
Recycled Poly(Ethylene Terephthalate) from Waste Textiles with Improved Thermal and Rheological Properties by Chain Extension


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
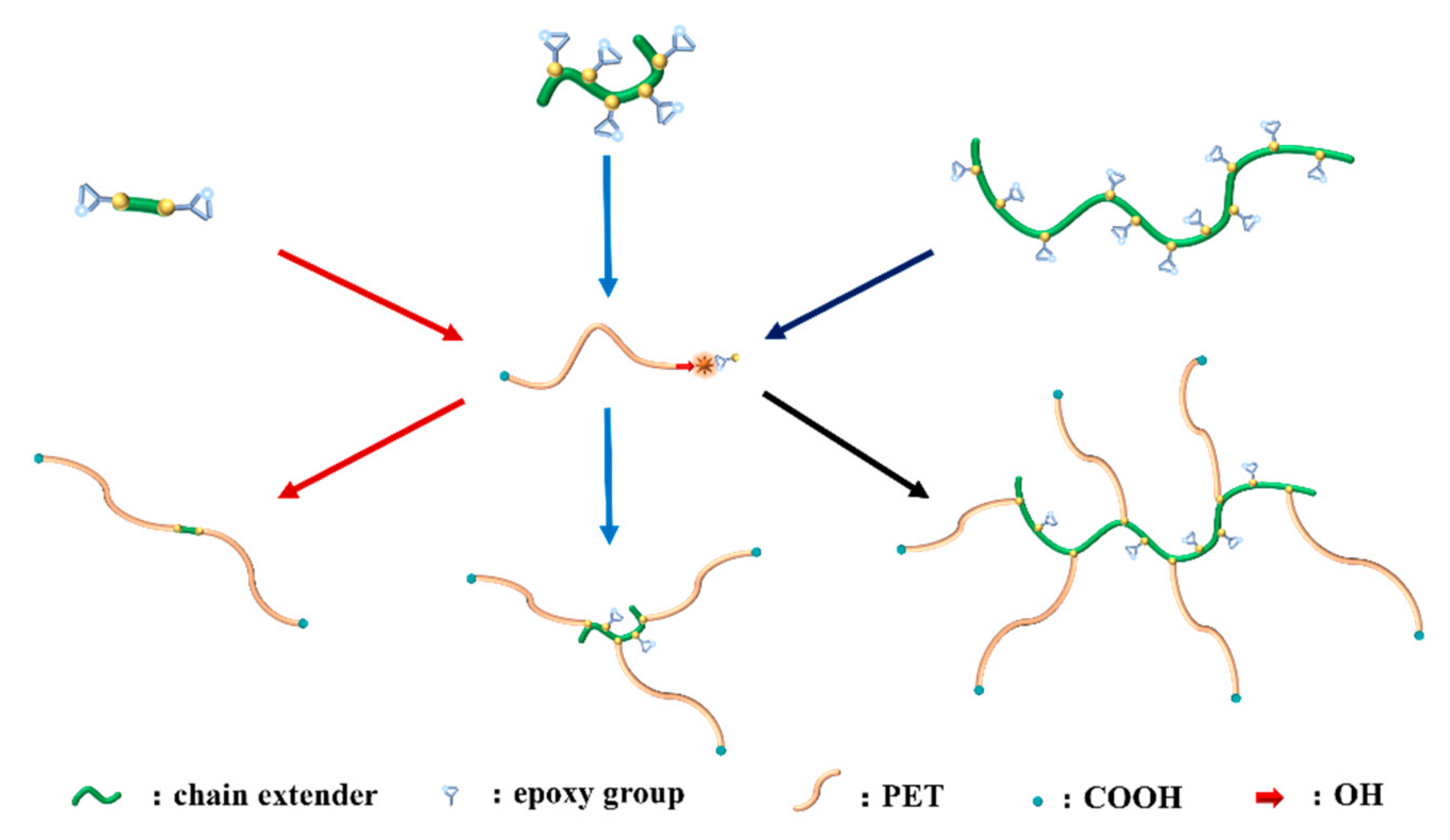
2.2. Synthesis of Copolymers
2.3. Blending Process of PET with Chain Extenders
2.4. The Melt Flow Rate
2.5. Intrinsic Viscosity
2.6. Gel Permeation Chromatography
2.7. Nuclear Magnetic Resonance Spectroscopy
2.8. Thermal Gravimetric Analysis
2.9. Differential Scanning Calorimetry
2.10. Dynamic Rheological Measurements
3. Results
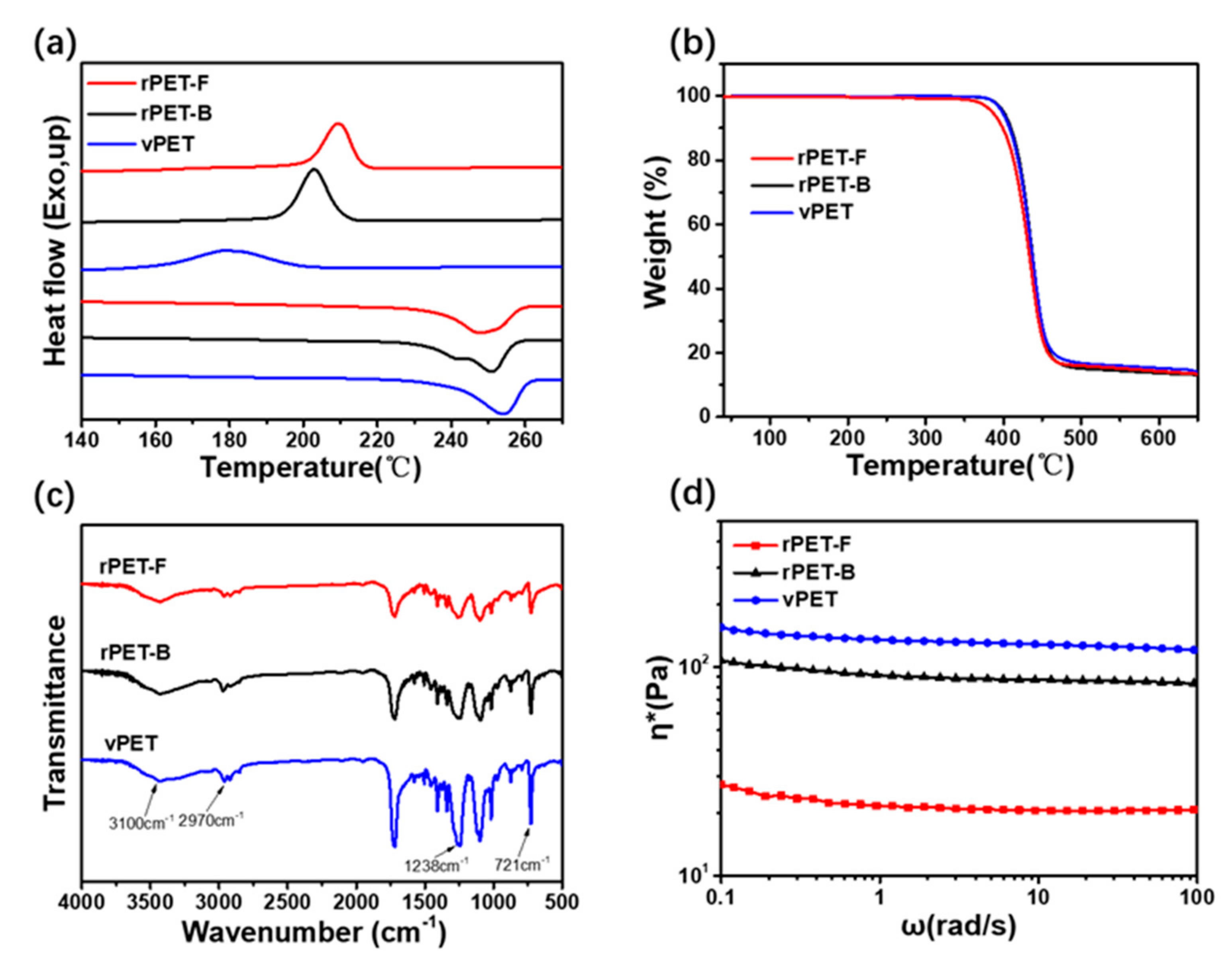
3.1. Properties of the rPET-F, rPET-B and vPET
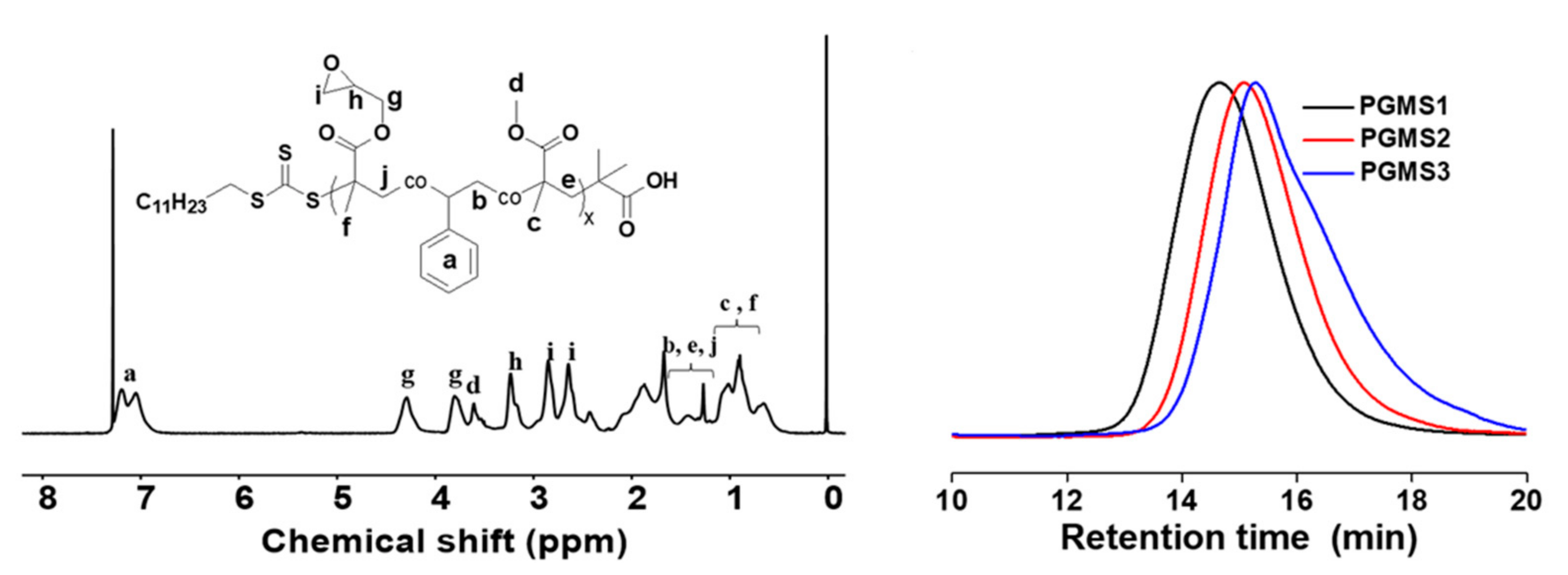
3.2. Synthesis and Characterization of Chain Extender
3.3. IV and MFI of the Chain Extended PET Samples
3.4. Thermal Characterization of the PET Samples
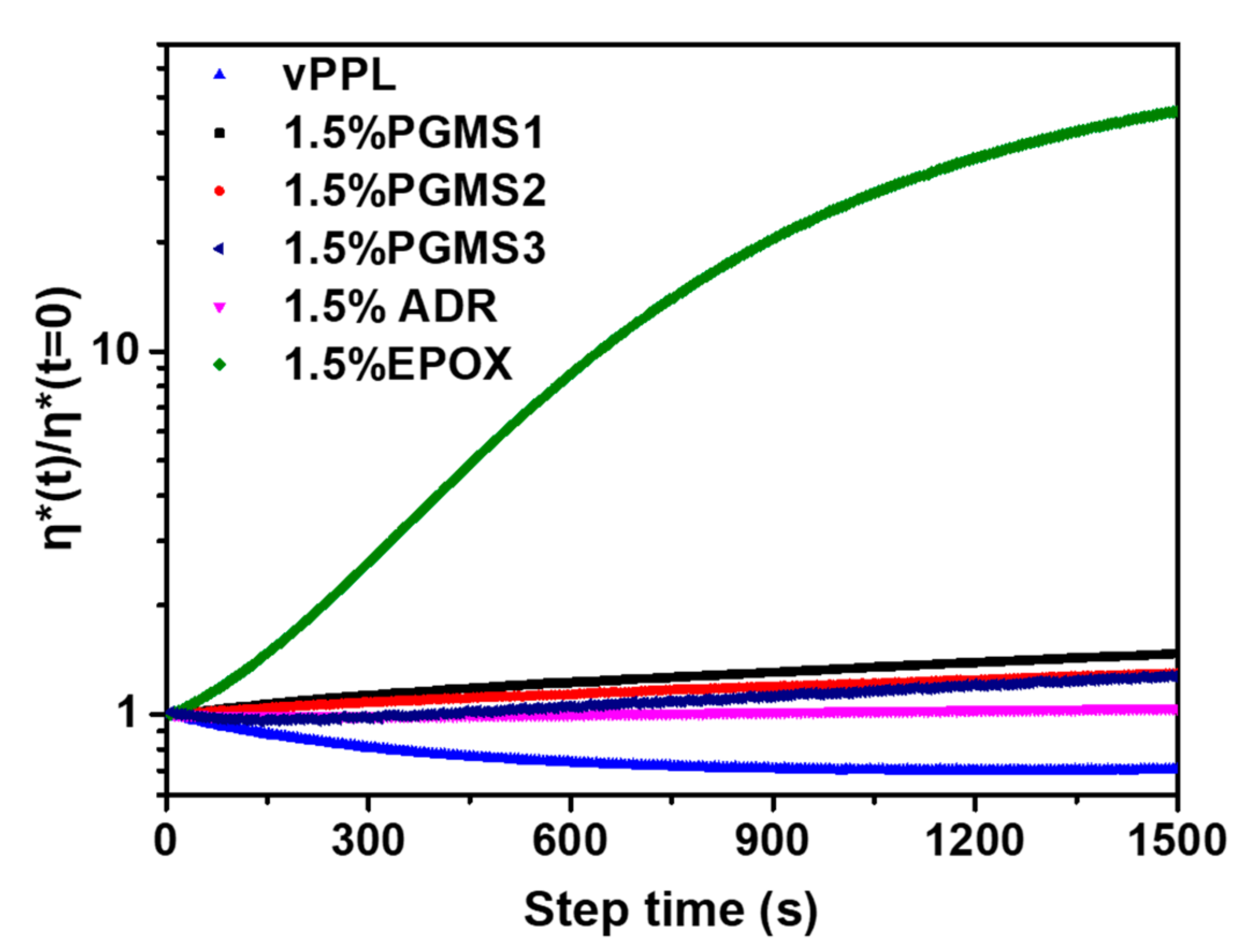
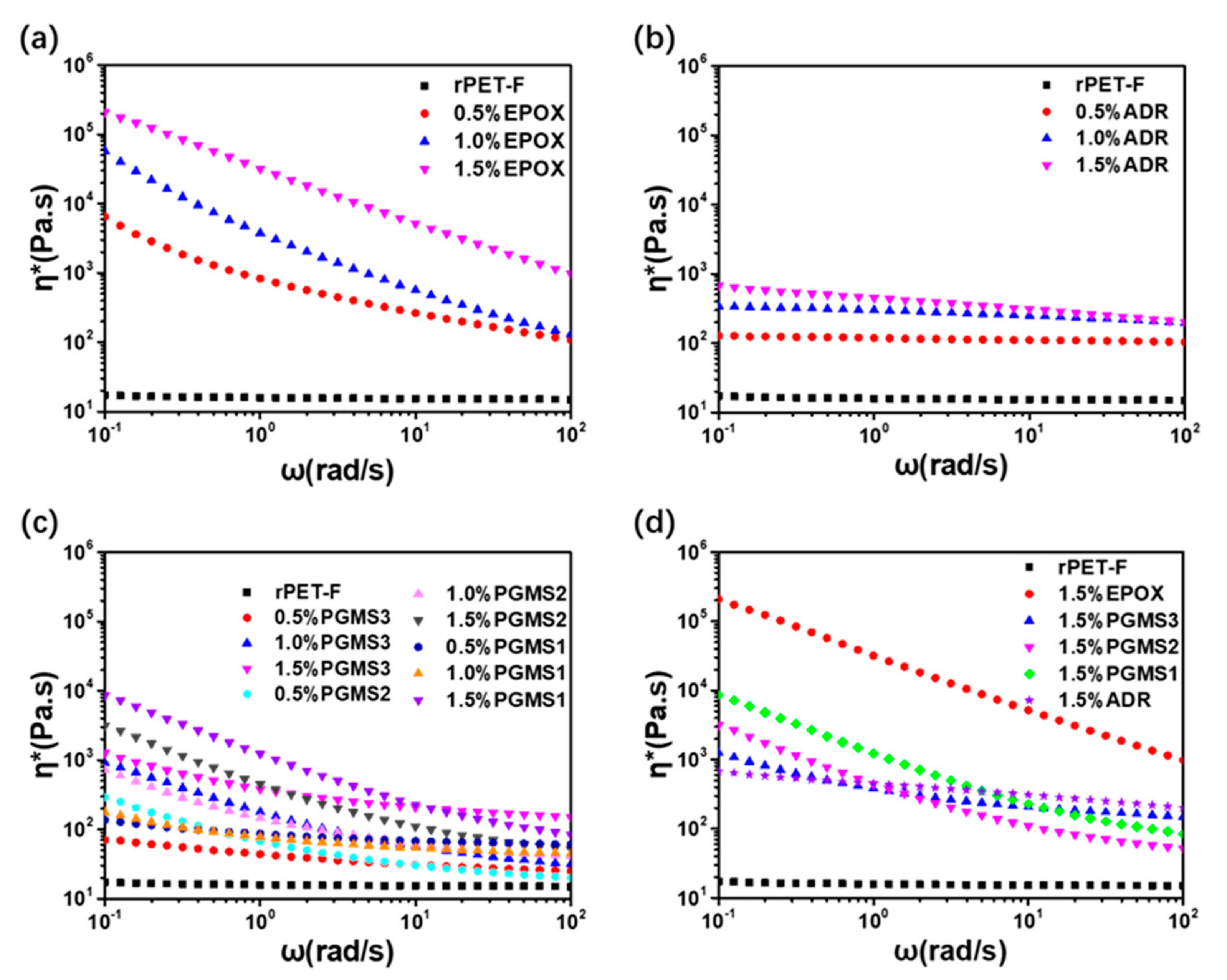
3.5. Rheological Behavior of the PET Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Damayanti, D.; Wulandari, L.A.; Bagaskoro, A.; Rianjanu, A.; Wu, H.S. Possibility routes for textile recycling technology. Polymers 2021, 13, 3834. [Google Scholar] [CrossRef] [PubMed]
- Al-Salem, S.M.; Lettieri, P.; Baeyens, J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manag. 2009, 29, 2625–2643. [Google Scholar] [CrossRef] [PubMed]
- Aizenshtein, E.M. Polyester fibres in the post-crisis period. Fibre Chem. 2011, 42, 341–349. [Google Scholar] [CrossRef]
- Zou, Y.; Reddy, N.; Yang, Y. Reusing polyester/cotton blend fabrics for composites. Compos. Part B Eng. 2011, 42, 763–770. [Google Scholar] [CrossRef]
- Mäkelä, M.; Rissanen, M.; Sixta, H. Machine vision estimates the polyester content in recyclable waste textiles. Resour. Conserv. Recycl. 2020, 161, 105007. [Google Scholar] [CrossRef]
- Sandin, G.; Peters, G.M. Environmental impact of textile reuse and recycling—A review. J. Clean. Prod. 2018, 184, 353–365. [Google Scholar] [CrossRef]
- Harmsen, P.; Scheffer, M.; Bos, H. Textiles for circular fashion: The logic behind recycling options. Sustainability 2021, 13, 9714. [Google Scholar] [CrossRef]
- Keßler, L.; Matlin, S.A.; Kümmerer, K. The contribution of material circularity to sustainability—Recycling and reuse of textiles. Curr. Opin. Green Sustain. Chem. 2021, 32, 100535. [Google Scholar] [CrossRef]
- Taniguchi, I.; Yoshida, S.; Hiraga, K.; Miyamoto, K.; Kimura, Y.; Oda, K. Biodegradation of PET: Current status and application aspects. ACS Catal. 2019, 9, 4089–4105. [Google Scholar] [CrossRef]
- Fortuna, L.M.; Diyamandoglu, V. Optimization of greenhouse gas emissions in second-hand consumer product recovery through reuse platforms. Waste Manag. 2017, 66, 178–189. [Google Scholar] [CrossRef]
- Thiounn, T.; Smith, R.C. Advances and approaches for chemical recycling of plastic waste. J. Polym. Sci. 2020, 58, 1347–1364. [Google Scholar] [CrossRef] [Green Version]
- Shamsi, R.; Abdouss, M.; Sadeghi, G.M.M.; Taromi, F.A. Synthesis and characterization of novel polyurethanes based on aminolysis of poly(ethylene terephthalate) wastes, and evaluation of their thermal and mechanical properties. Polym. Int. 2009, 58, 22–30. [Google Scholar] [CrossRef]
- Burgess, S.K.; Leisen, J.E.; Kraftschik, B.E.; Mubarak, C.R.; Kriegel, R.M.; Koros, W.J. Chain mobility, thermal, and mechanical properties of poly(ethylene furanoate) ccompared to poly(ethylene terephthalate). Macromolecules 2014, 47, 1383–1391. [Google Scholar] [CrossRef]
- Ghisellini, P.; Cialani, C.; Ulgiati, S. A review on circular economy: The expected transition to a balanced interplay of environmental and economic systems. J. Clean. Prod. 2016, 114, 11–32. [Google Scholar] [CrossRef]
- Mao, Y.; Li, Q.; Wu, C. Surface modification of PET fiber with hybrid coating and its effect on the properties of PP composites. Polymers 2019, 11, 1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.O. The circular economy opportunity for urban & industrial innovation in China. Circ. Econ. Perspect. Ser. 2018. Available online: https://ellenmacarthurfoundation.org/urban-and-industrial-innovation-in-china (accessed on 18 September 2018).
- Standau, T.; Nofar, M.; Dörr, D.; Ruckdäschel, H.; Altstädt, V. A review on multifunctional epoxy-based Joncryl® ADR chain extended thermoplastics. Polym. Rev. 2021, 1–55. [Google Scholar] [CrossRef]
- Cardi, N.; Po, R.; Giannotta, G.; Occhiello, E.; Garbassi, F.; Messina, G. Chain extension of recycled poly(ethylene terephthalate) with 2,2′-Bis (2-oxazoline). J. Appl. Polym. Sci. 1993, 50, 1501–1509. [Google Scholar] [CrossRef]
- Karayannidis, G.P.; Psalida, E.A. Chain extension of recycled poly(ethylene terephthalate) with 2,2′-(1,4-phenylene) Bis (2-oxazoline). J. Appl. Polym. Sci. 2000, 77, 2206–2211. [Google Scholar] [CrossRef]
- Kruse, M.; Wagner, M.H. Rheological and molecular characterization of long-chain branched poly(ethylene terephthalate). Rheol. Acta 2017, 56, 887–904. [Google Scholar] [CrossRef]
- Härth, M.; Kaschta, J.; Schubert, D.W. Shear and elongational flow properties of long-chain branched poly(ethylene terephthalates) and correlations to their molecular structure. Macromolecules 2014, 47, 4471–4478. [Google Scholar] [CrossRef]
- Kruse, M.; Wang, P.; Shah, R.S.; Wagner, M.H. Analysis of high melt-strength poly(ethylene terephthalate) produced by reactive processing by shear and elongational rheology. Polym. Eng. Sci. 2019, 59, 396–410. [Google Scholar] [CrossRef]
- Forsythe, J.S.; Cheah, K.; Nisbet, D.R.; Gupta, R.K.; Lau, A.; Donovan, A.R.; O’Shea, M.S.; Moad, G. Rheological properties of high melt strength poly(ethylene terephthalate) formed by reactive extrusion. J. Appl. Polym. Sci. 2006, 100, 3646–3652. [Google Scholar] [CrossRef]
- Daver, F.; Gupta, R.; Kosior, E. Rheological characterisation of recycled poly(ethylene terephthalate) modified by reactive extrusion. J. Mater. Process. Technol. 2008, 204, 397–402. [Google Scholar] [CrossRef]
- Jacques, B.; Devaux, J.; Legras, R.; Nield, E. Investigation on model molecules of the reactions induced by triphenyl phosphite addition during polyester processing. Macromolecules 1996, 29, 3129–3138. [Google Scholar] [CrossRef]
- Bimestre, B.H.; Saron, C. Chain extension of poly(ethylene terephthalate) by reactive extrusion with secondary stabilizer. Mater. Res. 2012, 15, 467–472. [Google Scholar] [CrossRef]
- Cicero, J.A.; Dorgan, J.R.; Dec, S.F.; Knauss, D.M. Phosphite stabilization effects on two-step melt-spun fibers of polylactide. Polym. Degrad. Stab. 2002, 78, 95–105. [Google Scholar] [CrossRef]
- Jacques, B.; Devaux, J.; Legras, R.; Nield, E. Reactions induced by triphenyl phosphite addition during melt mixing of PET/PBT blends: Chromatographic evidence of a molecular weight increase due to the creation of bonds of two different natures. Polymer 1997, 38, 5367–5377. [Google Scholar] [CrossRef]
- Guo, B.; Chan, C. Chain Extension of Poly(butylene terephthalate) by reactive extrusion. J. Appl. Polym. Sci. 1999, 71, 1827–1834. [Google Scholar] [CrossRef]
- Haralabakopoulos, A.A.; Tsiourvas, D.; Paleos, C.M. Chain extension of poly(ethylene terephthalate) by reactive blending using diepoxides. J. Appl. Polym. Sci. 1999, 71, 2121–2127. [Google Scholar] [CrossRef]
- Achilias, D.S.; Bikiaris, D.N.; Karavelidis, V.; Karayannidis, G.P. Effect of silica nanoparticles on solid state polymerization of poly(ethylene terephthalate). Eur. Polym. J. 2008, 44, 3096–3107. [Google Scholar] [CrossRef]
- Villalobos, M.; Awojulu, A.; Greeley, T.; Turco, G.; Deeter, G. Oligomeric chain extenders for economic reprocessing and recycling of condensation plastics. Energy 2006, 31, 3227–3234. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, H.; Qian, Q.; Jiang, X.; Liu, X.; Huang, B.; Chen, Q. Molecular and structural analysis of epoxide-modified recycled poly(ethylene terephthalate) from rheological data. Polym. Eng. Sci. 2012, 52, 2127–2133. [Google Scholar] [CrossRef]
- Makkam, S.; Harnnarongchai, W. Rheological and mechanical properties of recycled PET modified BY reactive extrusion. Energy Procedia 2014, 56, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Awaja, F.; Pavel, D. Recycling of PET. Eur. Polym. J. 2005, 41, 1453–1477. [Google Scholar] [CrossRef]
- Chen, C.W.; Liu, P.H.; Lin, F.J.; Cho, C.J.; Wang, L.Y.; Mao, H.I.; Chiu, Y.C.; Chang, S.H.; Rwei, S.P.; Kuo, C.C. Influence of different molecular weights and concentrations of poly(glycidyl methacrylate) on recycled poly(ethylene terephthalate): A thermal, mechanical, and rheological study. J. Polym. Environ. 2020, 28, 2880–2892. [Google Scholar] [CrossRef]
- Tan, Z.; Liu, S.; Cui, X.; Sun, S.; Zhang, H. Application of macromolecular chain extender and contribution to the toughening of poly(ethylene terephthalate). J. Thermoplast. Compos. Mater. 2016, 29, 833–849. [Google Scholar] [CrossRef]
- Montava-Jorda, S.; Lascano, D.; Quiles-Carrillo, L.; Montanes, N.; Boronat, T.; Martinez-Sanz, A.V.; Ferrandiz-Bou, S.; Torres-Giner, S. Mechanical recycling of partially bio-based and recycled polyethylene terephthalate blends by reactive extrusion with poly(styrene-co-glycidyl methacrylate). Polymers 2020, 12, 174. [Google Scholar] [CrossRef] [Green Version]
- Benvenuta Tapia, J.J.; Tenorio-López, J.A.; Martínez-Estrada, A.; Guerrero-Sánchez, C. Application of RAFT-synthesized reactive tri-block copolymers for the recycling of post-consumer R-PET by melt processing. Mater. Chem. Phys. 2019, 229, 474–481. [Google Scholar] [CrossRef]
- Benvenuta-Tapia, J.J.; González-Coronel, V.J.; Soriano-Moro, G.; Martínez-De la Luz, I.; Vivaldo-Lima, E. Recycling of poly(ethylene terephthalate) by chain extension during reactive extrusion using functionalized block copolymers synthesized by RAFT polymerization. J. Appl. Polym. Sci. 2018, 135, 46771. [Google Scholar] [CrossRef]
- Benvenuta-Tapia, J.J.; Vivaldo-Lima, E.; Guerrero-Santos, R. Effect of copolymers synthesized by nitroxide-mediated polymerization as chain extenders of postconsumer poly(ethylene terephthalate) waste. Polym. Eng. Sci. 2019, 59, 2255–2264. [Google Scholar] [CrossRef]
- Benvenuta-Tapia, J.J.; Vivaldo-Lima, E. Reduction of molar mass loss and enhancement of thermal and rheological properties of recycled poly(lactic acid) by using chain extenders obtained from RAFT chemistry. React. Funct. Polym. 2020, 153, 104628. [Google Scholar] [CrossRef]
- Benvenuta Tapia, J.J.; Hernández Valdez, M.; Cerna Cortez, J.; Díaz García, V.M.; Landeros Barrios, H. Improving the rheological and mechanical properties of recycled PET modified by macromolecular chain extenders synthesized by controlled radical polymerization. J. Polym. Environ. 2018, 26, 4221–4232. [Google Scholar] [CrossRef]
- Vargas, M.A.; Benvenuta Tapia, J.J.; Sánchez, A.; Manero, O. Asphalt modified with reactive tri-block polymers obtained by via reversible addition-fragmentation chain transfer polymerization. J. Appl. Polym. Sci. 2019, 136, 47201. [Google Scholar] [CrossRef]
- Benvenuta-Tapia, J.J.; Champagne, P.; Tenorio-López, J.A.; Vivaldo-Lima, E.; Guerrero-Santos, R. Improving recycled poly(Lactic acid) biopolymer properties by chain extension using block copolymers synthesized by nitroxide-mediated polymerization (nmp). Polymers 2021, 13, 2791. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.T.; Filla, D.; Shea, R. Functional polymers from novel carboxyl-terminated trithiocarbonates as highly efficient RAFT agents. Macromolecules 2002, 35, 6754–6756. [Google Scholar] [CrossRef]
- Allen, N.S.; Edge, M.; Mohammadian, M.; Jones, K. Physicochemical aspects of the environmental degradation of poly(ethylene terephthalate). Polym. Degrad. Stab. 1994, 43, 229–237. [Google Scholar] [CrossRef]
- Bremner, T.; Rudin, A.; Cook, D.G. Melt flow index values and molecular weight distributions of commercial thermoplastics. J. Appl. Polym. Sci. 1990, 41, 1617–1627. [Google Scholar] [CrossRef]
- Włochowicz, A.; Eder, M. Distribution of lamella thicknesses in isothermally crystallized polypropylene and polyethylene by differential scanning calorimetry. Polymer 1984, 25, 1268–1270. [Google Scholar] [CrossRef]
- Wood-Adams, P.M.; Dealy, J.M.; DeGroot, A.W.; Redwine, O.D. Effect of molecular structure on the linear viscoelastic behavior of polyethylene. Macromolecules 2000, 33, 7489–7499. [Google Scholar] [CrossRef]
- Nofar, M.; Zhu, W.; Park, C.B.; Randall, J. Crystallization kinetics of linear and long-chain-branched polylactide. Ind. Eng. Chem. Res. 2011, 50, 13789–13798. [Google Scholar] [CrossRef]
- Nofar, M.; Zhu, W.; Park, C.B. Effect of dissolved CO2 on the crystallization behavior of linear and branched PLA. Polymer 2012, 53, 3341–3353. [Google Scholar] [CrossRef]
- Nofar, M. Synergistic effects of chain extender and nanoclay on the crystallization behaviour of polylactide. Int. J. Mater. Sci. Res. 2018, 1, 1–8. [Google Scholar] [CrossRef]
- Nofar, M.; Oğuz, H. Development of PBT/recycled-PET blends and the influence of using chain extender. J. Polym. Environ. 2019, 27, 1404–1417. [Google Scholar] [CrossRef]
- Hung, C.Y.; Wang, C.C.; Chen, C.Y. Enhanced the thermal stability and crystallinity of polylactic acid (PLA) by incorporated reactive PS-b-PMMA-b-PGMA and PS-b-PGMA block copolymers as chain extenders. Polymer 2013, 54, 1860–1866. [Google Scholar] [CrossRef]
- Härth, M.; Dörnhöfer, A.; Kaschta, J.; Münstedt, H.; Schubert, D.W. Molecular structure and rheological properties of a poly(ethylene terephthalate) modified by two different chain extenders. J. Appl. Polym. Sci. 2021, 138, 1–14. [Google Scholar] [CrossRef]
- Arayesh, H.; Golshan Ebrahimi, N.; Khaledi, B.; Khabazian Esfahani, M. Introducing four different branch structures in PET by reactive processing––A rheological investigation. J. Appl. Polym. Sci. 2020, 137, 49243. [Google Scholar] [CrossRef]
- Chen, J.; Wei, W.; Qian, Q.; Xiao, L.; Liu, X.; Xu, J.; Huang, B.; Chen, Q. The structure and properties of long-chain branching poly(trimethylene terephthalate). Rheol. Acta 2014, 53, 67–74. [Google Scholar] [CrossRef]
- Graebling, D. Synthesis of branched polypropylene by a reactive extrusion process. Macromolecules 2002, 35, 4602–4610. [Google Scholar] [CrossRef]
- Tian, J.; Yu, W.; Zhou, C. The preparation and rheology characterization of long chain branching polypropylene. Polymer 2006, 47, 7962–7969. [Google Scholar] [CrossRef]
- Trinkle, S.; Friedrich, C. Van Gurp-Palmen-plot: A way to characterize polydispersity of linear polymers. Rheol. Acta 2001, 40, 322–328. [Google Scholar] [CrossRef]
- Trinkle, S.; Walter, P.; Friedrich, C. Van Gurp-Palmen plot II—Classification of long chain branched polymers by their topology. Rheol. Acta 2002, 41, 103–113. [Google Scholar] [CrossRef]
- Yang, Z.; Xin, C.; Mughal, W.; Li, X.; He, Y. High-melt-elasticity poly(ethylene terephthalate) produced by reactive extrusion with a multi-functional epoxide for foaming. J. Appl. Polym. Sci. 2018, 135, 45805. [Google Scholar] [CrossRef]
- Liu, J.; Lou, L.; Yu, W.; Liao, R.; Li, R.; Zhou, C. Long chain branching polylactide: Structures and properties. Polymer 2010, 51, 5186–5197. [Google Scholar] [CrossRef]
- García-Franco, C.A.; Srinivas, S.; Lohse, D.J.; Brant, P. Similarities between gelation and long chain branching viscoelastic behavior. Macromolecules 2001, 34, 3115–3117. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, H.; Pan, H.; Ran, X.; Zhang, H. The effect of MBS on the heat resistant, mechanical properties, thermal behavior and rheological properties of PLA/EVOH blend. J. Polym. Res. 2018, 25, 1–9. [Google Scholar] [CrossRef]
- Vargas, M.A.; Herrera, R.; Manero, O. Modeling of the linear viscoelastic behavior of partially hydrogenated polymer-modified asphalts. Rubber Chem. Technol. 2007, 80, 340–364. [Google Scholar] [CrossRef]
- Li, Y.; Jia, S.; Du, S.; Wang, Y.; Lv, L.; Zhang, J. Improved properties of recycled polypropylene by introducing the long chain branched structure through reactive extrusion. Waste Manag. 2018, 76, 172–179. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | IV (dL/g) | MFR (g/10 min) | Tg (°C) | Tm (°C) | Tc (°C) | Tonset (°C) |
|---|---|---|---|---|---|---|
| rPET-F | 0.58 | 48.8 | 80.1 | 248.1 | 209.4 | 385.8 |
| rPET-B | 0.66 | 18.6 | 80.7 | 251.0 | 202.1 | 398.7 |
| vPET | 0.68 | 14.6 | 83.9 | 254.9 | 179.0 | 400.6 |
| Samples | GMA (mol%) | S (mol%) | MMA (mol%) | Nepoxy | Mn (g/mol) | Mw (g/mol) | Epoxide Number (mol/100 g) | PDI |
|---|---|---|---|---|---|---|---|---|
| PGMS1 | 76.0 1 | 11.0 1 | 13.0 1 | 152 | 63,000 2 | 99,000 2 | 0.24 | 1.58 2 |
| PGMS2 | 70.4 1 | 14.1 1 | 15.5 1 | 70 | 38,000 2 | 61,000 2 | 0.18 | 1.59 2 |
| PGMS3 | 60.6 1 | 19.4 1 | 20.0 1 | 38 | 28,000 2 | 51,000 2 | 0.14 | 1.80 2 |
| ADR-4468 | 30–50 | - | - | 5–9 | <2500 | 7250 | 0.17–0.3 | >3 |
| EPOX | - | - | - | 2 | 202 | - | 0.99 | - |
| Samples | MFR (g/10 min) | IV (dL/g) | Mn (g/mol) | Insoluble Content (%) |
|---|---|---|---|---|
| 0.5%EPOX/rPET-F | 50.2 | 0.62 | 17,100 | - |
| 1.0%EPOX/rPET-F | 44.6 | 0.66 | 18,400 | - |
| 1.5%EPOX/rPET-F | 24.2 | 0.70 | 19,800 | - |
| 0.5%ADR/rPET-F | 18.6 | 0.72 | 20,500 | - |
| 1.0%ADR/rPET-F | 15.5 | 0.78 | 22,600 | - |
| 1.5%ADR/rPET-F | 13.4 | 0.86 | 25,400 | - |
| 0.5%PGMS1/rPET-F | 16.3 | - | 6.7 | |
| 1.0%PGMS1/rPET-F | 13.6 | - | 10.7 | |
| 1.5%PGMS1/rPET-F | 10.4 | - | 14.7 | |
| 0.5%PGMS2/rPET-F | 16.4 | - | 5.3 | |
| 1.0%PGMS2/rPET-F | 14.8 | - | 10.4 | |
| 1.5%PGMS2/rPET-F | 11.0 | - | 13.1 | |
| 0.5%PGMS3/rPET-F | 16.3 | - | 4.0 | |
| 1.0%PGMS3/rPET-F | 13.3 | - | 8.9 | |
| 1.5%PGMS3/rPET-F | 13.2 | - | 10.6 | |
| Samples | Tc (°C) | Tm (°C) | Tg (°C) |
|---|---|---|---|
| 0.5%EPOX/rPET-F | 210.7 | 246.4 | 75.7 |
| 1.0%EPOX/rPET-F | 206.9 | 244.5 | 73.7 |
| 1.5%EPOX/rPET-F | 198.7 | 240.2 | 72.1 |
| 0.5%ADR/rPET-F | 208.0 | 247.2 | 77.5 |
| 1.0%ADR/rPET-F | 205.8 | 245.5 | 78.1 |
| 1.5%ADR/rPET-F | 204.5 | 245.7 | 77.5 |
| 0.5%PGMS1/rPET-F | 215.5 | 250.4 | 78.5 |
| 1.0%PGMS1/rPET-F | 215.5 | 249.9 | 77.5 |
| 1.5%PGMS1/rPET-F | 214.7 | 249.5 | 77.9 |
| 0.5%PGMS2/rPET-F | 215.7 | 248.9 | 77.3 |
| 1.0%PGMS2/rPET-F | 214.5 | 250.2 | 79.1 |
| 1.5%PGMS2/rPET-F | 214.3 | 249.0 | 77.5 |
| 0.5%PGMS3/rPET-F | 215.5 | 250.4 | 76.9 |
| 1.0%PGMS3/rPET-F | 213.8 | 249.7 | 78.1 |
| 1.5%PGMS3/rPET-F | 213.4 | 249.0 | 77.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.-J.; Sun, X.-L.; Chen, Q.; Qian, Q. Recycled Poly(Ethylene Terephthalate) from Waste Textiles with Improved Thermal and Rheological Properties by Chain Extension. Polymers 2022, 14, 510. https://doi.org/10.3390/polym14030510
Wu W-J, Sun X-L, Chen Q, Qian Q. Recycled Poly(Ethylene Terephthalate) from Waste Textiles with Improved Thermal and Rheological Properties by Chain Extension. Polymers. 2022; 14(3):510. https://doi.org/10.3390/polym14030510
Chicago/Turabian StyleWu, Wen-Jun, Xiao-Li Sun, Qinghua Chen, and Qingrong Qian. 2022. "Recycled Poly(Ethylene Terephthalate) from Waste Textiles with Improved Thermal and Rheological Properties by Chain Extension" Polymers 14, no. 3: 510. https://doi.org/10.3390/polym14030510
APA StyleWu, W.-J., Sun, X.-L., Chen, Q., & Qian, Q. (2022). Recycled Poly(Ethylene Terephthalate) from Waste Textiles with Improved Thermal and Rheological Properties by Chain Extension. Polymers, 14(3), 510. https://doi.org/10.3390/polym14030510