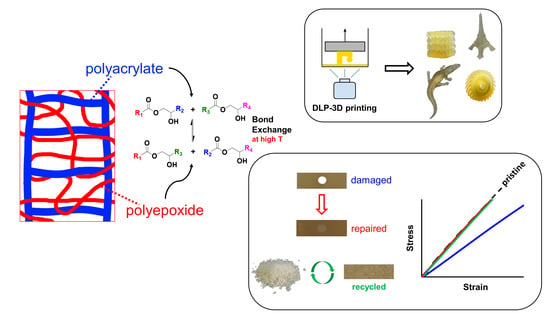
1. Introduction
Conceived in the 1980s and initially intended merely for prototyping, additive manufacturing (or 3D printing) has garnered industrial and academic interest during the last decade. This is mainly due to the versatility and speed of processing that it entails, making it invaluable in an era driven by customized, small-scale, and make-to-order type industrial practices. There are excellent papers in the literature reviewing the different 3D printing technologies and their products for industries such as biotechnology, automotive and aerospace, sporting goods, and food [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10].
Several thermoplastic- or thermoset-based polymer systems can be processed using different 3D printing methods. Apart from these conventional materials, a relatively new class of polymer materials, namely vitrimers, can also be used as building blocks for additive manufacturing. These materials combine the ease of processing of thermoplastics with the mechanical performance of thermosets [
11,
12,
13,
14,
15]. This capability stems from their covalent but dynamic bonds that can reshuffle given appropriate stimuli, such as heat. The presence of such dynamic bonds, given sufficient temperature and time, permits even fully crosslinked polymer networks to disintegrate and allows for macroscale recycling through facile processes, such as by grinding and molding. The use of vitrimers in the realm of 3D-printing is somewhat new. Intended mostly for sterolithographic applications, such as digital light processing (DLP) printing, photosensitive resins are formulated from vitrimeric monomers that feature the aforementioned dynamic bonds based on chemistries such as disulfides [
16,
17] or esters [
18,
19] that can undergo bond exchange reactions when heated. These exchange reactions enable the repair and reprocessing of printed parts, using simple welding or hot pressing procedures.
In this work, we expose parameters that should be controlled in order to optimize an acrylate-epoxy hybrid and vitrimeric photoresin formulation so that a three-way balance is achieved between ease of (photo) processing, mechanical properties, and repairability/reprocessability. Starting from a vitrimeric photoresin that was characterized previously [
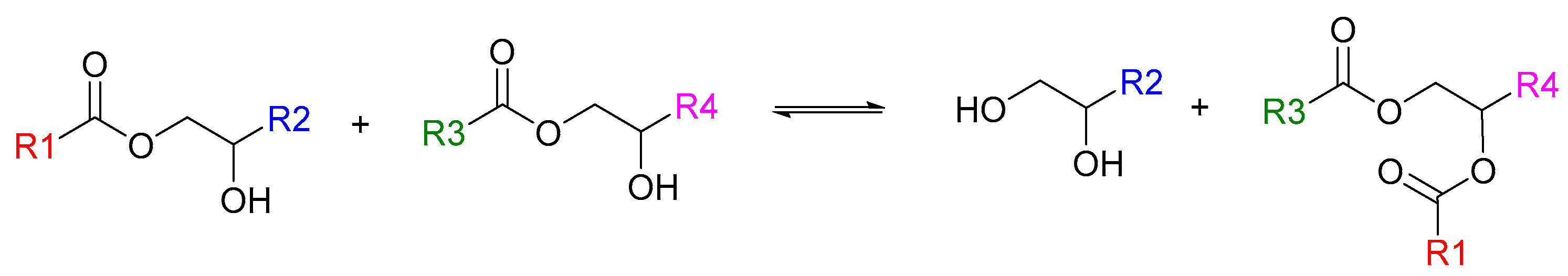
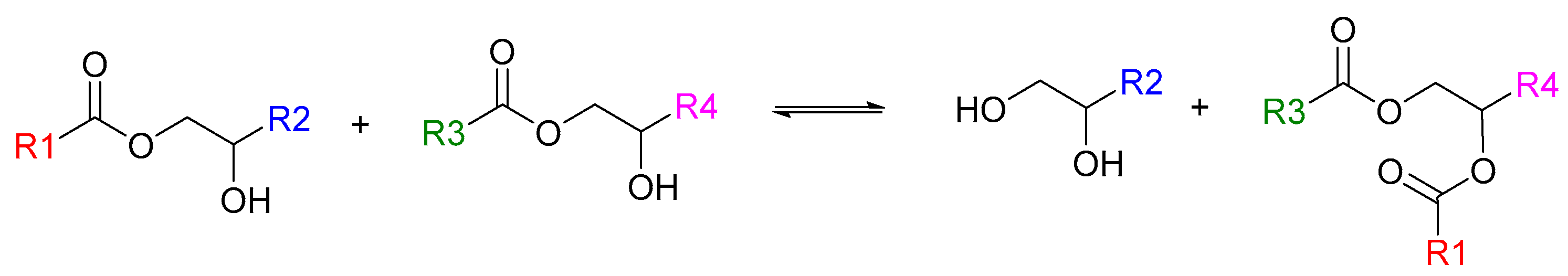
20], we obtain an improved formulation by exploiting the leverage points revealed by our investigation. We then conduct mechanical tests on printed and repaired/recycled samples with this improved formulation. The acrylate-epoxy polymer contains a high concentration of β-hydroxyesters that can undergo transesterification when a suitable catalyst, such as a zinc salt, is used [
21,
22,
23,
24] (
Scheme 1).
This enables the facile repair and reprocessing of the cured material. Furthermore, the hybrid formulation confers added versatility to the production process since the acrylate:epoxy composition can be changed so as to tailor the thermomechanical properties of the final material [
20,
25]. Moreover, this hybrid formulation can be cured sequentially, wherein a quick photocuring process is followed by thermal curing.
2. Materials and Methods
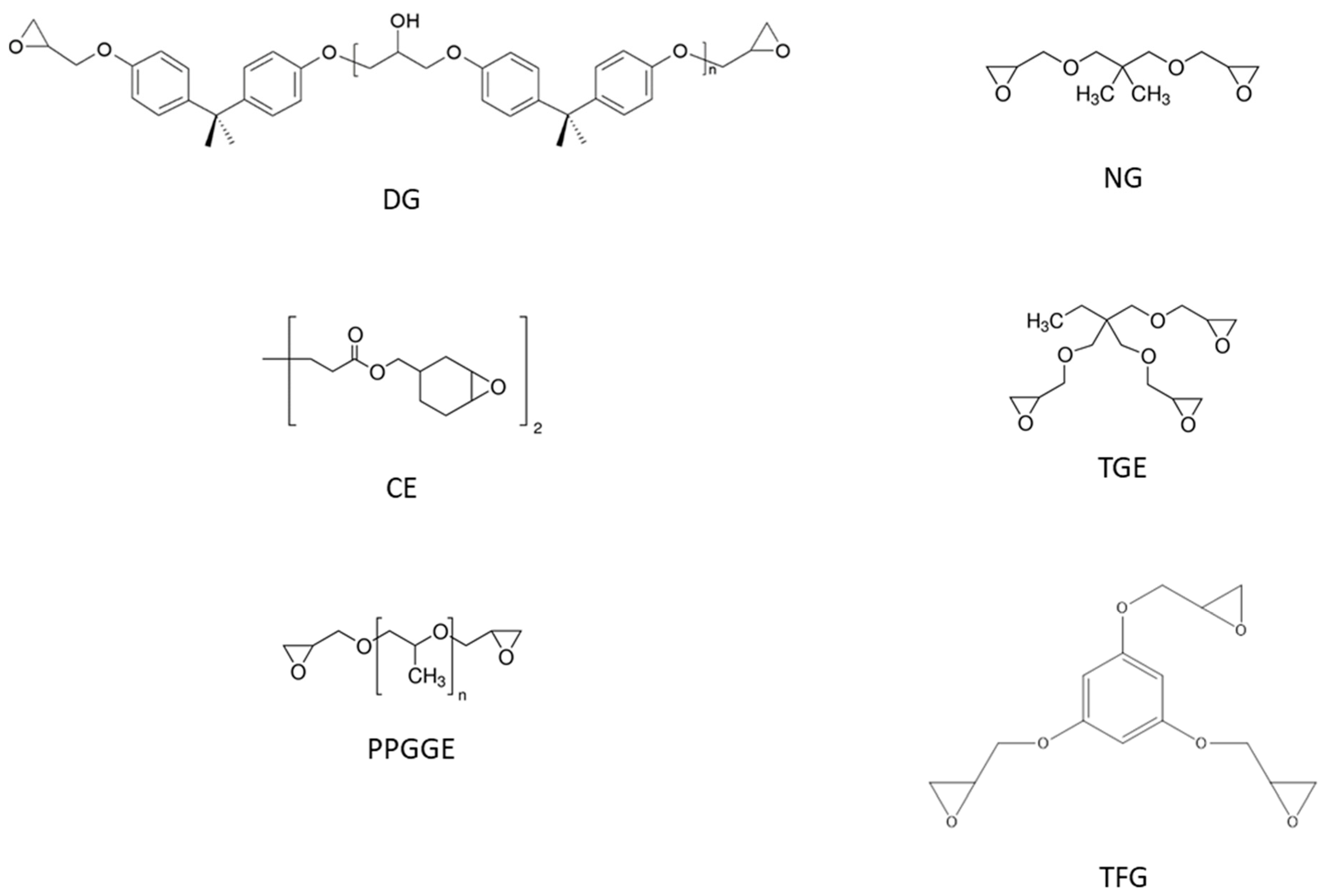
The acrylate part of the basic hybrid formulation consists of a 1:1 mixture (by weight) of glycerol 1,3-diglycerolate diacrylate (GLYDA) and poly(ethylene glycol) methacrylate (PEGMA). In the improved formulation optimized for 3D printing, a different methacrylate, namely 2-Phenoxyethyl methacrylate (FEMA), was used. The epoxy component of the basic formulation is a DGEBA-type epoxy resin (DG) with an epoxy equivalent weight of 187 g/ee. Other epoxides used in the study are neopentyl glycol diglycidyl ether (NG), bis(3,4.epoxycyclohexylmethyl) adipate (CE), trimethylolpropane triglycidyl ether (TGE), poly(propylene glycol) diglycidyl ether with an average molecular weight of 640 g/mol (PPGGE) or 380 g/mol (PPGGEs), and phloroglucinol triglycidyl ether (TFG). For epoxy-acid curing, glutaric acid (GLU) is used. To compatibilize the acrylate and epoxy parts, glycidyl methacrylate (GMA), which bears both acrylate and epoxide functionalities, is used as a coupling agent. In the basic formulation, the total mass of acrylates is the same as the mass of DG. Furthermore, the number of acrylate groups contributed by GMA is the same as the acrylate groups contributed by GLYDA. Lastly, GLU is added in stoichiometry with the total epoxides. As a photoinitiator, diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) was used. As a transesterification catalyst (to confer the vitrimer behavior), zinc acetoacetonate (Zn) was used. For further details about this formulation and the sample preparation procedure in general, refer to our previous paper [
20]. All monomer materials, except for DG and TFG, were purchased from Sigma Aldrich (Madrid, Spain) and used without purification. DG was kindly supplied by Po.Int.Er Srl. (Valfenera, Italy). TFG was supplied by Specific Polymers (Castries, France).
The formulations were either photocured using a Vilber-Lourmat UV oven (Vilber Lourmat Sté., Collégien, France), inside hand-made transparent molds with polytetrafluoroethylene (PTFE) spacers to obtain rectangular samples, or 3D printed using an Asiga UV Max385 DLP printer (Asiga, Sydney, Australia). In the latter process, the layer thickness was 100 µm, and the wavelength of irradiation was 385 nm. Each layer received an irradiation of 87.5 mJ·cm
−2, except the initial layer, which received 175 mJ·cm
−2, to avoid cohesive failure between the build platform and the printed part. After that, the samples were thermally treated to carry out the epoxy-acid reaction at 120 °C for 12 h in a Memmert convection oven. Complete cure of materials was confirmed using a temperature-controlled Brucker Vertex 70 FTIR spectrometer equipped with an attenuated total reflection (ATR) accessory (GoldenGate™, Specac Ltd., Orpington, UK). The FTIR spectra obtained during the two curing steps are given in
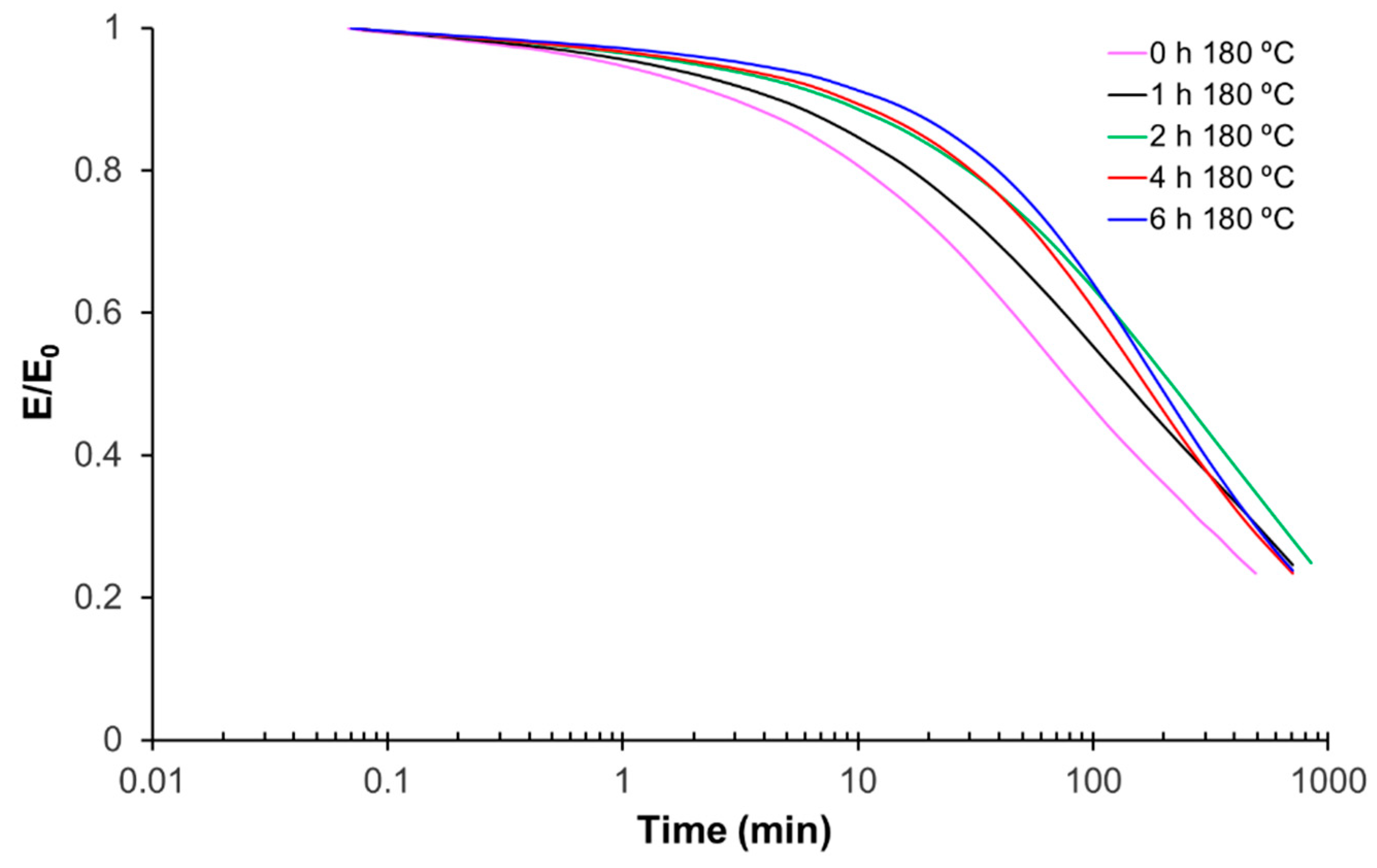
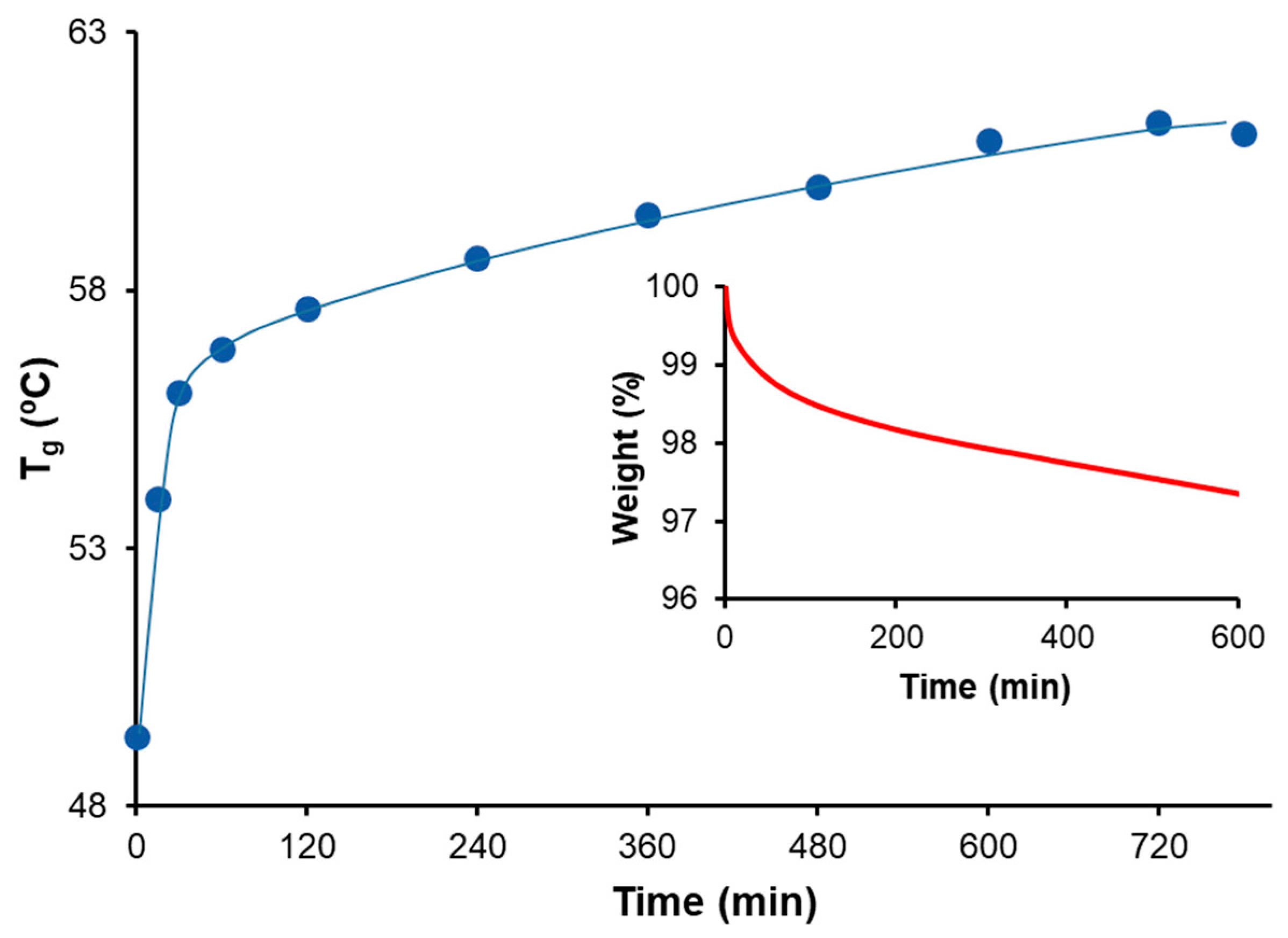
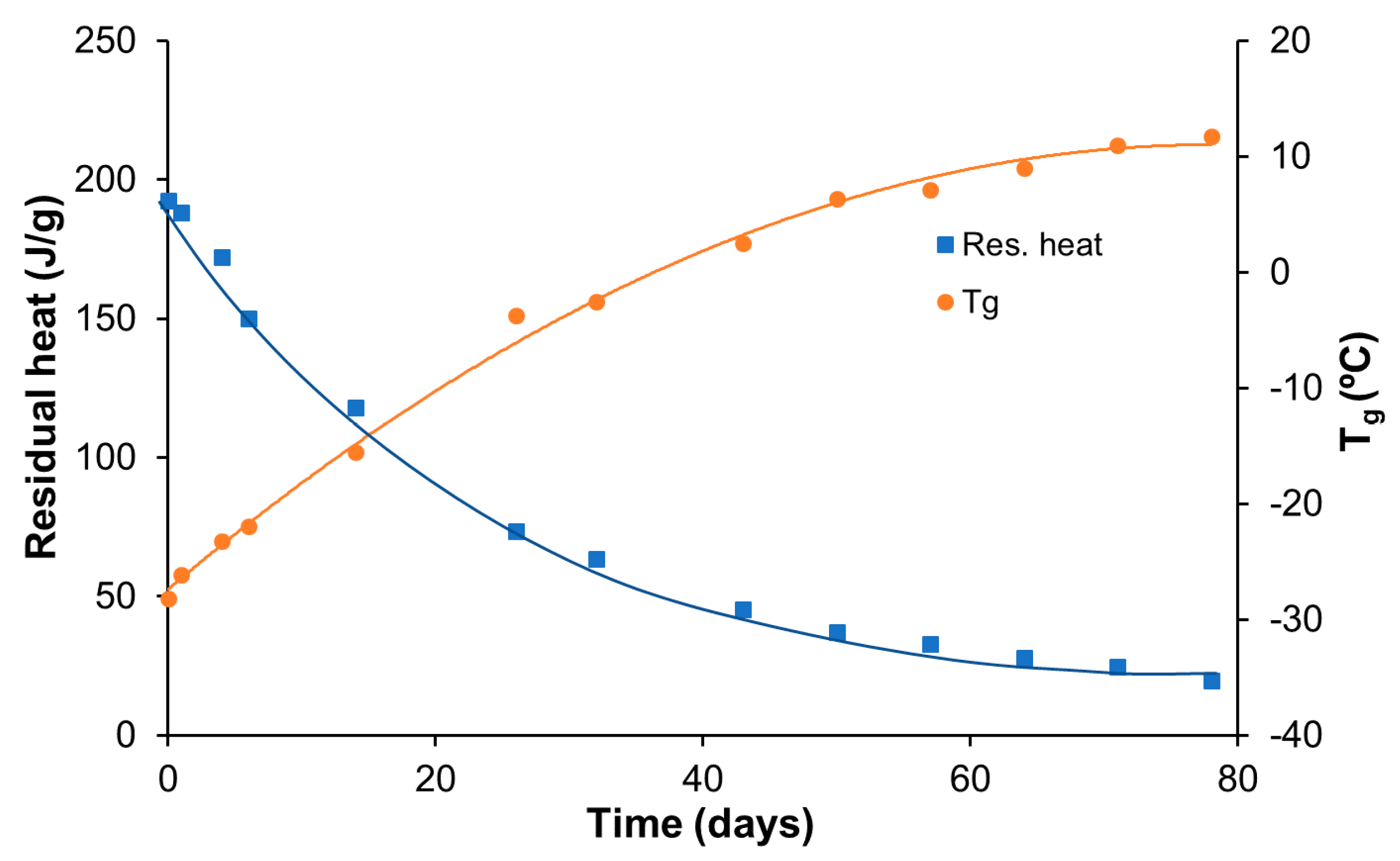
Figures S1 and S2 (supporting information). A final thermal post-treatment at 180 °C was carried out, also in the same oven, for an additional 4 h to ensure the equilibrium of transesterification reactions.
A Mettler DSC3+ calorimeter (Mettler-Toledo, Greifensee, Switzerland) was used to measure both the and the polymerization heats. Photocuring experiments were performed using a Hamamatsu LC5 light source (Hamamatsu Photonics K.K., Hamamatsu, Japan) equipped with a Hg-Xe mid-pressure lamp adapted to a Mettler DSC821 calorimeter. The UV light intensity was 36 mW cm−2, measured at 365 nm using a radiometer (Hamamatsu Light Power Meter, Model C6080-03, Hamamatsu Photonics K.K., Hamamatsu, Japan). To quantify the storage stability, conversions were calculated using , where is the heat evolved during the thermal cure after UV irradiation of a sample stored for a known duration of time, and is the total reaction heat of a freshly prepared and photocured sample.
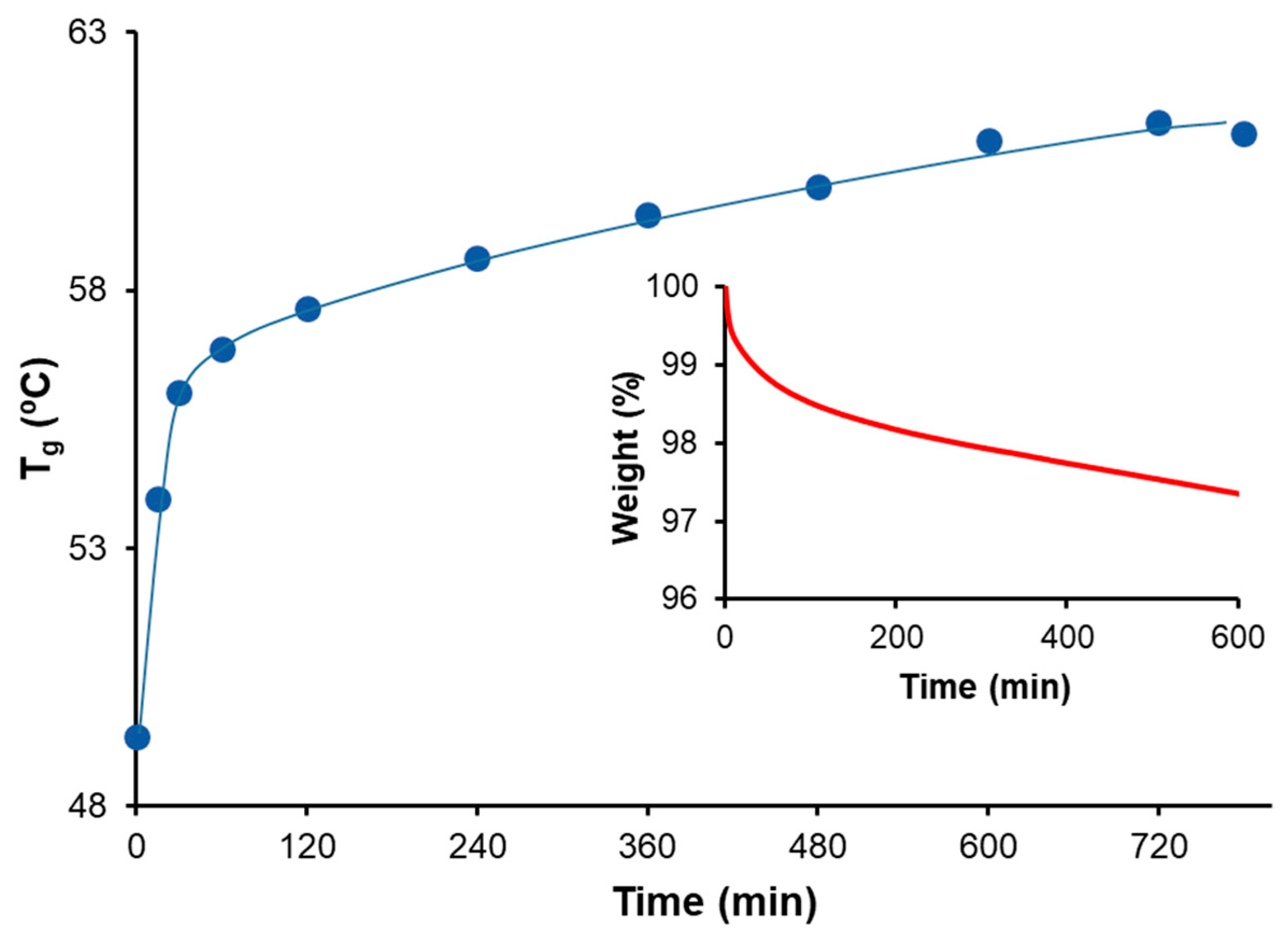
The cured material was taken as the halfway point of the heat capacity step observed in a scan at 10 °C min−1, following the DIN 51007 standard method.
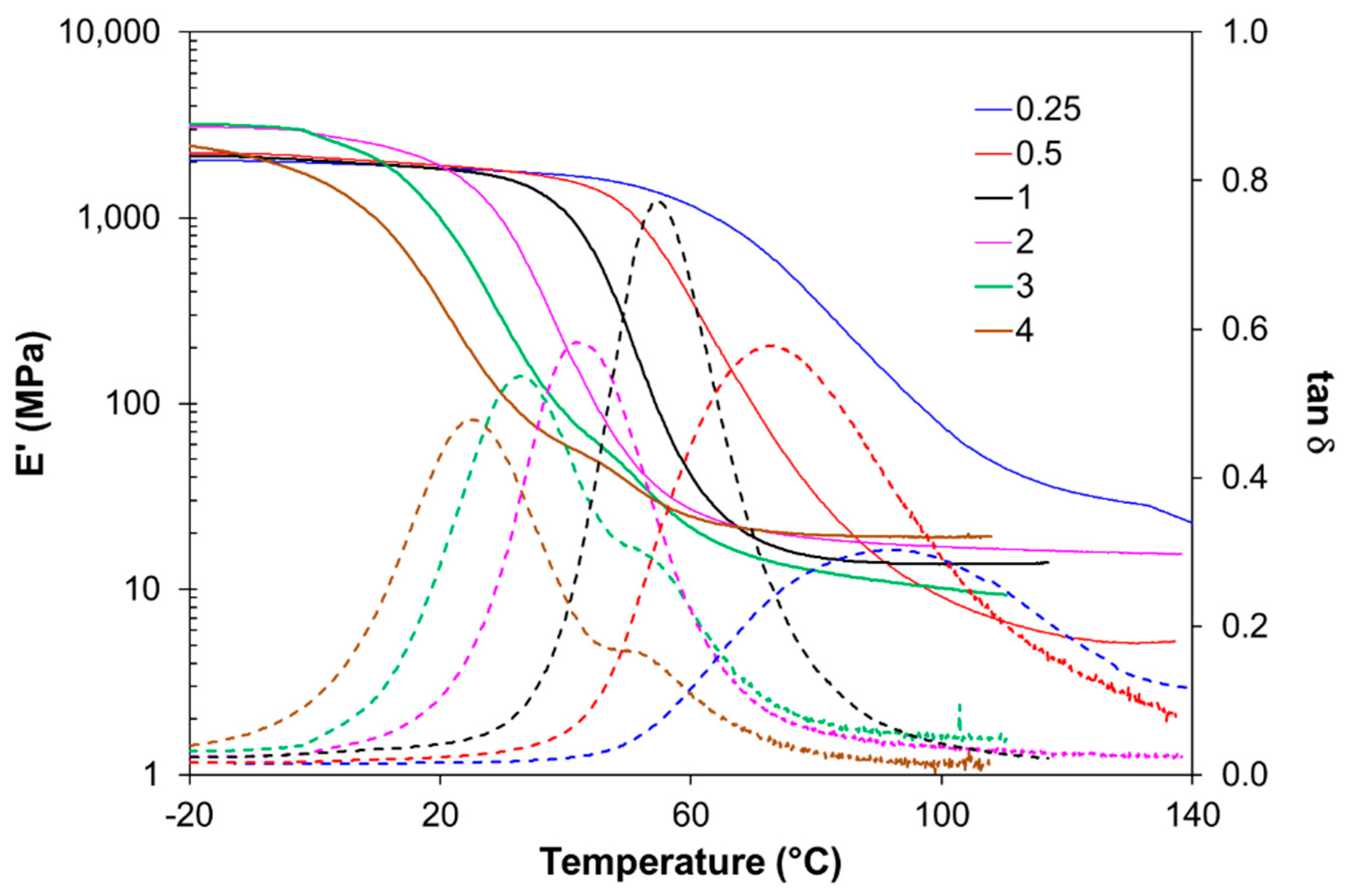
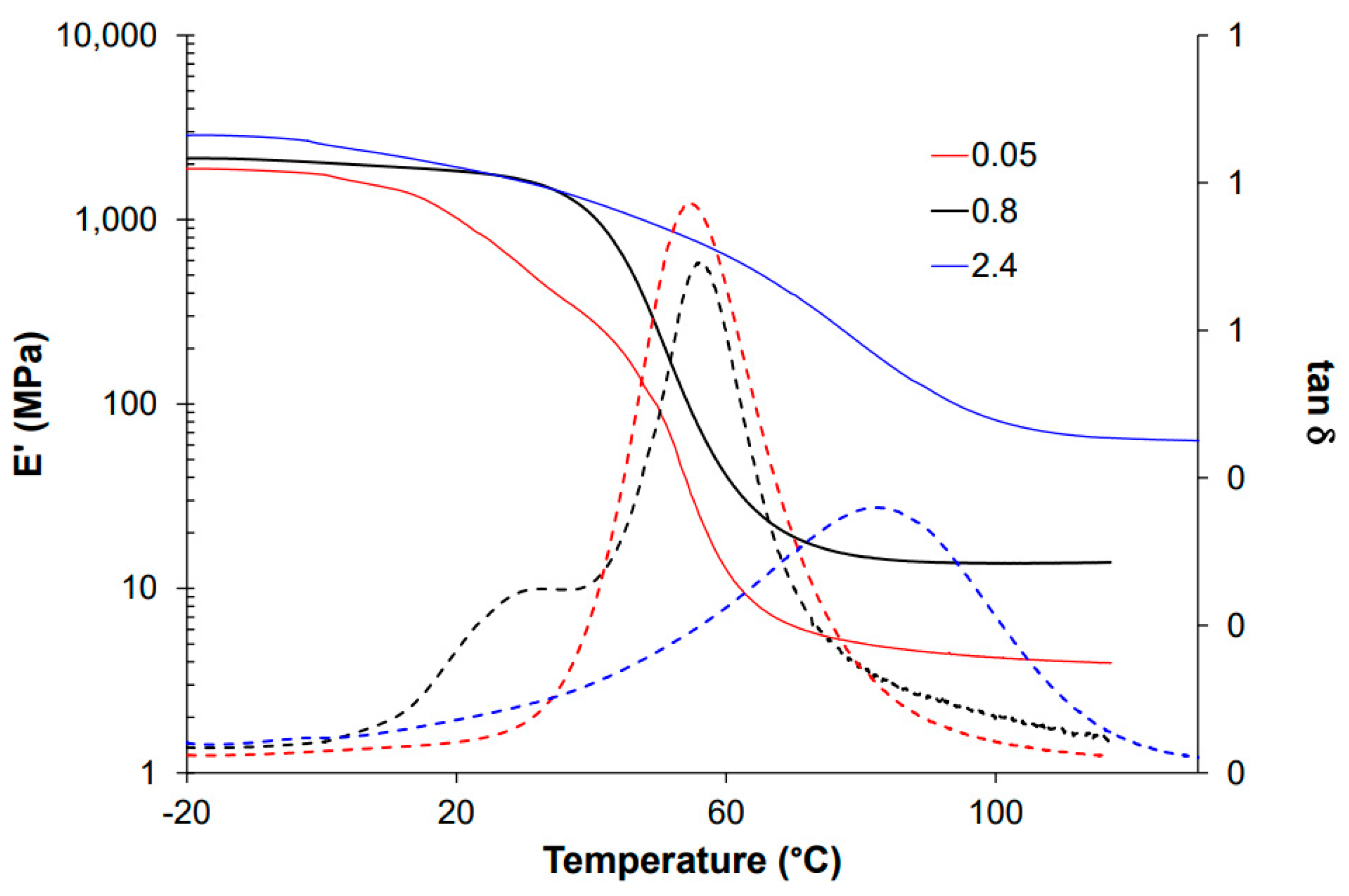
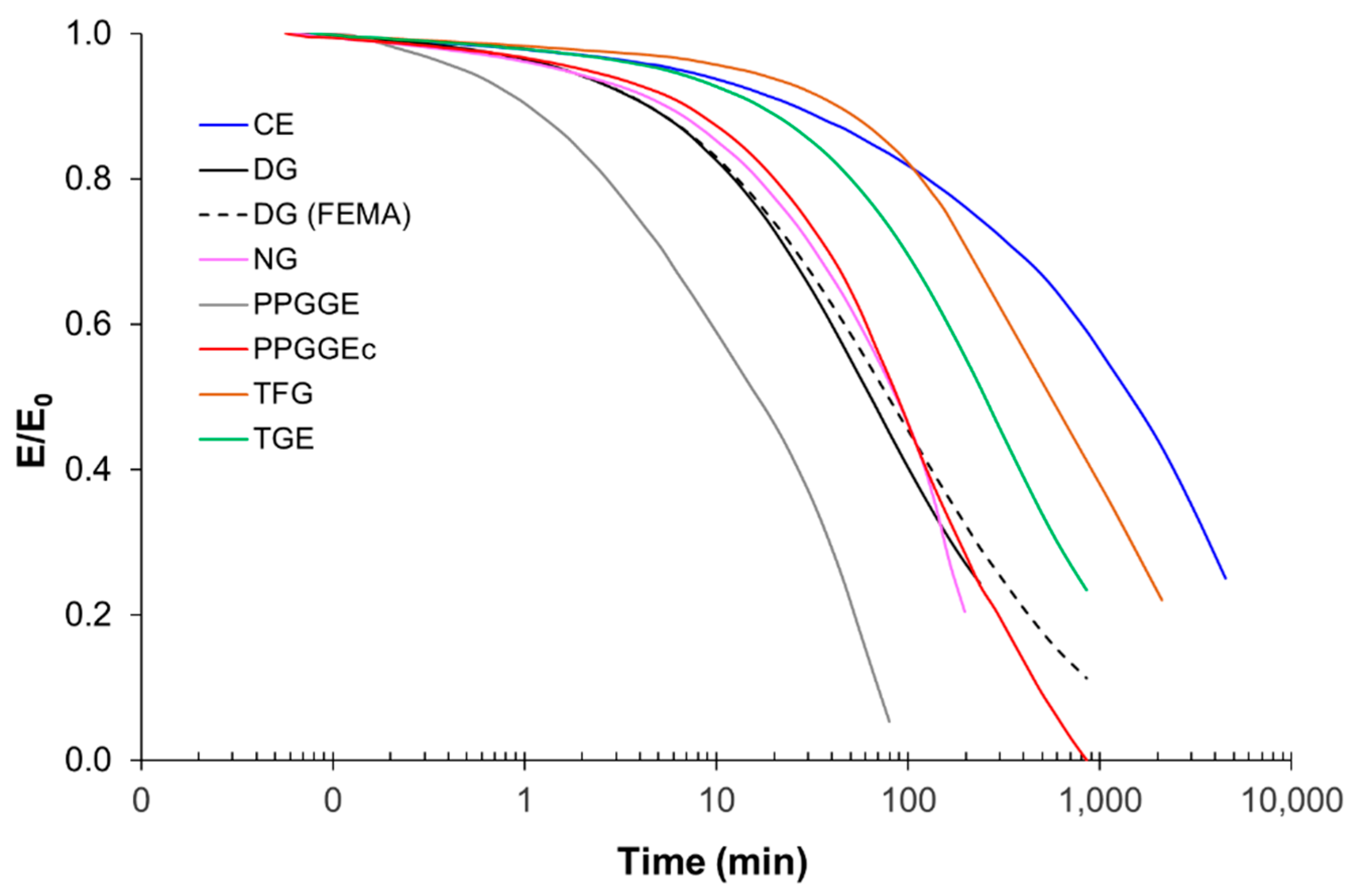
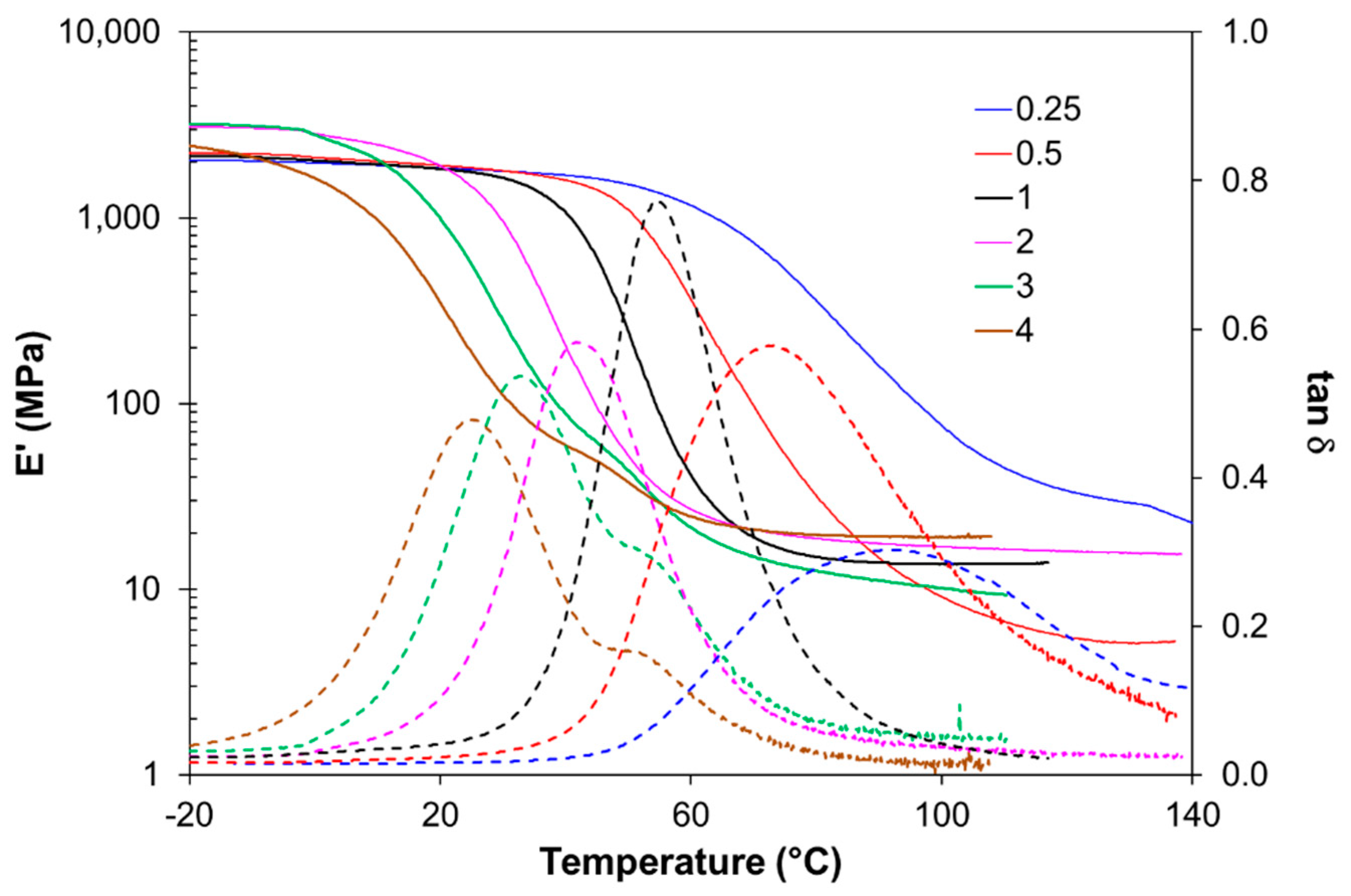
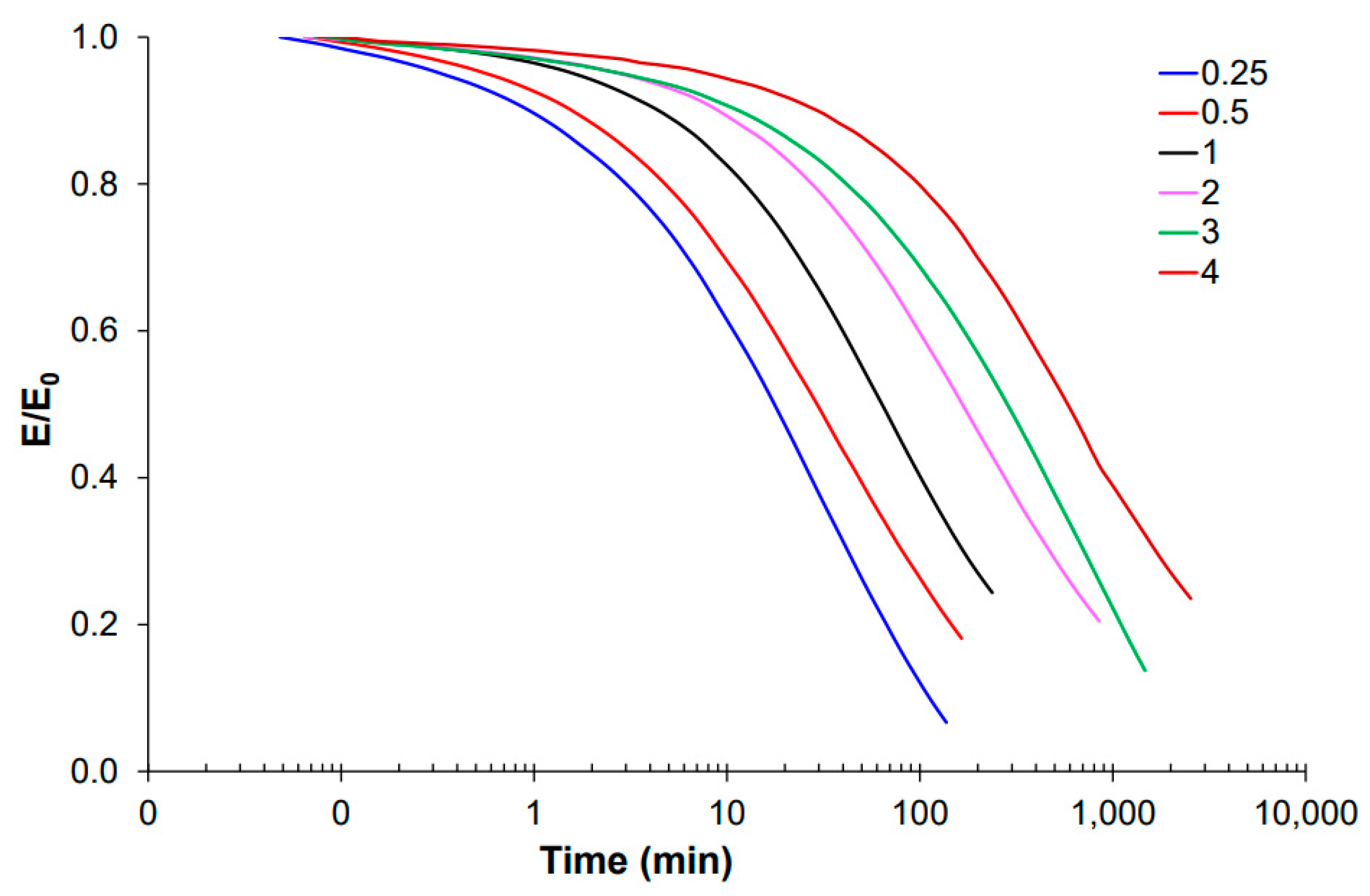
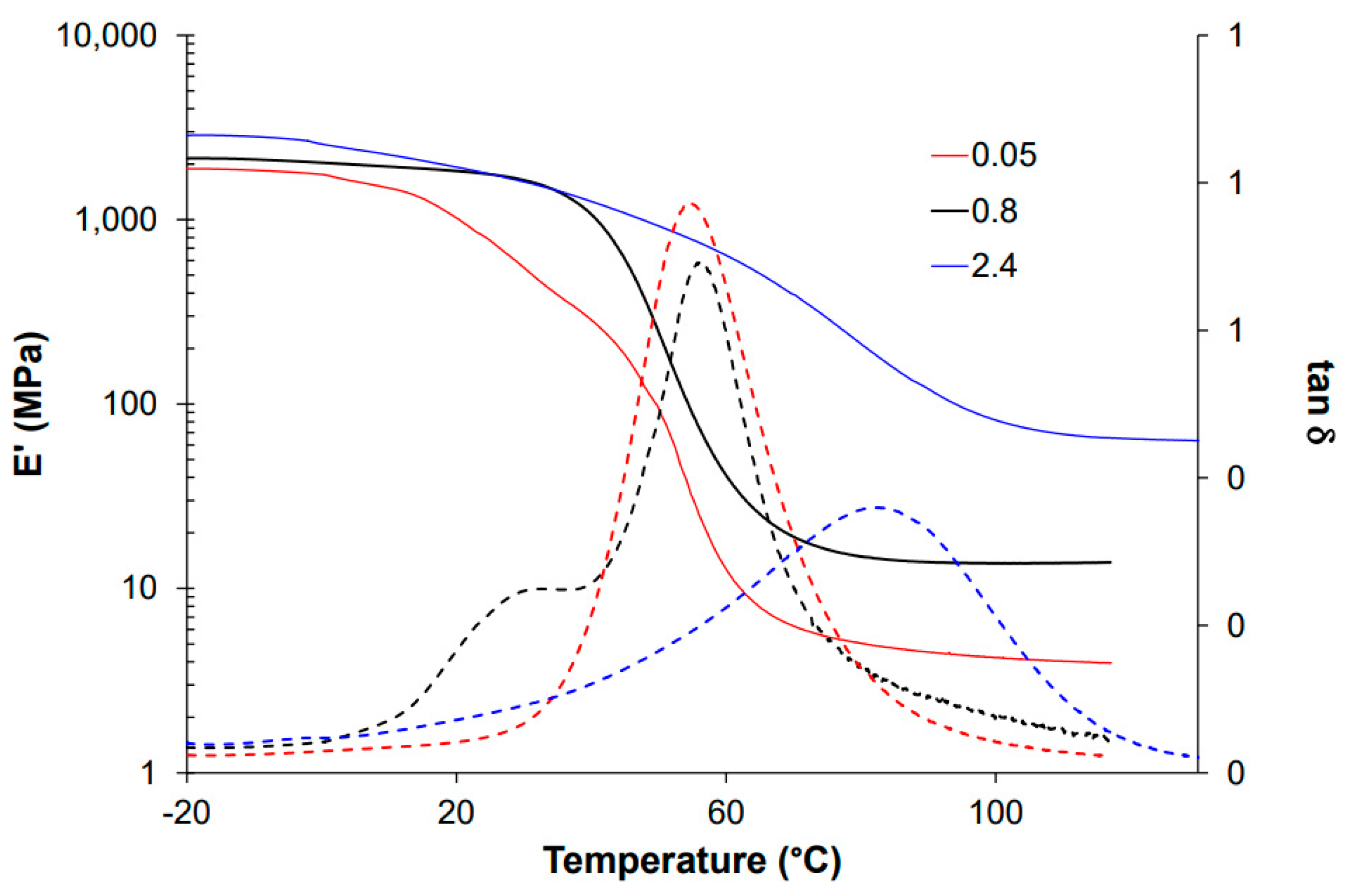
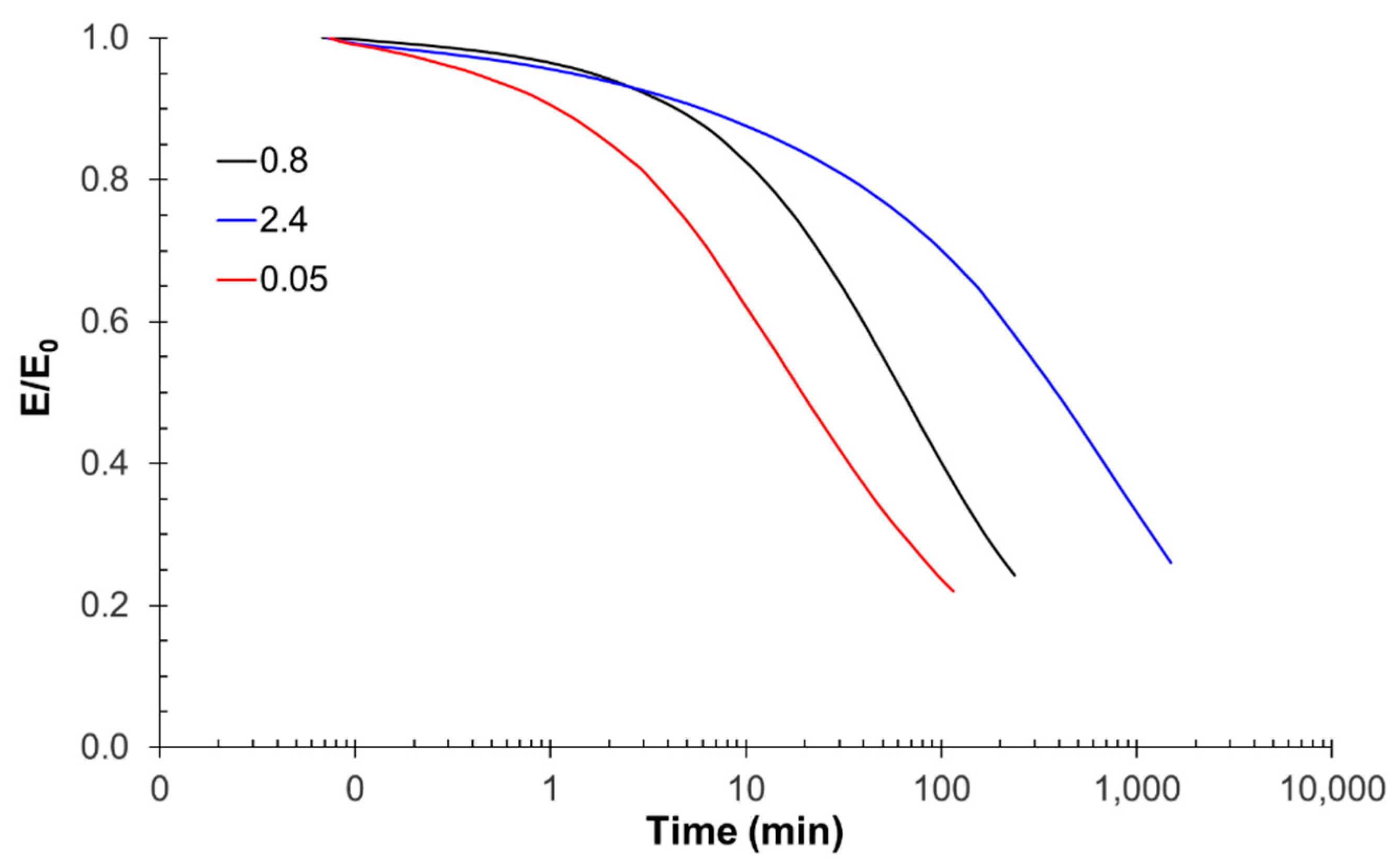
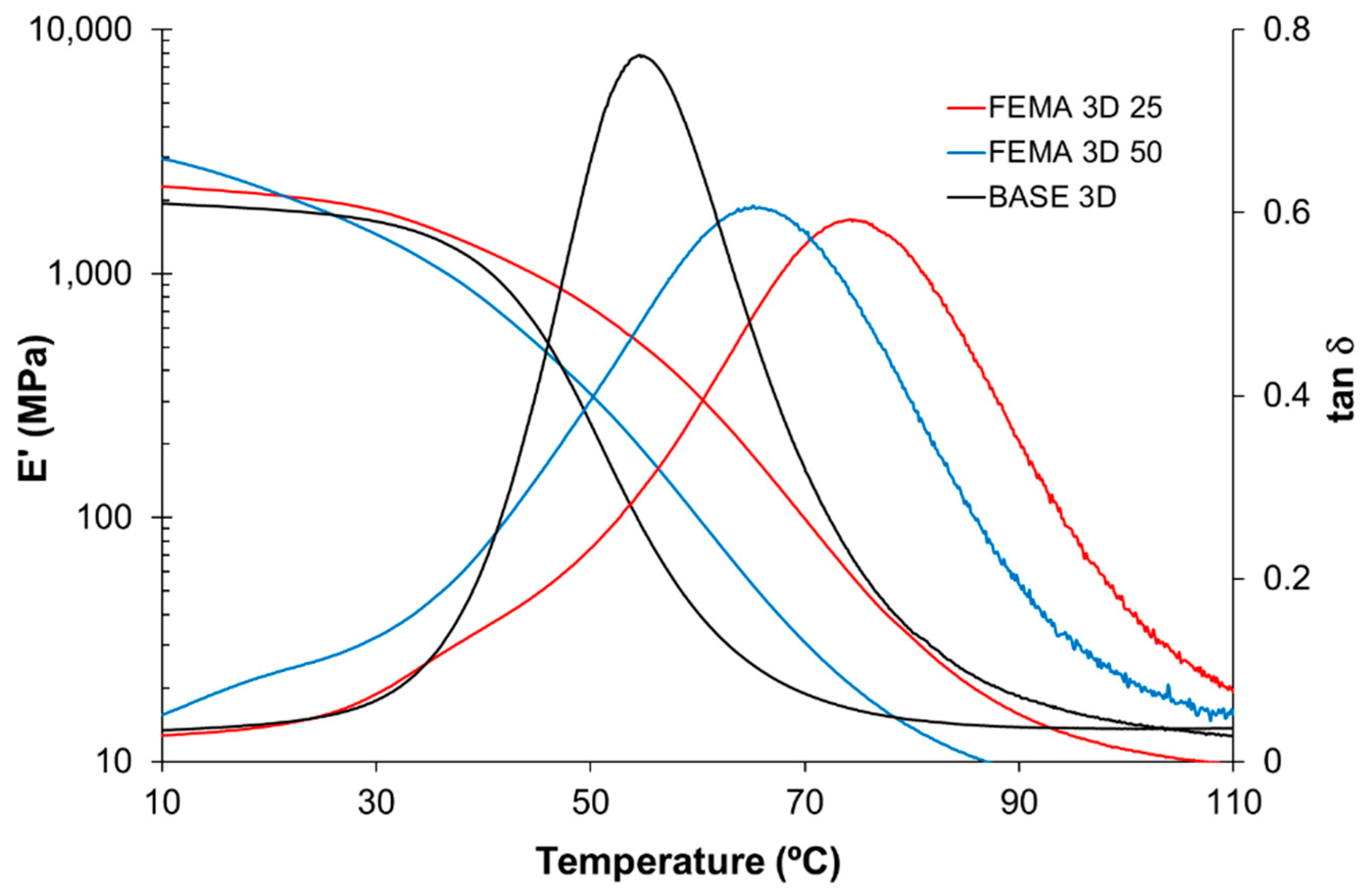
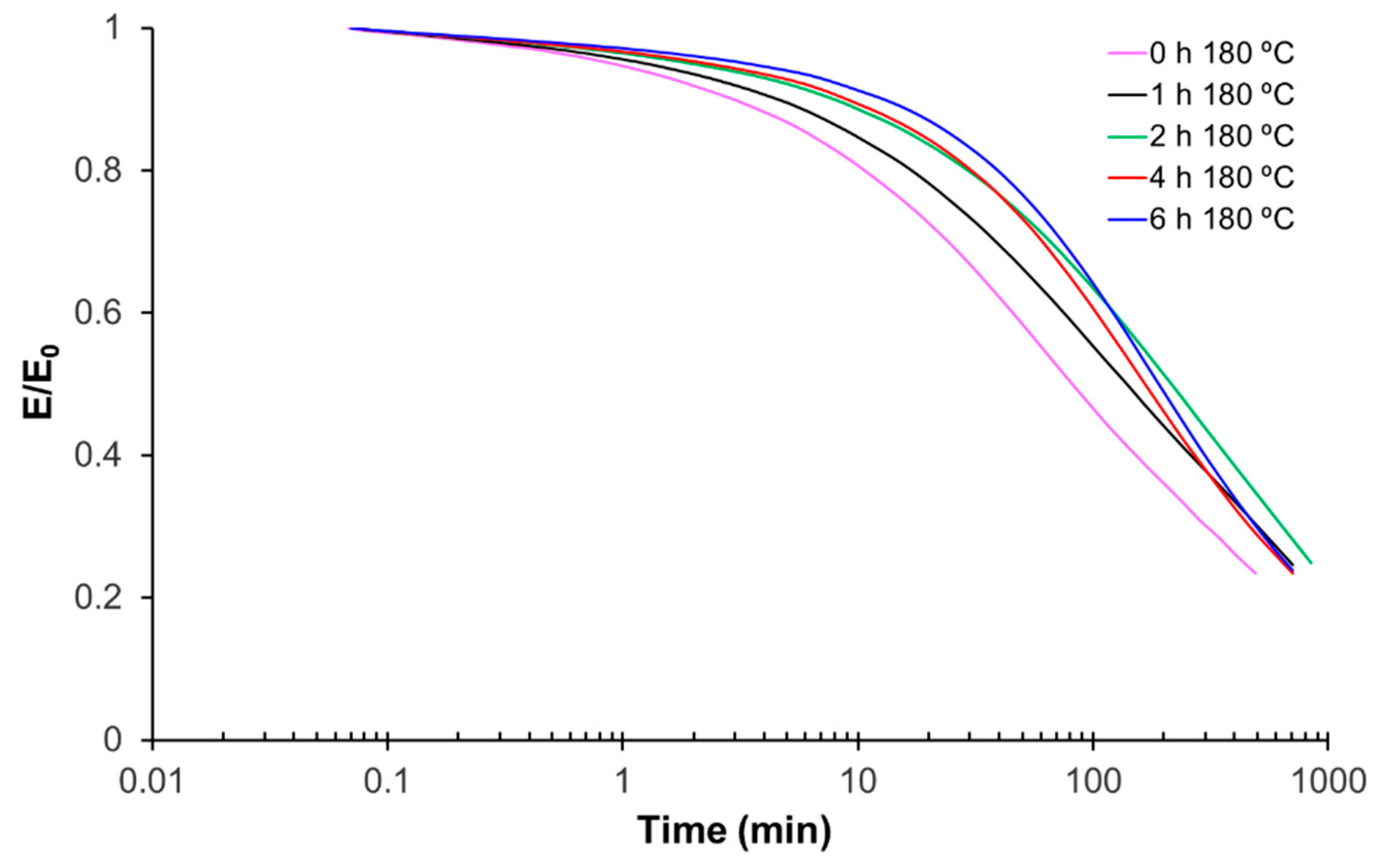
For thermomechanical analysis, a TA Instruments DMA Q800 device (TA Instruments, New Castle, DE, USA) was used. Prismatic samples with dimensions of 1.5 × 10 × 30 mm3 (thickness × width × length) were analyzed using a single cantilever clamp with a free length of 10 mm at a frequency of 1 Hz and an amplitude of 15 µm at 3 °C min−1 from −50 °C up to the rubbery state to measure the storage modulus, loss modulus, and tan δ. Stress relaxation experiments were performed on samples with dimensions of 1.5 × 10 × 20 mm3 using a 3-point bending configuration with a preload force of 0.01 N and a fixed strain of 1%. The flexural modulus was determined at 25 °C using a 3-point bending configuration with the same dimensions as used in the stress relaxation experiments, using the same preload force and a force ramp of 3 N/min.
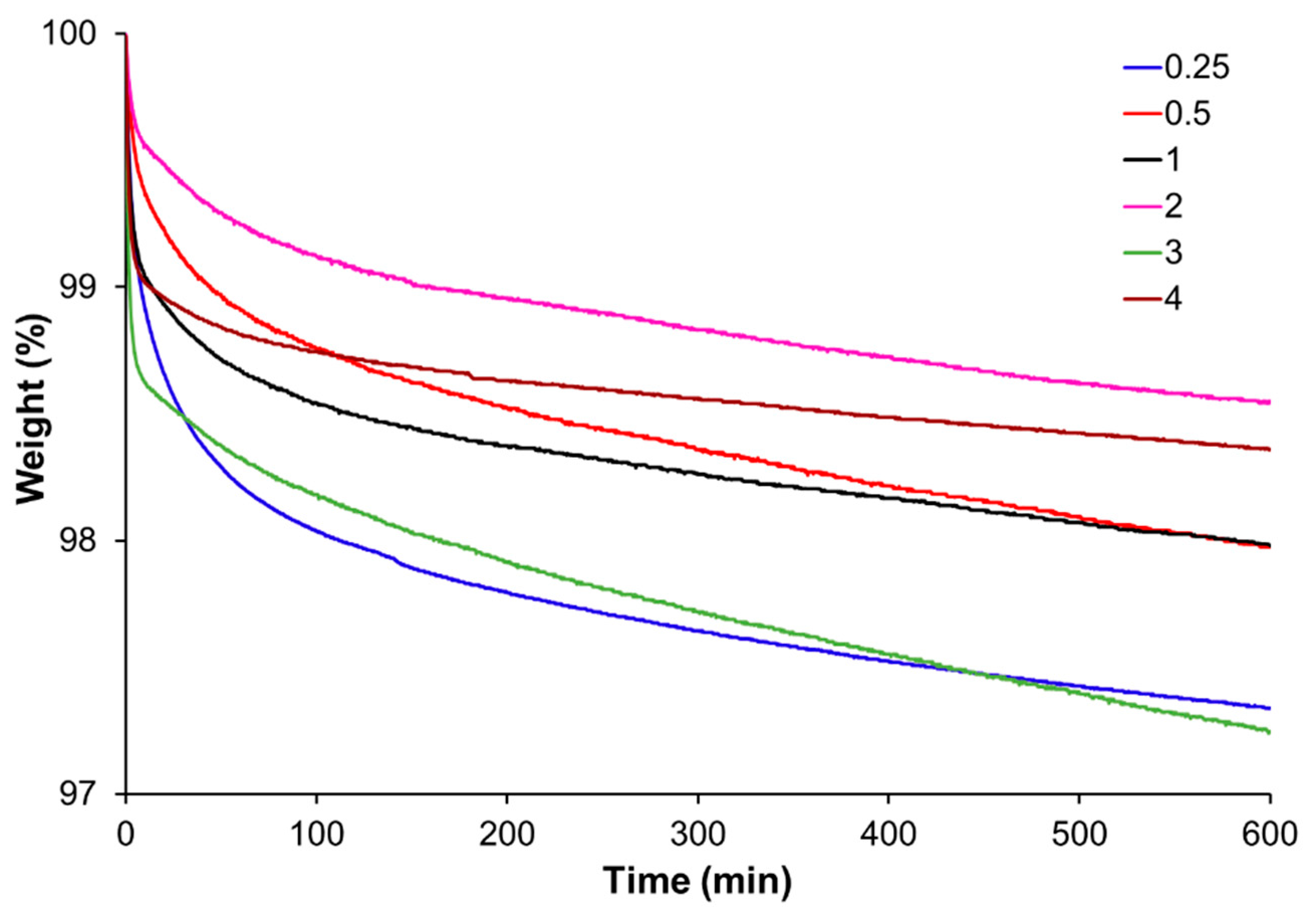
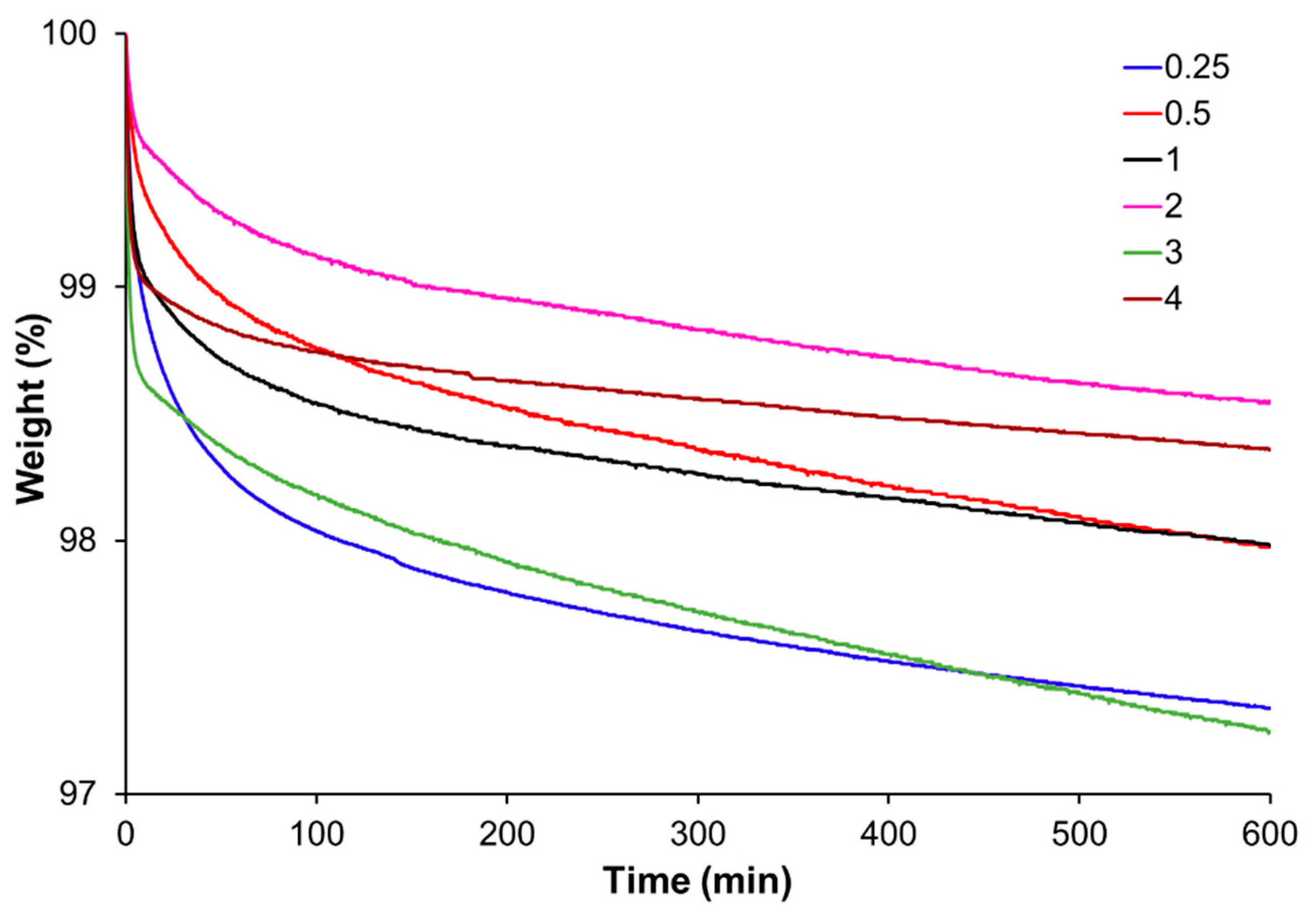
Thermogravimetric analysis (TGA) was performed using a Mettler TGA/SDTA 851e/LF/1100 thermobalance (Mettler-Toledo, Greifensee, Switzerland). Fully cured samples were analyzed under isothermal conditions at 180 °C and under 50 cm3 min−1 of nitrogen purge.
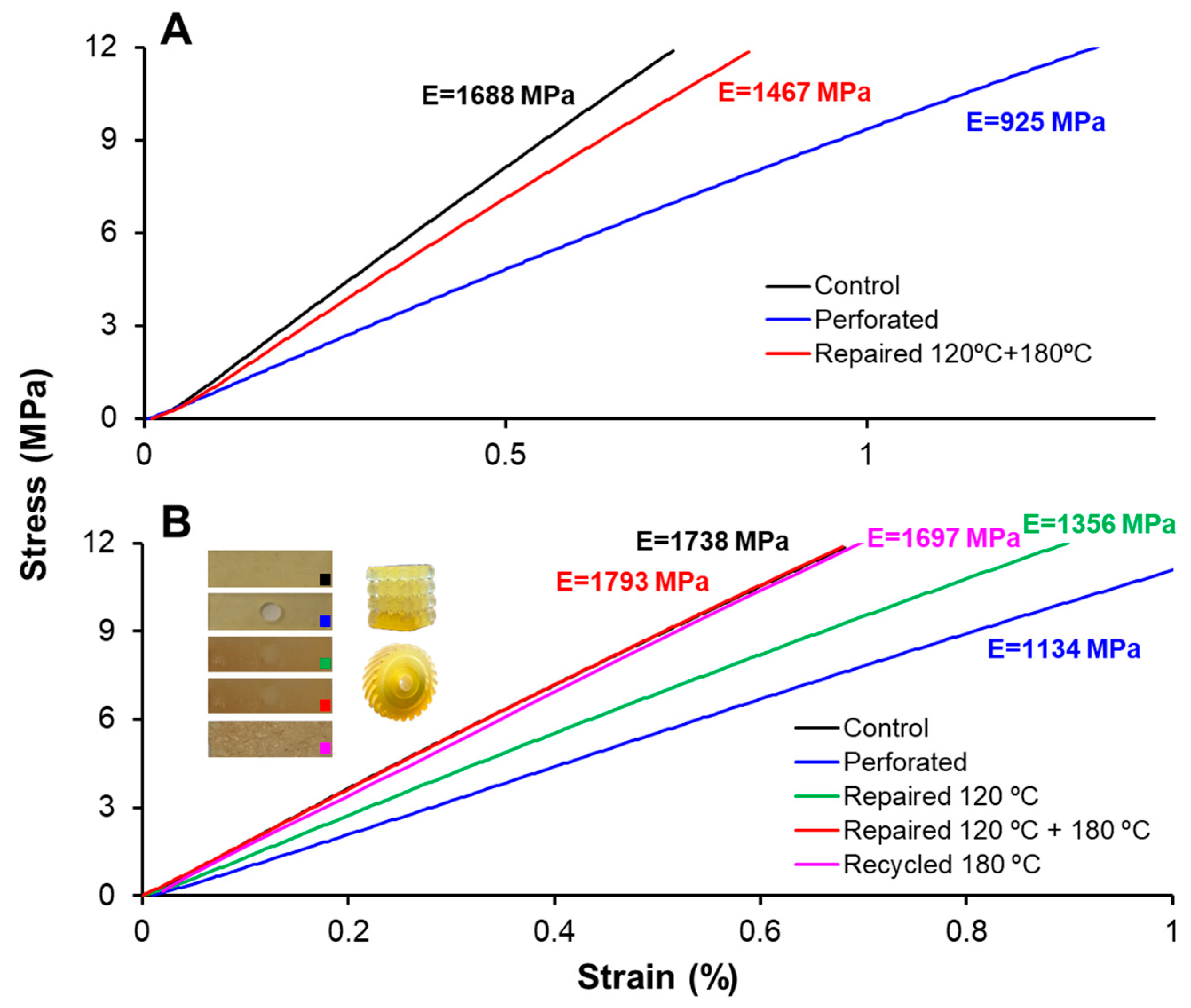
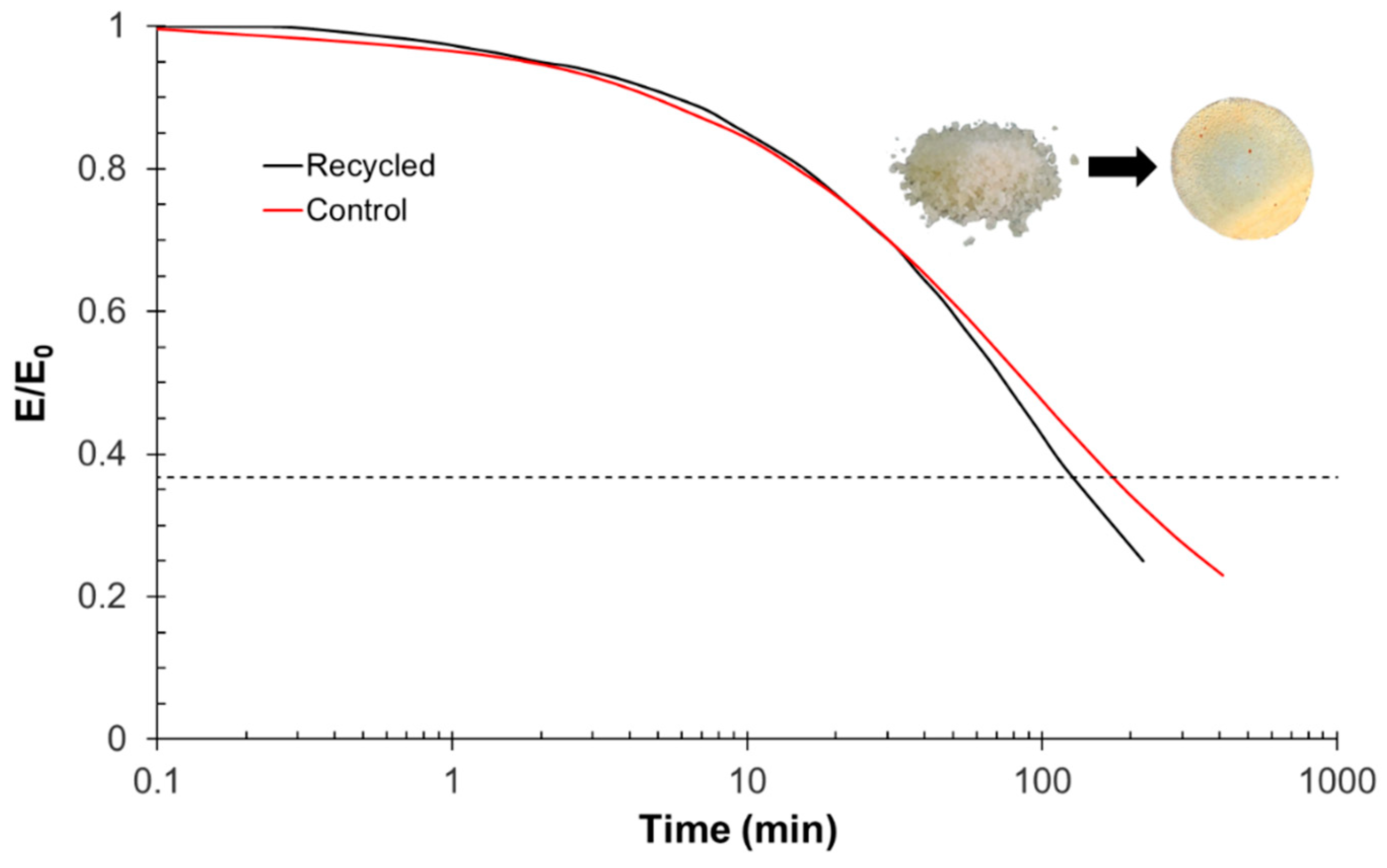
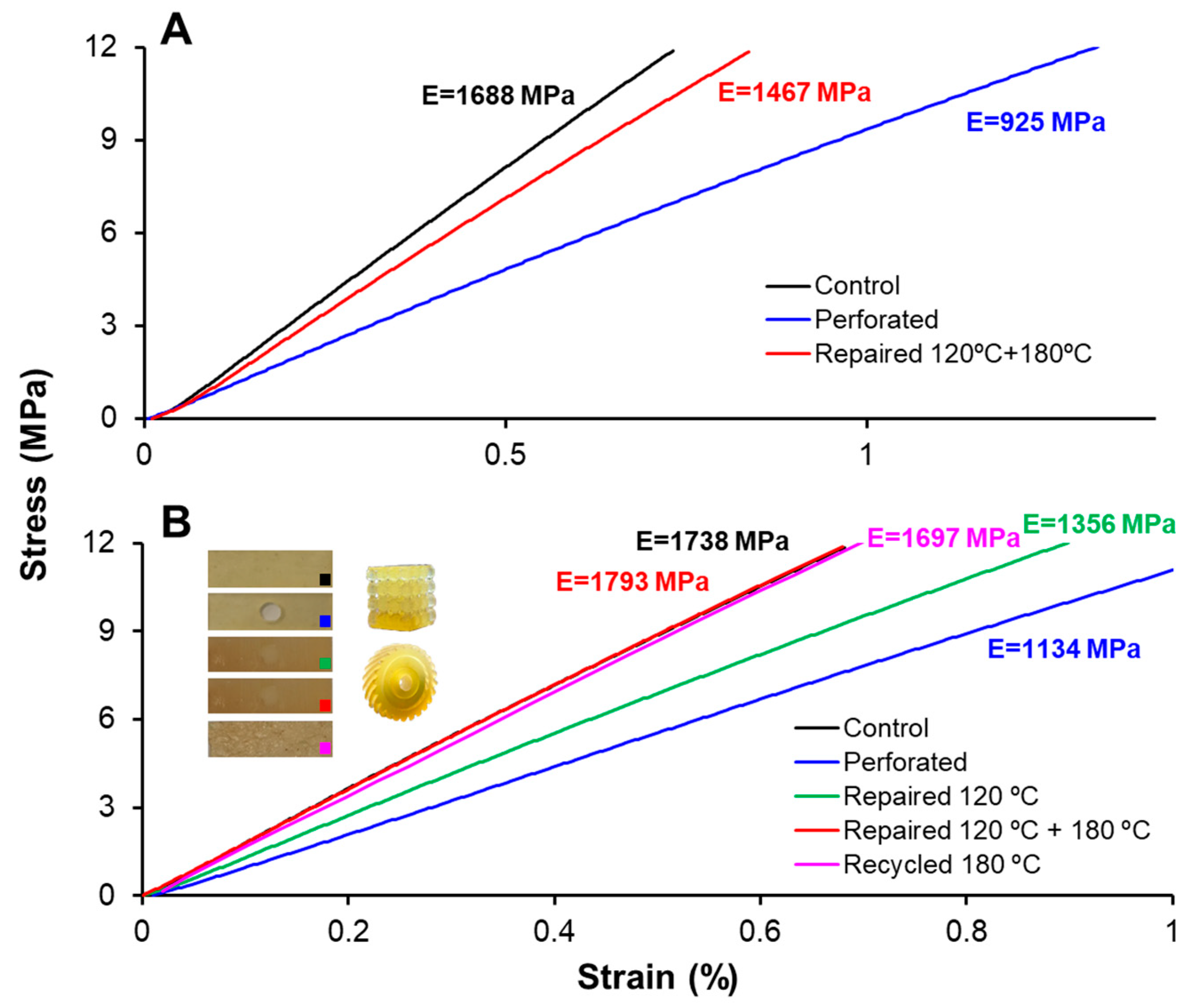
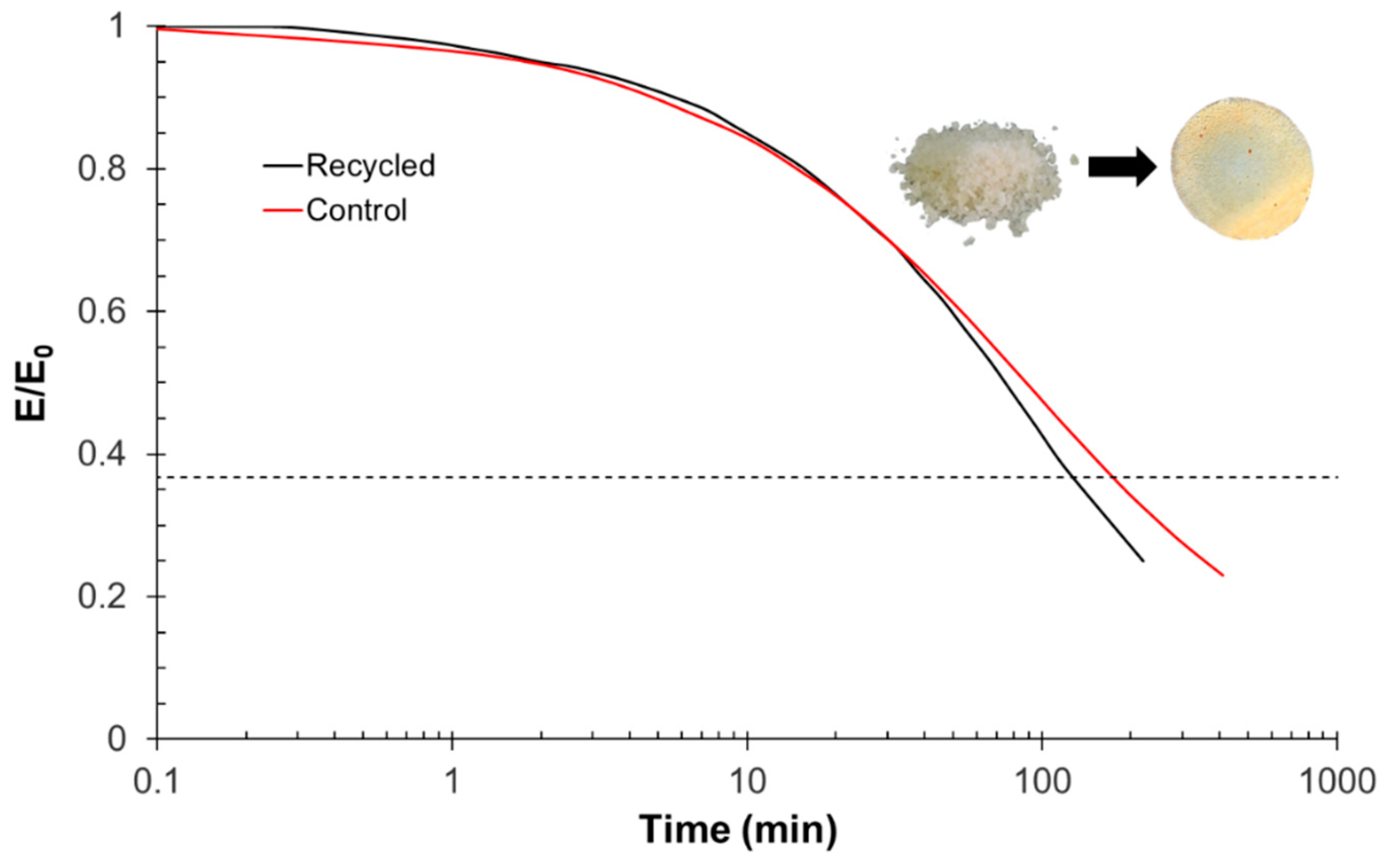
The recycling of finely chopped samples was performed using a Specac Atlas manual 15T hydraulic hot-press (Specac Ltd., Orpington, UK) at a pressure of 12 MPa in an aluminum mold at 180 °C for 8 h.
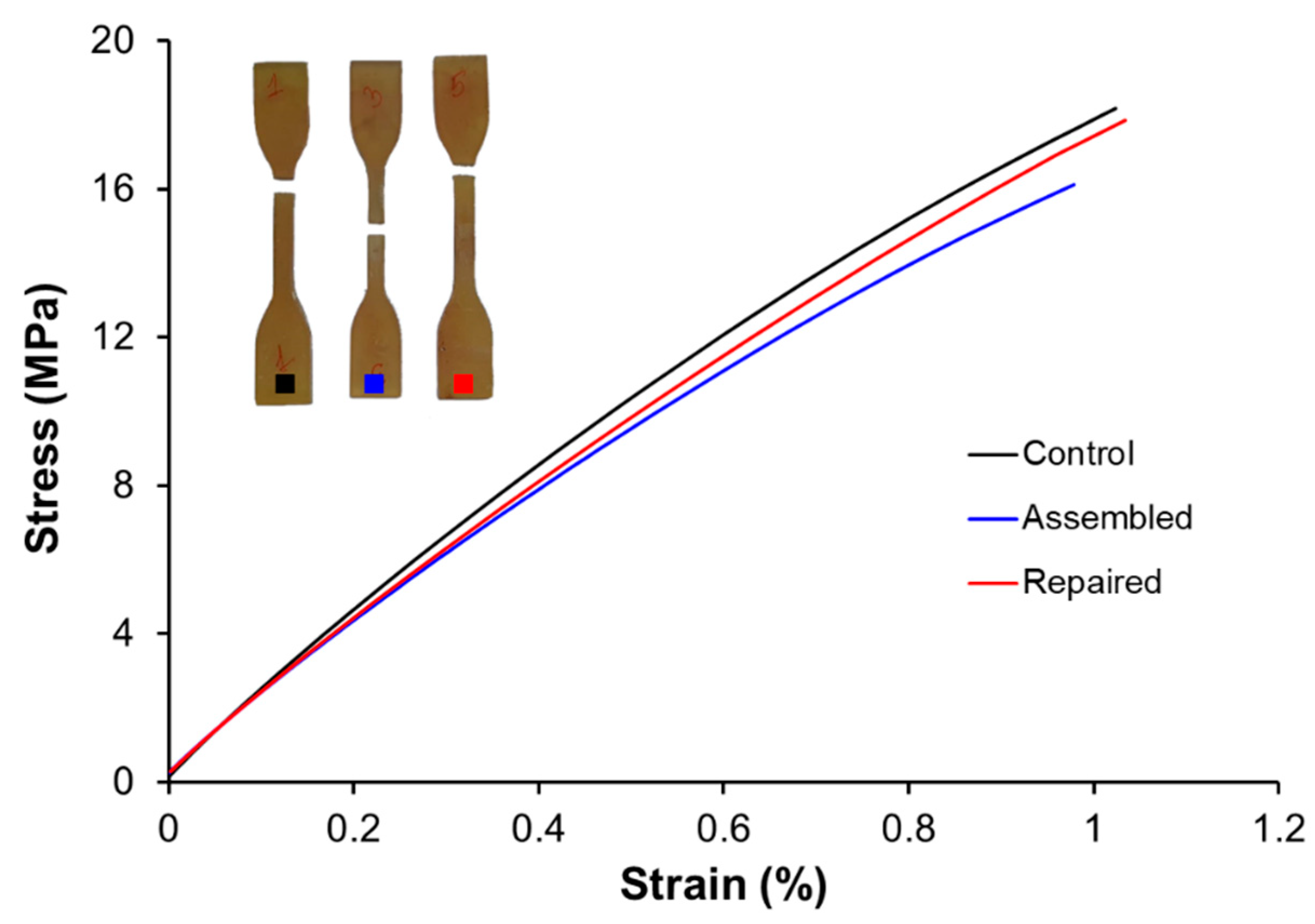
Tensile tests on type-IV standard (ASTM D638-14) dog-bone samples were performed using an Instron 3366 Universal Testing Machine (Instron, Norwood, MA, USA) equipped with an extensometer. The test was performed at a constant displacement rate of 2 mm·min−1 until specimen failure. Young’s modulus was calculated for each sample using the linear regime of its stress–strain response.
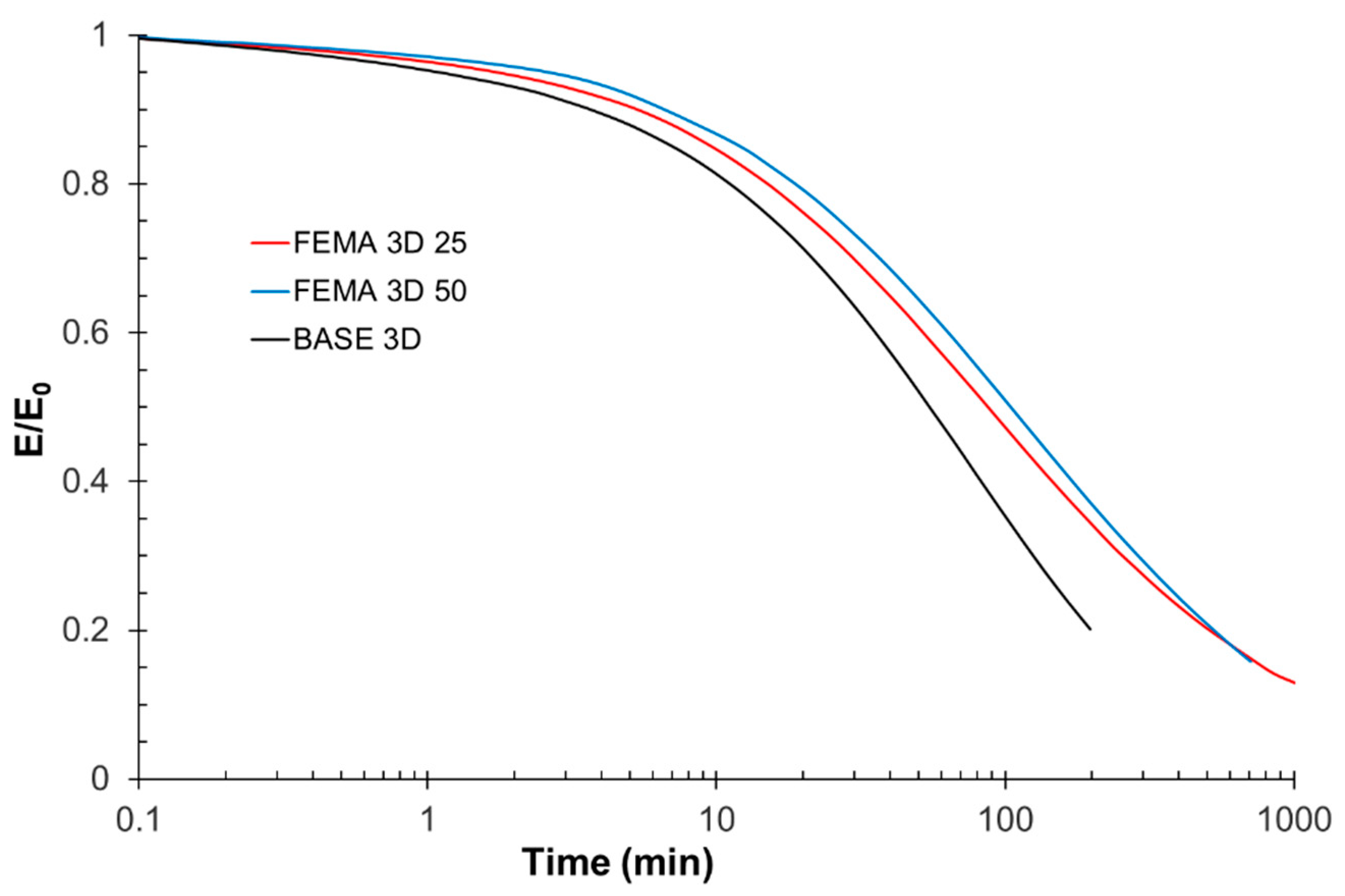
The viscosity of a particular formulation, namely FEMA 3D 25 (vide infra), was measured with a Brookfield RST Rheometer (AMETEK Brookfield, Middleboro, MA, USA) with a cone/plate accessory using a shear rate of 10 to 1000 s−1 at 25 °C.



 ,
, 



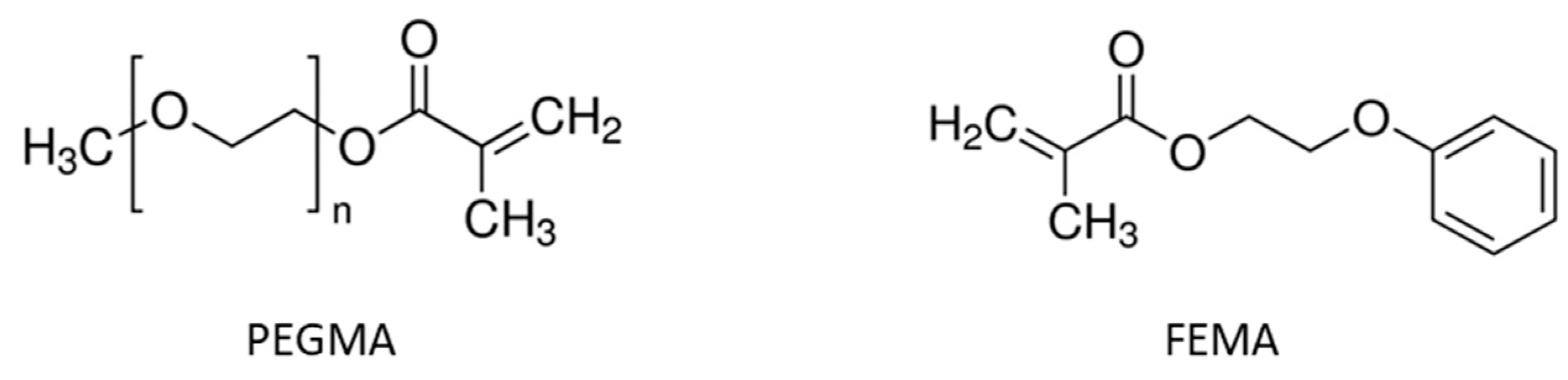




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
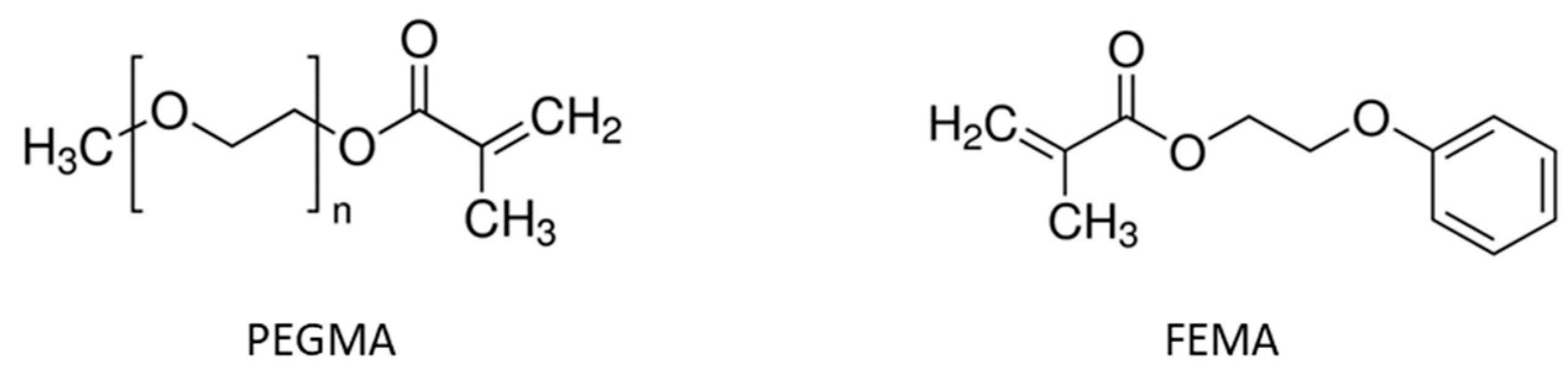{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}