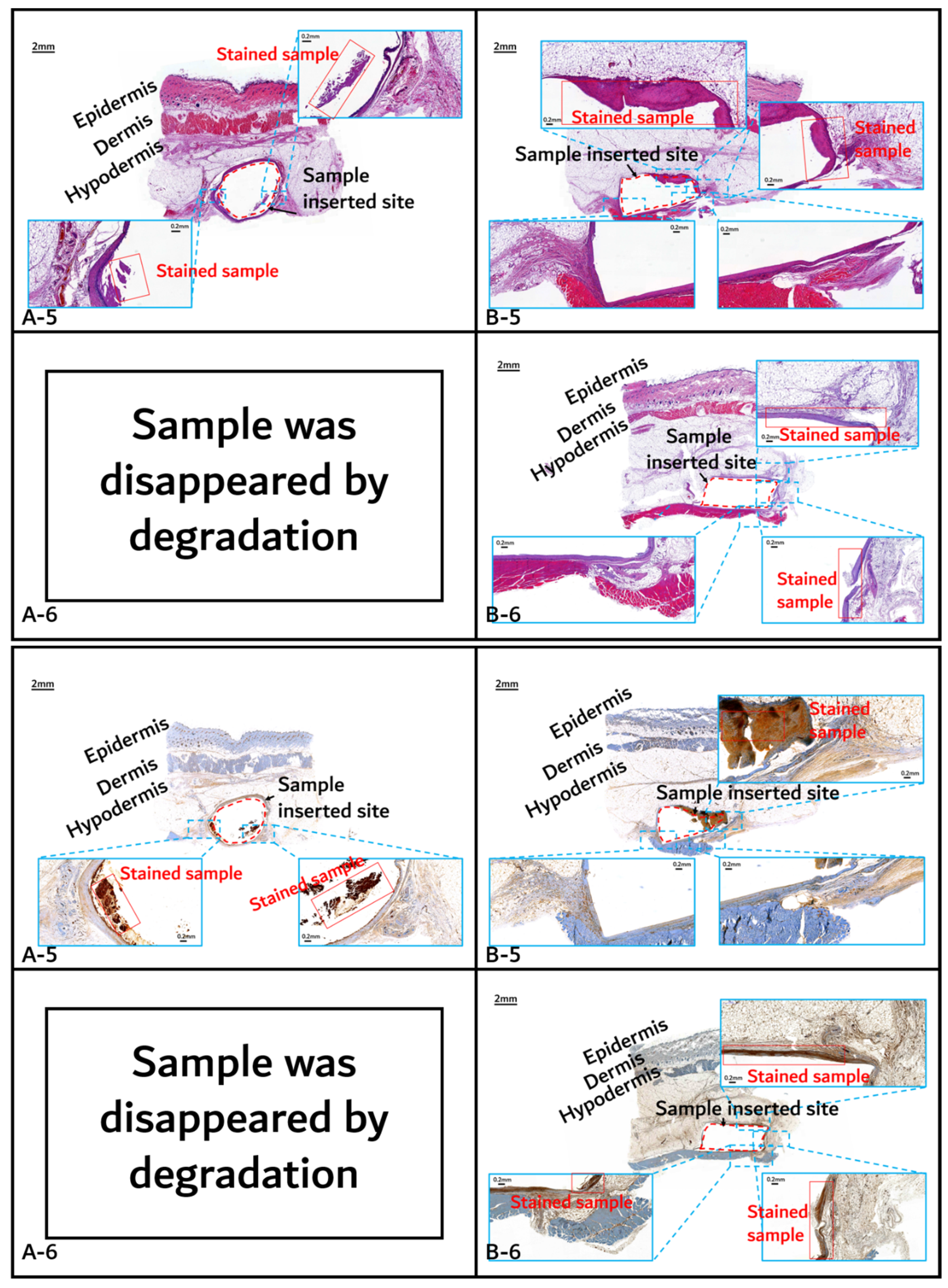
3.2. In vivo Degradation Test
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Pistner, H.; Bendi, D.R.; Muhling, J.; Reuther, J.F. Poly(L-lactide): A long-term degradation study in vivo; Part III. Analytical characterization. Biomaterials 1993, 14, 291–298. [Google Scholar] [CrossRef]
- De Jong, W.H.; Bergsma, J.E.; Robinson, J.E.; Bos, R.R.M. Tissue response to partially in vitro predegraded poly-L-lactide implants. Biomaterials 2005, 26, 1781–1791. [Google Scholar] [CrossRef]
- Waris, E.; Konttinen, Y.T.; Ashammakhi, N.; Suuronen, R.; Santavirta, S. Bioabsorbable fixation devices in trauma and bone surgery: Current clinical standing. Expert Rev. Med. Devices 2004, 1, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Webster, T.J. Mechanical properties of dispersed ceramic nanoparticles in polymer composites for orthopedic applications. Int. J. Nanomed. 2010, 5, 299–313. [Google Scholar]
- Armentano, I.; Dottori, M.; Fortunati, E.; Mattioli, S.; Kenny, J.M. Biodegradable polymer matrix nanocomposites for tissue engineering: A review. Polym. Degrad. Stabil. 2010, 95, 2126–2146. [Google Scholar] [CrossRef]
- Yoon, S.-K.; Yang, J.-H.; Lim, H.-T.; Chang, Y.-W.; Ayyoob, M.; Yang, X.; Kim, Y.-J.; Ko, H.-S.; Jho, J.-Y.; Chung, D.-J. In vitro and in vivo biosafety analysis of resorbable polyglycolic acid-polylactic acid block copolymer composites for spinal fixation. Polymers 2021, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y. Molecular, structure, and material design of bio-based polymers. Polym. J. 2009, 41, 797–807. [Google Scholar] [CrossRef]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Ayyoob, H.; Lee, S.-M.; Kim, Y.-J. Well-definded high molecular weight polyglycolide-b-poly(L-)lactide-b-polyglycolide triblock copolymers: Synthesis, characterization and microstructural analysis. J. Polym. Res. 2020, 27, 109–121. [Google Scholar] [CrossRef]
- Ramdhanie, L.I.; Aubuchon, S.R.; Boland, E.D.; Knapp, D.C.; Barnes, C.P.; Simpson, D.G.; Wnek, G.E.; Bowlin, G.L. Thermal and mechanical characterization of electrospun blends of poly(lactic acid) and poly(glycolic acid). Polym. J. 2006, 38, 1137–1145. [Google Scholar] [CrossRef]
- Meduri, A.; Fuoco, T.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Versatile copolymerization of glycolide and rac-lactide by dimethyl(salicylaldiminato)aluminum compounds. Macromoecules 2014, 47, 534–543. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, X.; Li, B.; Hou, Y.; Cai, Z.; Yang, J.; Li, Y. Paclitaxel-loaded PLGA microspheres with a novel morphology to facilitate drug delivery and antitumor efficiency. RSC Adv. 2018, 47, 3274–3285. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Liang, Z.; Zhu, B.; Dong, T.; Inoue, Y. Blending effects on polymorphic crystallization of poly(L-lactide). Macromolecules 2009, 42, 3374–3380. [Google Scholar] [CrossRef]
- Feng, P.; Guo, X.; Gao, C.; Gao, D.; Xiao, T.; Shuai, X.; Shuai, C.; Peng, S. Diopside modified porous polyglycolide scaffolds with improved properties. RSC Adv. 2015, 5, 54822–54829. [Google Scholar] [CrossRef]
- Oca, H.M.; Farrar, D.F.; Ward, I.M. Degradation studies on highly oriented poly(glycolic acid) fibers with different lamellar structures. Acta Biomater. 2011, 7, 1535–1541. [Google Scholar]
- Bakary, M.A.E.; Farahaty, K.A.E.; Sayed, N.M.E. In vitro degradation characteristics of polyglycolic/polycaprolactone (PGA/PCL) copolymer material using machzehnder interferometer. Mater. Res. Express 2019, 6, 105374–105385. [Google Scholar] [CrossRef]
- Zong, X.; Ran, S.; Kim, K.-S.; Fang, D.; Hsiao, B.S.; Chu, B. Structure and morphology changes during in vitro degradation of electrospun poly(glycolide-co-lactide) nanofiber membrane. Biomacromolecules 2003, 4, 416–423. [Google Scholar] [CrossRef]
- Gopferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar] [CrossRef]
- Zong, X.-H.; Wang, Z.-G.; Hsiao, B.S.; Chu, B.; Zhou, J.J.; Jamiolkowski, D.D.; Muse, E.; Dormier, E. Structure and morphology changes in absorbable poly(glycolide) and poly(glycolide-colactide) during in vitro degradation. Macromolecules 1999, 32, 8107–8114. [Google Scholar] [CrossRef]
- Luo, Y.; Lin, Z.; Guo, G. Biodegradation assessment of poly(lactic acid) filled with functionalized titania nanoparticles (PLA/TiO2) under compost conditions. Nanoscale Res. Lett. 2019, 14, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Park, S.-H.; Kim, S.-H. Surface alkylation of cellulose nanocrystals to enhance their compatibility with polylactide. Polymers 2020, 12, 178. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.Z.; Yang, X.M.; Li, Q.F. Influences of terminal POSS on crystallization and degradation behavior of PCL-PLLA block copolymer. Polym. Cryst. 2018, 1, e10019. [Google Scholar] [CrossRef]
- Hanzi, A.C.; Gerber, I.; Schinhammer, M.; Loffler, J.F.; Uggowitezer, P.J. On the in vitro and in vivo degradation performance and biological response of new biodegradable Mg-Y-Zn alloys. Acta Biomater. 2010, 6, 1824–1833. [Google Scholar] [CrossRef]
- Vollkommer, T.; Henningsen, A.; Friedrich, R.E.; Felthaus, O.H.; Eder, F.; Morsczeck, C.; Smeets, R.; Gehmert, S.; Gosau, M. Extent of inflammation and foreign body reaction to porous polyethylene in vitro and in vivo. In Vivo 2019, 33, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Incio, J.; Soares, R. Angiogenesis and chronic inflammation: Cause or consequence? Angiogenesis 2007, 10, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-J.; Chon, J.-W.; Yoo, H.; Kim, J.-H.; Chung, D.-J. Cytotoxicity and in vivo biosafety studies of the poly(alkylphenol) derivatives as vulcanizing agents. Macromol. Res. 2019, 27, 1081–1088. [Google Scholar] [CrossRef]
- Chon, J.-W.; Jang, I.-K.; Bae, K.-W.; Suh, S.-W.; Chung, I.-K. Cytotoxicity and in vivo biosafety of the polylactide based bone composite materials. Macromol. Res. 2017, 25, 648–655. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Tg (°C) a | Tm (°C) a | Molecular Weight b (Mw) | Flexural Strength c (MPa) | ||
|---|---|---|---|---|---|---|
| PLA | PGA | PLA | PGA | |||
| PGA–PLA block copolymer | 40.44 | 58.87 | 167.84 | 218.11 | 159,000 | 137 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, S.-K.; Chung, D.-J. In Vivo Degradation Studies of PGA-PLA Block Copolymer and Their Histochemical Analysis for Spinal-Fixing Application. Polymers 2022, 14, 3322. https://doi.org/10.3390/polym14163322
Yoon S-K, Chung D-J. In Vivo Degradation Studies of PGA-PLA Block Copolymer and Their Histochemical Analysis for Spinal-Fixing Application. Polymers. 2022; 14(16):3322. https://doi.org/10.3390/polym14163322
Chicago/Turabian StyleYoon, Seung-Kyun, and Dong-June Chung. 2022. "In Vivo Degradation Studies of PGA-PLA Block Copolymer and Their Histochemical Analysis for Spinal-Fixing Application" Polymers 14, no. 16: 3322. https://doi.org/10.3390/polym14163322
APA StyleYoon, S.-K., & Chung, D.-J. (2022). In Vivo Degradation Studies of PGA-PLA Block Copolymer and Their Histochemical Analysis for Spinal-Fixing Application. Polymers, 14(16), 3322. https://doi.org/10.3390/polym14163322