1. Introduction
Three-dimensional (3D) polymeric structures with their swelling agent being water are known as hydrogels and are commonly used for biomedical applications such as targeted drug delivery, tissue engineering, and smart biosensors, as well as in 3D and four-dimensional (4D) printing [
1,
2,
3]. Such applications introduce sensitive components such as cells or the need of complex material shapes. Macroscopic structuring (e.g., by an auxetic structure) can add a further dimension of functionalization to a material. Auxetic structures exhibit a negative Poisson’s ratio and thus, for example, react with a construction in certain spatial directions upon the application of mechanical stress. In comparison to unstructured objects of the same material, the introduction of an auxetic structure can improve mechanical properties such as shear resistance, fracture toughness, and resilience [
4,
5,
6,
7].
In general, hydrogels can be produced by physical or chemical crosslinking of a monomer source [
8]. As one possibility, unmodified proteins can be used to obtain dityrosine- crosslinked hydrogels by crosslinking phenolic hydroxy groups via enzymatic, fenton-like, or photoinitiated reactions [
9]. In the field of 3D printing, a common photoinitiator for dityrosine crosslinking is tris(2,2′-bipyridyl)dichlororuthenium(II) (Ru(bpy)
3Cl
2), which is induced by visible light in the presence of an electron acceptor such as ammonium or sodium persulfate [
10,
11,
12,
13]. Using this approach, stimuli-responsive hydrogels can be obtained [
14,
15]. In particular, these hydrogels change their structural and volume phase transition as a response to external stimuli [
16]. To enhance the fabrication window in extrusion 3D printing, thickeners such as the linear and highly charged polysaccharide sodium alginate are widely used to increase the structural integrity of ink formulations [
17,
18]. Due to its high biocompatibility and ease of handling, alginate is also widely applied as an extrudable bioink hydrogel material itself [
19], as in the presence of multivalent metal cations or in acid solutions, hydrogelation of sodium alginate is propagated by ionic crosslinking [
20]. Thus, with additional crosslinking of sodium alginate, a so-called double-network hydrogel can be obtained.
Since water is an integral part of hydrogels, an improved understanding of swelling degree and mechanical stability should be obtained [
21]. Current approaches to determine the swelling degree and water uptake mainly focus on weight or volume analysis of dried or as-prepared hydrogels in comparison to soaked hydrogel samples. Weight measurements can thereby easily be executed, although estimating the volume of the complex geometries is not always feasible. Thus, weight-based swelling ratios are usually reported in the literature [
21,
22]. However, these methods do not provide insight into the time-dependent, spatially resolved hydration of the inner hydrogel structure and require the full removal of excess liquids without withdrawing liquid from the hydrogel network.
Therefore, an easily transferable, site-resolving, nondestructive 3D analytical method is desired for material development. Magnetic resonance imaging (MRI) is a 3D imaging technique that is used in medical diagnostics, material sciences, and food research [
23]. The image contrast thereby often relies on the NMR relaxation properties and spin density differences in a material [
24,
25,
26]. Recently, it was shown to be applicable to the investigation of complex 3D objects obtained by 3D printing [
27] and of different common bioink materials [
28]. Setting aside the high initial costs and complexity of operation, the main advantage of MRI is its independence from the analyzed material and from optical transparency, enabling volumetric time and spatially resolved longitudinal studies on the same object [
23,
29].
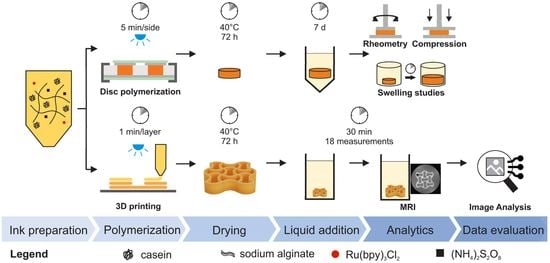
Initially, traditional material characterization of the hydrogel material—based on sodium alginate and dityrosine-crosslinked casein [
30]—was performed by swelling studies as well as rheometric analysis and uniaxial compression of casted discs using four different liquid systems. Following this, macroscopic auxetic structuring introduced by 3D printing was used for further functionalization of the protein-based hydrogel. In order to assess the now-complex structure of the hydrogel, we applied MRI, assessing time-dependent external and internal stimuli behavior. As a proof of principle, the 2D-crosssections of an auxetic geometry were analyzed with regard to their changes in the two-dimensional plane. Monitoring included the observation of visual structural changes of the lattice network as well as the estimation of area ratios of the hydrogel cross sections by using image analysis.
2. Materials and Methods
2.1. Buffer and Stock Solutions
The formulation buffer for material fabrication consisted of 20 mM sodium phosphate buffer (SPB) pH 8 containing 4 M urea. All liquid systems were prepared with ultrapure water (PURELAB Ultra, ELGA LabWater, Lane End, UK). At a temperature of 22 °C, the pH was adjusted using a 4 M sodium hydroxide solution and the buffer filtered through an 0.45 µm membrane (Pall Corporation, New York, NY, USA). The photoinitiator tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate (Ru(bpy)3Cl2·6H2O) was diluted in the formulation buffer with a concentration of 5 mM and stored at 4 °C as a stock solution. A 2 M stock solution of the oxidant ammonium persulfate (APS, chemical formula: (NH4)2S2O8) was prepared in the formulation buffer, stored as aliquots at −20 °C, and thawed directly prior to use.
To examine swelling or the effect of external stimuli in different systems, four different liquid systems for immersion of the dried hydrogel were prepared. These liquids were as follows:
System 1: ultrapure water;
System 2: ultrapure water + 0.1 M CaCl2;
System 3: ultrapure water + 0.1 M CaCl2 + 4 M urea;
System 4: ultrapure water + 20 mM SPB + 4 M urea.
For all liquids, pH (pH Meter HI 3220-2, Hanna Instruments, Woonsocket, RI, USA, equipped with the pH electrode Sentix
® 62, Xylem Inc., Rye Brook, NY, USA) and conductivity using a conductivity meter CDM230 (Radiometer Analytica SAS, Lyon, France) were determined (see
Supplementary Information S1). All chemicals reported in this manuscript were purchased from Merck KGaA (Darmstadt, Germany).
2.2. Ink Preparation
The ink formulation consisted of 10 wt% casein, 0.25 mM Ru(bpy)3Cl2, 75 mM APS, 3.5 wt% sodium alginate (SA), and the formulation buffer described above. Therefore, SA was dissolved in 60% of the total buffer volume in a dual asymmetric centrifuge (DAC, SpeedMixer® DAC 150.1 FVZ-K, Hauschild GmbH & Co. KG, Hamm, Germany) at 2500 rpm. Subsequently, the remaining formulation buffer, casein powder, and photoinitiator stock solution were added and mixed in the DAC (2500 rpm, 5 min). To enable the induction of the photopolymerization reaction, the precursor solution of the ink was finalized by addition of the APS stock solution directly prior to polymerization using the DAC (2500 rpm, 2 min).
2.3. Disc Polymerization
After the addition of APS, the precursor solution of the ink was transferred into a cylindrical polytetrafluoroethylene mold (diameter 10 mm, height 3 mm) that was covered on top and bottom with a transparent hydrophobic layer and acrylic glass. The precursor solution was irradiated for 5 min from above and below using a blue light-emitting diode (LZ4-00B208, LED Engin Inc., San Jose, CA, USA) with a radiant flux of 3.9 W at a distance of 7.5 cm. The discs were released from the mold and dried for 72 h at 40 °C (T6120, Heraeus Holding GmbH, Hanau, Germany).
2.4. Extrusion Printing
An auxetic structure was 3D-printed using a BioScaffolder 3.1 (GeSiM mbH, Radeberg, Germany) after adding the APS stock solution. The scaffold design consisted of nine point-symmetrically arranged re-entrant honeycomb unit cells with curved edges and had a total height of 3 mm (see
Supplementary Information S2). The printer was equipped with a pneumatic cartridge dispenser (GeSiM mbH), light-blocking cartridges (10 mL, Nordson Corporation, Westlake, OH, USA), and conic ultraviolet light-blocking nozzles (0.41 mm inner diameter, VIEWEG GmbH, Kranzberg, Germany). A velocity of 1.6 mm/s, strand height of 0.1 mm, start pause of 0.3 s, end pause of 0.1 s, and vertical tear off with an applied pressure of 19 kPa were set as printing parameters. Photopolymerization was initiated after each layer by 60 s illumination using the blue light-emitting diode, as described in
Section 2.3.
2.5. Magnetic Resonance Imaging (MRI)
The 3D-printed auxetic structures were dried for 72 h at 40 °C, and the time-dependent swelling behavior was characterized by MRI using an Avance HD III SWB 200 MHz tomograph (Bruker BioSpin MRI GmbH, Ettlingen, Germany) applying fast low-angle shot (FLASH) pulse sequence (experiment parameters are summarized in
Supplementary Information S3). To speed up the measurement time for a meaningful time resolution, a compromise between resolution and measurement time was found by measuring a stack of 2D measurements (referred to as 2.5 D MRI). FLASH was chosen as a fast and reliable pulse sequence that allows contrast generation mainly along T1 and T2*. The image contrast was found to be adequate for the purpose of the manuscript after optimization of the MRI parameters for the investigated samples.
A total number of 10 slices along the z axis without a distance between the cross sections were simultaneously imaged. Each slice summarizes the signal of a z-axial distance of 0.6 mm. In all cases, the hydrogel specimens were placed in a flat-bottom glass vial (25 mm diameter) containing 3 mL of the respective liquid system. Subsequently, the glass vial was manually positioned into the magnet, and correct sample positioning was tested by an initial measurement (<10 s). The measurement sequence (t0–t17) was initiated 60–120 s after liquid addition, with the first measurement being referred to as t0. The time series consisted of 18 measurements with a time interval of 1.7 min each.
To optimize the liquid-to-hydrogel contrast, hydrogel specimens immersed in 0.1 M CaCl
2 within the magnet were additionally measured by applying rapid acquisition with relaxation enhancement (RARE) pulse sequences with the same time interval (experimental parameters are summarized in
Supplementary Information S4) in the images. Due to the slightly different chemical composition, this step was essential for being able to apply the subsequent data analysis steps with high enough reliability. The chosen MRI parameters provide in this case a combined weighting by T1 and T2, while the T1 weighting is reduced compared to the FLASH parameters. For the numerical details, see
Table S3. In all cases, the
1H spin signal of all 10 slices was transferred into a grayscale representation for visualization purposes.
2.6. Image Analysis
MRI data of the fifth slice in the z-direction were processed in MATLAB
® R2021b (The MathWorks, Natick, MA, USA). An overview of the image processing is shown in
Figure 1. All pixel intensities were normalized to the maximum intensity in the slice. The image resolution was aligned to the highest resolution measured in the data set (function “imresize”, bilinear method) to minimize statistical errors during the following edge detection and overall pixel counting. Due to the small liquid-to-hydrogel contrast in some images, two edge detection methods were performed for each file (function “edge”, methods “canny” and “zerocross”). An overlay of the resulting images was used to generate and fill structures. If necessary, non-continuous edges were manually connected to generate an enclosed area. In subsequent postprocessing, small objects (<2000 px) were removed, small holes (<150 px) in the structure were filled, and the structure edges were smoothed. Finally, the areas of the lattice and interstices were determined by counting the pixels of interest multiplied with the image resolution. The obtained area ratio (AR) is defined as follows:
where
AreaSystem,t describes the area of either the hydrogel lattice, interstices, or the overall hydrogel area being a combination of the area determined for lattice and interstices (see
Figure 1, Data Evaluation) depending on the four liquid phases (System 1 to System 4) at distinctive time points
t and
t0, respectively (number of replicates (
n) = 2).
Distances (in the
Section 3 and
Section 3.4) were estimated with ImageJ V1.53k (NIH, Bethesda, MD, USA) of distinct measurements, and their length ratios (LR) are defined as follows:
where
DistanceSystem,t refers to the length of longitudinal and transverse cross section of the overall structure or the inner unit cell, depending on the four liquid systems at distinctive time points
t and
t0 (
n = 2).
2.7. Additional Hydrogel Characterization Methods
2.7.1. Weight-Based Swelling Studies
The hydrogel discs were weighted as prepared (m0) after being dried for 72 h (m_dry0), soaked in liquid (m1), and being dried again for 72 h (m_dry1). These weights were used to determine the swelling ratio mrel = m1/m0 as well as the dry-weight ratio m_dryrel = m_dry1/m_dry0 (n = 3).
2.7.2. Oscillatory Rheometry
Prior to analytics, previously dried hydrogel discs (preparation see
Section 2.3) were stored for 7 d in each of the four liquid systems (see
Section 2.1) with a 100-fold liquid excess for monitoring of the swollen state. Amplitude sweeps (angular frequency ω = 1 and 25 rad·s
−1, shear stress τ = 5–10.000 Pa,
n = 2) and frequency sweeps (τ = 10 Pa and ω = 1–25 rad·s
−1,
n = 3) were performed on a Physica MCR 301 plate rheometer equipped with a plate–plate geometry (PP10, diameter 10 mm, all Anton Paar GmbH, Graz, Austria) at 22 °C. To reduce the influence of potential outliers, the storage modulus G′ and the loss factor tan δ (=G″/G′ with G″ being the loss modulus) of a hydrogel were determined by averaging all data points obtained by the frequency sweep measurements after excluding outliers, which were identified by a difference of three scaled median absolute deviations from the median.
2.7.3. Uniaxial Compression Tests
Sample preparation for uniaxial compression tests was executed along the lines of the rheometry tests (see
Section 2.7.2). Accordingly, hydrogel discs were dried and stored in each liquid system for 7 d prior to analysis of the swollen state. Uniaxial compression tests were performed either on a universal testing machine (zwickiLine Z0.5TN or Z2.5) equipped with either a Xforce HP 100 N (all ZwickRoell GmbH & Co. KG, Ulm, Germany) for forces ≤ 100 N or a KAP-Z 1 kN (Angewandte System Technik GmbH, Dresden, Germany) load cell and stainless-steel compression platen (diameter 30 mm, ZwickRoell GmbH & Co. KG, Ulm, Germany) for forces > 100 N. Initial hydrogel length was determined at a preforce of 0.2 N, with a uniform velocity of 2 mm/min being applied until sample fracture to determine the compressive strength σ
max and the fracture strain ε
max (
n = 5).
4. Discussion
Magnetic resonance imaging for hydrogel characterization. Buffer substances such as urea or salts in general can account for more than half of the as-prepared hydrogel dry weight. By using different liquid phases with special characteristics with and without salts and urea being present, commonly applied weight-based swelling ratios are thus inapplicable, as diffusion of buffer substances into or out of the hydrogel network cannot be neglected. In addition, weight-based swelling ratios are mostly determined using simple geometric structures, such as cylinders or cubes. Thus, the analysis of a more complex auxetic structure with interstices filled with liquid causes difficulties in reproducible removal of excess water.
To overcome these disadvantages, MRI was applied as a new tool to describe the time-dependent stimuli and thus swelling behavior caused by liquid diffusion into an auxetic, protein-based hydrogel network. Using this approach, both the liquid front development as such but also the swelling behavior of outer and inner structures could be traced in a time- and spatially resolved way. Please be aware that potential scaffold defects, such as small holes within the structure, are smoothed during image processing, which can lead to misinterpretations of the liquid penetration time and swelling behavior. This is especially important for production processes, which tend to lead to many structural defects. Since no printing defects were observed during visual inspection of the printed hydrogels (see exemplarily
Figure S2)—apart from the still visible incomplete strands in Sample #1, illustrated in
Figure 2C, and thus the different hydrogel-to-liquid ratio of this sample—this effect was not further considered in the context of this manuscript. Within the scope of this manuscript, it was possible to trace three different liquids with one analysis method, as shown by a high liquid-to-hydrogel contrast in
Figure 2 for liquid systems 1, 3, and 4. By changing the MRI pulse sequence, an optimized liquid-to-hydrogel contrast (
Figure 3) allowed the monitoring of hydrogel specimens placed in a fourth liquid (System 2) that could not be previously analyzed using the proposed image analysis method. Overall, these results are indicative of a good transferability of the approach used for the screening of new materials. Further, we can envision the adaption of this method to study short-time diffusional effects of core-shell microcapsules, hydrogel shells which exemplarily encapsulate a liquid core containing bioactive molecules. In the area of tissue engineering, the concept could be suitable for the long-term monitoring of hydrogels containing cells [
31] or for liquid diffusion monitoring (e.g., vascularization) and swelling behavior of functional hydrogels.
Stimuli responsiveness of the used protein-based hydrogel formulation. In order to discuss the stimuli responsiveness of the hydrogel formulations used, several effects which are responsible for the formation and expansion of a hydrogel network have to be considered, taking into account a more detailed study of the influencing parameters during hydrogel formation published previously [
15]. The network formation is caused by the chemical crosslinking of casein by the induced photoreaction, so the main proportion of casein is expected to be incorporated into the hydrogel network. In contrast, the used thickening agent, sodium alginate, is soluble in aqueous solutions and thus may diffuse out of the hydrogel network. However, it can be physically crosslinked by ionic interactions in the presence of divalent cations, such as those introduced by 0.1 M CaCl
2 in the liquid phase. This double crosslinking was verified by an increasing storage modulus in the rheometric analysis, whereas elasticity decreased upon CaCl
2 addition and was even further decreasing for liquids without urea with an opposite effect for the compressive strength and maximum elongation.
In addition to the formation of the hydrogel network by chemical crosslinking or a combination of the chemical dityrosine crosslinking of casein and physical ionic crosslinking of sodium alginate, interactions inside the hydrogel network are responsible for its expansions and mechanical properties. These interactions are based on a complicated interplay between the crosslinked protein (and sodium alginate) and the formulation buffer components that remain in the (dried) hydrogel specimen, as well as the composition of the immersion liquid system. Concentration differences in the liquid systems and within the hydrogel result in diffusion of the various components into or out of the hydrogel.
Figure 6 illustrates the uptake and decrease of chemical substances in the hydrogel specimens, which are either incorporated into the hydrogel network or present in the liquid phase, depending on the immersing liquid. Since the spatially resolved concentration and thus contribution of the individual components to the swelling behavior of the hydrogel were not in the focus of this study and thus not analyzed on a molecular basis, the course of the time-dependent swelling will not be discussed in detail.
In general, an overall volume increase is explained by the liquid immersion into a dry hydrogel network, causing an association of the polymer molecules with water, explaining the initial increase in the overall area for all liquid systems. Thus, intermolecular effects are gaining importance, with increasing liquid content affecting the hydrogel characteristics. The chaotropic agent urea—which is part of the formulation buffer since it enhances the solubility of the hydrophobic protein casein—decreases hydrophobic interactions [
32]. Upon drying, together with sodium phosphate as the other formulation buffer substance, it will remain in the hydrogel structure. Therefore, just looking at the urea content in the hydrogel specimens, if no urea is present in the immersing liquid, urea may slowly diffuse out of the hydrogel network. This leads to increasing intermolecular interactions and thus an overall decrease in lattice area and the overall network dimensions. Additionally, due to the presence of salts such as sodium phosphate and CaCl
2, the ionic strength is altered depending on the liquid system. The higher the ionic strength, which is correlated with the solution’s conductivity, the more ionic interactions cause a volume decrease in hydrogels. Moreover, the pH, and thus the surface net charge of the protein, depends on the used liquid system. For micellar casein with a pI around pH 4.6 [
33], a lower protein repulsion is expected in the urea-containing liquid systems (≈pH 8) compared to the non-urea-containing liquids (<pH 7). The combination of these effects results in a complex time and stimuli-dependent behavior of the overall area covered by the hydrogel dimension, as well as the lattice and interstitial area. However, to address the exact reason for the observed time-dependent behavior, the hydrogel structure and its reaction to each stimulus applied should be further understood (e.g., by using advanced material characterization techniques).
Macroscopic auxetic structuring. Besides the induction of stimuli-dependent behavior on a micromolecular level, the protein-based hydrogel was functionalized by macroscopic auxetic structuring. This geometric pattern enables site-directed macromolecular responses. Auxetic structures exhibit a negative Poisson’s ratio, meaning that upon an external force in a uniaxial, longitudinal direction (stretching), their perpendicular cross section increases [
6], which is opposite to most of the material structures known. As the liquid uptake into a dried structure exhibits a non-uniaxial force, one would not inevitably expect an auxetic behavior upon liquid immersion. However, this was observed upon liquid uptake from the outside, and the cross section as well as the overall dimension increased further for the center unit cell (
Figure 5). Dominating re-entrant parts compared to the longitudinal dimension bend the 3D printed object in one direction. Using extrusion-based 3D printing as a fabrication method generated an auxetic architecture with higher strand widths at the nodes compared to the strands, as the material is merging within a printed layer before light-induced crosslinking. The resulting longer diffusion paths at the nodes and the delayed wetting compared to the strands could be visualized in a site-dependent way by MRI. As the directed stretching is more pronounced at the center unit cell of the auxetic structure, which is influenced by diffusion as well as the directed stretching and swelling of the neighboring units, these effects may contribute to and enhance the overall auxetic behavior during liquid uptake.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}