
High-Performance Polyurethane Nanocomposite Membranes Containing Cellulose Nanocrystals for Protein Separation



Abstract
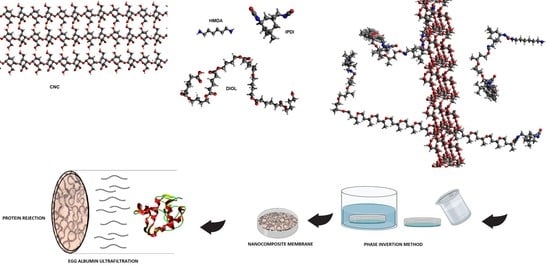
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Cellulose Nanocrystal
2.3. Synthesis of PU and Their Nanocomposites
2.4. Membrane Preparation
2.5. Microfiltration
2.6. Water Content and Porosity
2.7. Protein Rejection
2.8. Differential Scanning Calorimetry (DSC)
2.9. Mechanical Test
2.10. Infrared Spectroscopy (FTIR)
2.11. Optical Microscopy
2.12. Scanning Electronic Microscopy (SEM)
2.13. Water Contact Angle (WCA) Measurement
2.14. Carbon-13 Solid-State NMR
3. Results
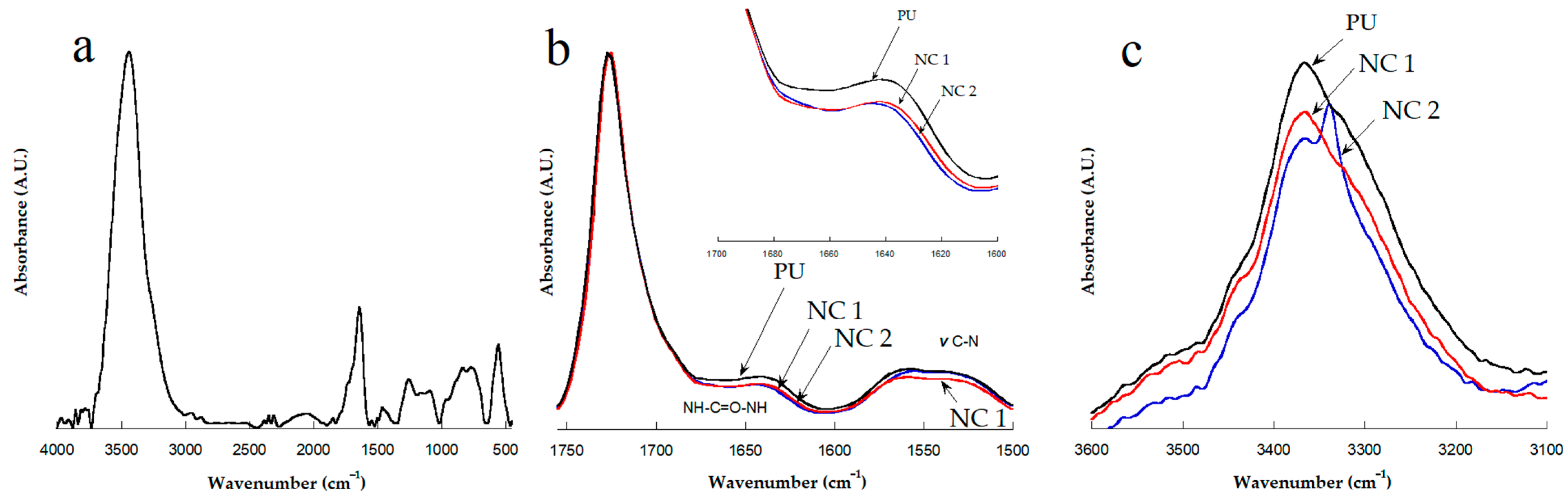
3.1. FTIR Characterization
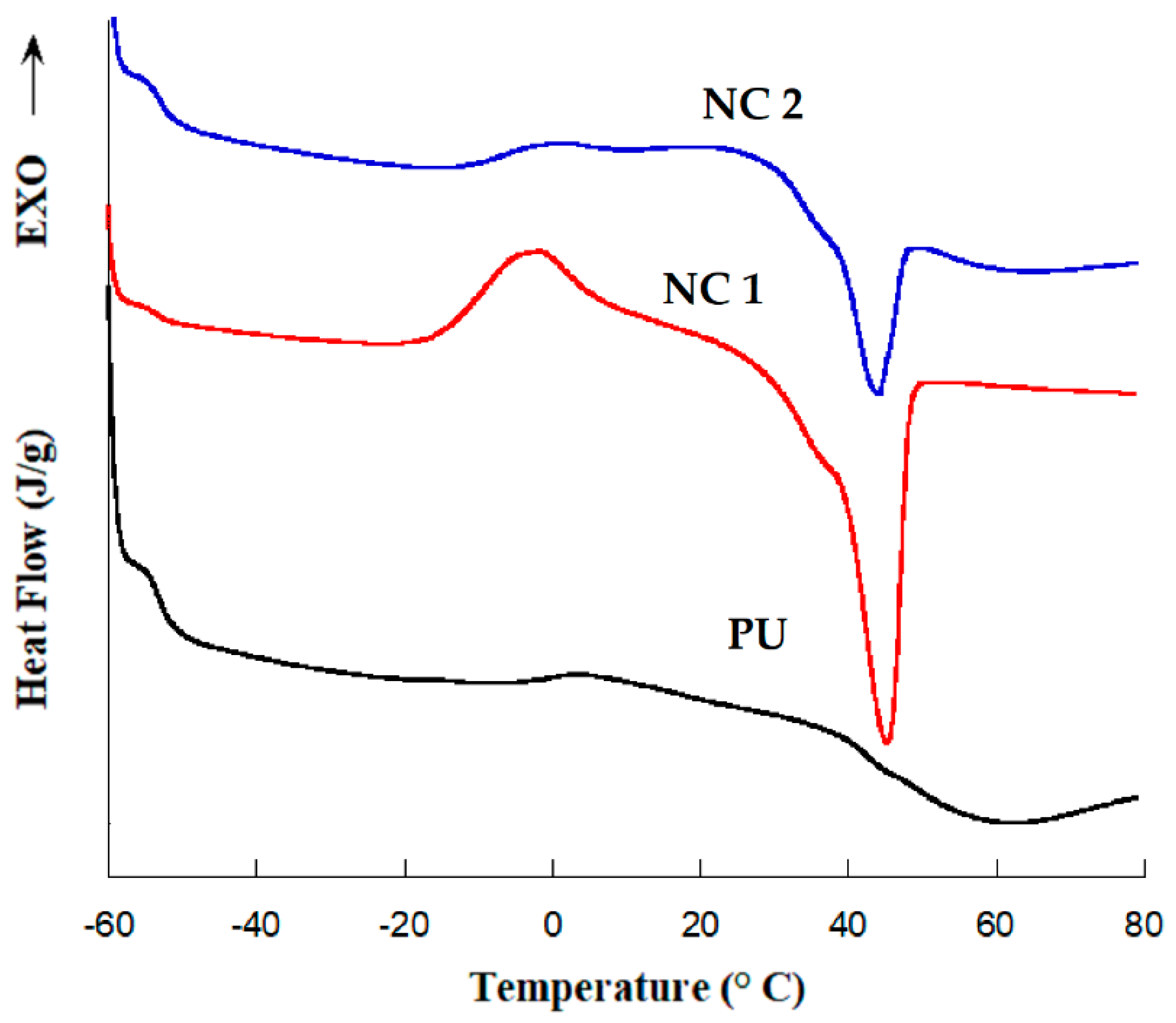
3.2. DSC Measurements
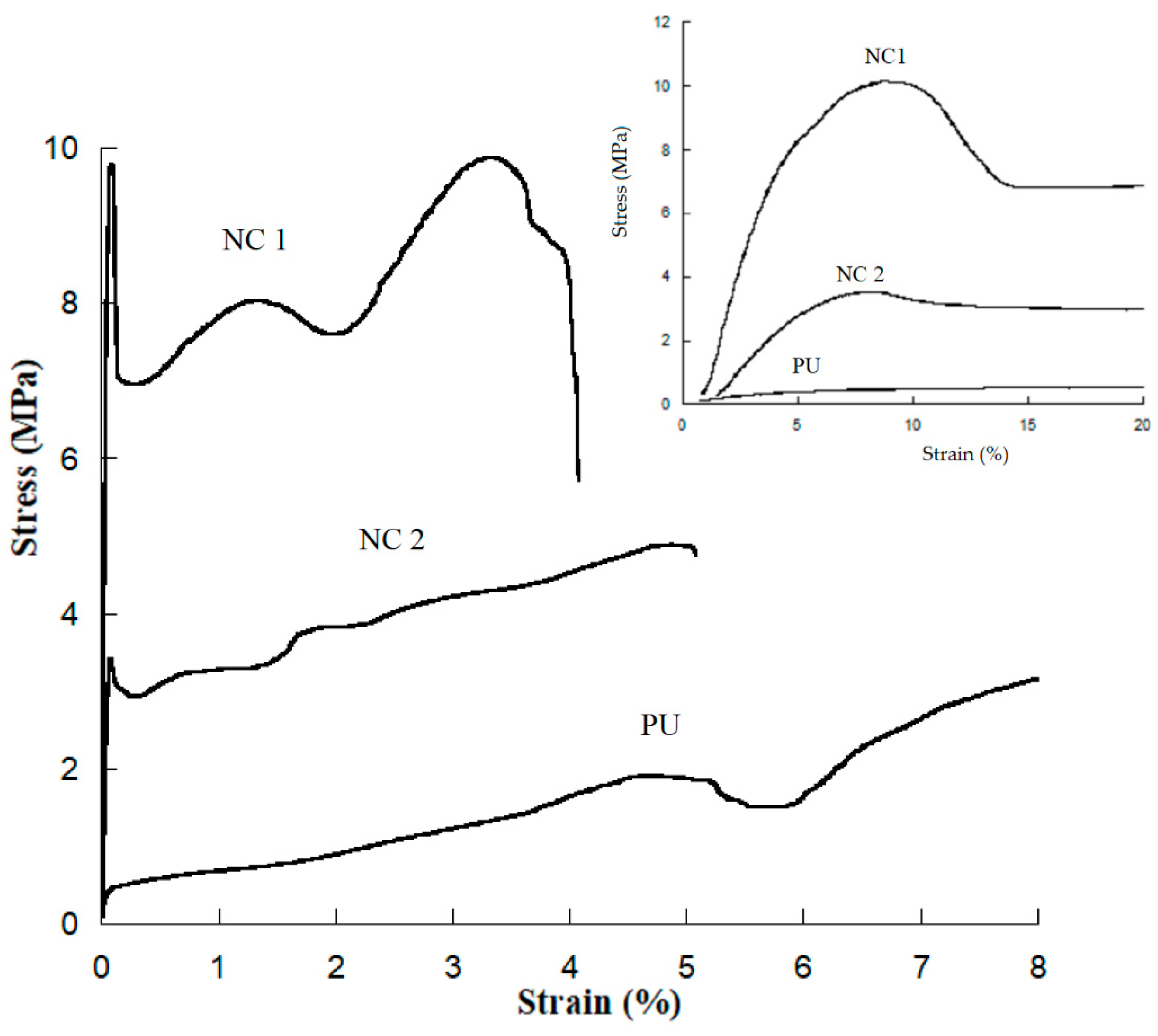
3.3. Stress–Strain Testing
3.4. Carbon-13 Solid-State NMR
3.5. Microfiltration
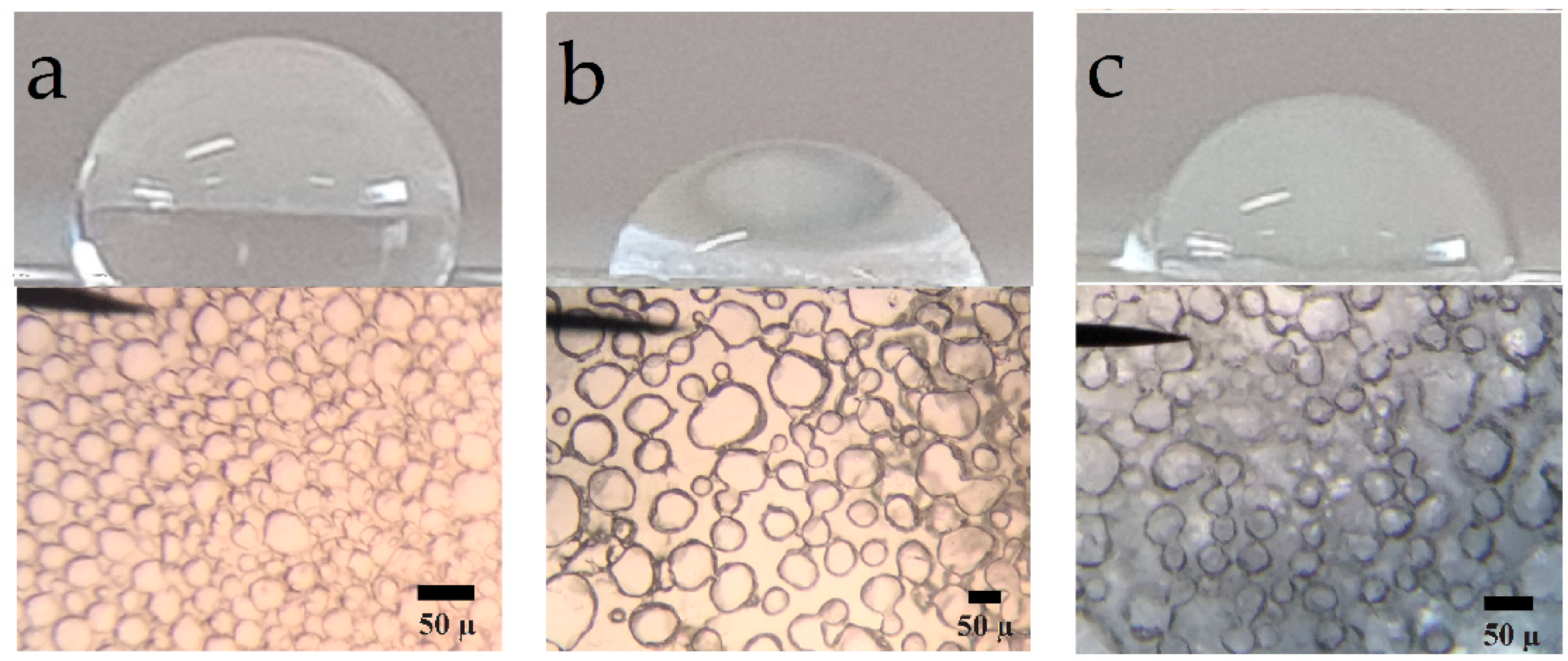
3.6. Photomicrography
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rzeszutek, K.; Chow, A. Transport of organic dyes through ether-type polyurethane membrane. Talanta 1999, 49, 757–771. [Google Scholar] [CrossRef]
- Sivakumar, M.; Mohan, D.; Rangarajan, R. Preparation and performance of cellulose acetate–polyurethane blend membranes and their applications. Part 1. Polym. Inter. 1998, 47, 311. [Google Scholar] [CrossRef]
- Malaisamy, R.; Mohan, D.; Rajendran, M. Polyurethane and sulfonated polysulfone blend ultrafiltration membranes: I. Preparation and characterization studies. Journal of colloid and interface science. J. Colloid Interf. Sci. 2002, 254, 129–140. [Google Scholar] [CrossRef]
- Podsiadlo, P.; Kaushik, A.K.; Arruda, E.M.; Waas, A.M.; Shim, B.S.; Xu, J.D.; Nandivada, H.; Pumplin, B.G.; Lahann, J.; Ramamoorthy, A.; et al. Ultrastrong and stiff layered polymer nanocomposites. Science 2007, 318, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Liff, S.M.; Kumar, N.; McKinley, G.H. High-performance elastomeric nanocomposites via solvent-exchange processing. Nat. Mater. 2007, 6, 76–83. [Google Scholar] [CrossRef]
- Dong, X.M.; Revol, J.F.; Gray, D.G. Effect of microcrystallite preparation conditions on the formation of colloid crystals of cellulose. Cellulose 1998, 5, 19–32. [Google Scholar] [CrossRef]
- Eichhorn, S.J.; Dufresne, A.; Aranguren, M.; Marcovich, N.E.; Capadona, J.R.; Rowan, S.J.; Weder, C.; Thielemans, W.; Roman, M. Current international research into cellulose nanofibres and nanocomposites. J. Mater. Sci. 2010, 45, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose nanocrystals: Chemistry, self-assembly, and applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef]
- Samir, M.; Alloin, F.; Dufresne, A. Review of recent research into cellulosic whiskers, their properties and their application in nanocomposite field. Biomacromolecules 2005, 6, 612–626. [Google Scholar] [CrossRef]
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71–85. [Google Scholar] [CrossRef]
- O’Connell, D.W.; Birkinshaw, C.; O’Dwyer, T.F. Heavy metal adsorbents prepared from the modification of cellulose: A review. Bioresour. Technol. 2008, 99, 6709–6724. [Google Scholar] [CrossRef] [PubMed]
- Wan Ngah, W.S.; Hanafiah, M.A.K.M. Removal of heavy metal ions from wastewater by chemically modified plant wastes as adsorbents: A review. Bioresour. Technol. 2008, 99, 3935–3948. [Google Scholar] [CrossRef] [PubMed]
- Barzin, J.; Feng, C.; Khulbe, K.C.; Matsuura, T.; Madaeni, S.S.; Mirzadeh, H. Characterization of polyethersulfone hemodialysis membrane by ultrafiltration and atomic force microscopy. J. Membr. Sci. 2004, 237, 77–85. [Google Scholar] [CrossRef]
- Gejyo, F.; Yamada, T.; Odani, S.; Nakagawa, Y.; Arakawa, M.; Kunitomo, T.; Kataoka, H.; Suzuki, M.; Hirasawa, Y.; Shirahama, T.; et al. A new form of amyloid protein associated with chronic hemodialysis was identified as beta 2-microglobulin. Biochem. Biophys. Res. Commun. 1985, 129, 701–706. [Google Scholar] [CrossRef]
- Antolín-Cerón, V.H.; Altamirano-Gutiérrez, A.; Astudillo-Sánchez, P.D.; Barrera-Rivera, K.A.; Martínez-Richa, A. Development of novel nanocomposite polyurethane ultrafiltration membranes based on multiwalled carbon nanotubes functionalized with PAMAM dendrimer. Polym. Plast. Technol. Mater. 2021, 60, 974–993. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Morphological and optical characterization of polyelectrolyte multilayers incorporating nanocrystalline cellulose. Biomacromolecules 2006, 7, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Fortunati, E.; Armentano, I.; Zhou, Q.; Iannoni, A.; Saino, E.; Visai, L.; Berglund, L.A.; Kenny, J.M. Multifunctional bionanocomposite films of poly(lactic acid), cellulose nanocrystals and silver nanoparticles. Carbohydr. Polym. 2012, 87, 1596–1605. [Google Scholar] [CrossRef]
- Navarro-Baena, I.; Kenny, J.M.; Peponi, L. Thermally-activated shape memory behaviour of bionanocomposites reinforced with cellulose nanocrystals. Cellulose 2014, 21, 4231–4246. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, M.P.; López, J.; López, D.; Kenny, J.M.; Peponi, L. Biodegradable electrospun bionanocomposite fibers based on plasticized PLA–PHB blends reinforced with cellulose nanocrystals. Ind. Crops Prod. 2016, 93, 290–301. [Google Scholar] [CrossRef]
- Kim, Y.D.; Kim, J.Y.; Lee, H.K.; Kim, S.C. Formation of polyurethane membranes by immersion precipitation. II. Morphology formation. J. Appl. Polym. Sci. 1999, 74, 2124–2132. [Google Scholar] [CrossRef]
- Latha, C.S.; Shanthanalakshmi, D.; Mohan, D.; Balu, K.; Kumarasamy, M.D.K. Polyurethane and Carboxylated Polysulfone Blend, Ultrafiltration Membranes. I. Preparation and Characterization. J. Appl. Polym. Sci. 2005, 97, 1307–1315. [Google Scholar] [CrossRef]
- Melnig, V.; Apostu, M.O.; Turaa, V.; Ciobanu, C. Optimization of polyurethane membranes Morphology and structure studies. J. Membr. Sci. 2005, 267, 58–67. [Google Scholar] [CrossRef]
- Osada, V.; Nakagawa, I. Membrane Science Technology; Marcel Dekker Inc.: New York, NY, USA, 1992. [Google Scholar]
- Kutowy, O.; Sourirajan, S. Cellulose acetate ultrafiltration membranes. J. Appl. Polym. Sci. 1975, 19, 1449. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; McCarthy, M.J.; Grieves, R.B. Charged membrabe ultrafiltration of inorganic ions in single and multi-salt systems. AIChE J. 1974, 20, 1206. [Google Scholar] [CrossRef]
- Tamura, M.; Uragami, T.; Sugihara, M. Studies on syntheses and permeabilities of special polymer membranes: 30. Ultrafiltration and dialysis characteristics of cellulose nitrate-poly (vinyl pyrrolidone) polymer blend membranes. Polymer 1981, 22, 82. [Google Scholar] [CrossRef]
- Sotto, A.; Kim, J.; Arsuaga, J.M.; Del Rosario, G.; Martínez, A.; Nam, D.; Luise, P.; Van der Bruggen, B. Binary metal oxides for composite ultrafiltration membranes. J. Mater. Chem. A 2014, 19, 7054–7064. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, M.; Malaisamy, R.; Sajitha, C.J.; Mohan, D.; Mohan, V.; Rangarajan, R. Ultrafiltration application of cellulose acetate–polyurethane blend membranes. Eur. Polym. J. 1999, 35, 1647–1651. [Google Scholar] [CrossRef]
- Shojaeiarani, J.; Bajwa, D.S.; Chanda, S. Cellulose nanocrystal based composites: A review. Composites Part C 2021, 5, 100164. [Google Scholar] [CrossRef]
- Khalil, M.L.; Beliakova, M.K.; Aly, A.A. Preparation of some starch ethers using the semi-dry state process. Carbohydr. Polym. 2001, 46, 217–226. [Google Scholar] [CrossRef]
- Le Troedec, M.; Sedan, D.; Peyratout, C.; Bonnet, J.P.; Smith, A.; Guinebretiere, R.; Krausz, P. Influence of various chemical treatments on the composition and structure of hemp fibres. Compos. Part A Appl. Sci. Manuf. 2008, 39, 514–522. [Google Scholar] [CrossRef]
- Pappas, C.; Tarantilis, P.A.; Daliani, I.; Mavromoustakos, T.; Polissiou, M. Comparison of classical and ultrasound-assisted isolation procedures of cellulose from kenaf (Hibiscus cannabinus L.) and eucalyptus (Eucalyptus rodustrus Sm.). Ultrason. Sonochem. 2002, 1, 19–23. [Google Scholar] [CrossRef]
- Garside, P.; Wyeth, P. Identification of Cellulosic Fibres by FTIR Spectroscopy—Thread and Single Fibre Analysis by Attenuated Total Reflectance. Stud. Conserv. 2003, 48, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Marcos-Fernandez, A.; Lozano, A.E.; Gonzalez, L.; Rodrıguez, A. Hydrogen Bonding in Copolymer (ether-urea) and Its Relationship with the Physical Properties. Macromolecules 1997, 3, 3584–3592. [Google Scholar] [CrossRef] [Green Version]
- Santamaria-Echart, A.; Ugarte, L.; García-Astrain, C.; Arbelaiz, A.; Corcuera, M.A.; Eceiza, A. Cellulose nanocrystals reinforced environmentallyfriendly waterborne polyurethane nanocomposites. Carbohydr. Polym. 2016, 151, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.M.; Chen, J.; Huo, S.P.; Liu, G.F.; Kong, Z.W. Thermoset nanocomposites from two-component waterborne polyurethanes and cellulose whiskers. Carbohydr. Polym. 2014, 105, 207–213. [Google Scholar] [CrossRef]
- Habibi, Y.; Goffin, A.; Schiltz, N.; Duquesne, E.; Dubois, P.; Dufresne, A. Bionanocomposites based on poly (ε-caprolactone)-grafted cellulose nanocrystals by ring-opening polymerization. J. Mater. Chem. 2008, 18, 5002–5010. [Google Scholar] [CrossRef]
- Tian, C.; Fu, S.; Habibi, Y.; Lucia, L.A. Polymerization topochemistry of cellulose nanocrystals: A function of surface dehydration control. Langmuir 2014, 30, 14670–14679. [Google Scholar] [CrossRef]
- Tian, C.; Fu, S.Y.; Meng, Q.J.; Lucia, A. New insights into the material chemistry of polycaprolactone-grafted cellulose nanofibrils/polyurethane nanocomposites. Cellulose 2016, 23, 2457–2473. [Google Scholar] [CrossRef]
- Antolín-Cerón, V.H.; Barrera-Rivera, K.A.; Fuentes-García, M.A.; Nuño-Donlucas, S.M.; Martinez-Richa, A. Preparation and Characterization of Nanocomposites Made from Chemoenzymatically Prepared Polyester Urethanes and Functionalized Multiwalled Carbon Nanotubes. Polym. Compos. 2018, 39, E697–E709. [Google Scholar] [CrossRef]
- Pei, A.; Malho, J.M.; Ruokolainen, J.; Zhou, Q.; Berglund, L.A. Strong Nanocomposite Reinforcement Effects in Polyurethane Elastomer with Low Volume Fraction of Cellulose Nanocrystals. Macromolecules 2011, 44, 4422–4427. [Google Scholar] [CrossRef]
- Ivdre, A.; Mucci, V.; Stefani, P.M.; Aranguren, M.I.; Cabulis, U. Nanocellulose reinforced polyurethane obtained from hydroxylated soybean oil. IOP Conf. Ser. Mater. Sci. Eng. 2011, 111, 012011. [Google Scholar] [CrossRef]
- Lee, M.; Heo, M.H.; Lee, H.H.; Kim, Y.W.; Shin, J. Tunable softening and toughening of individualized cellulose nanofibers-polyurethane urea elastomer composites. Carbohydr. Polym. 2017, 159, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Klemm, D.; Kramer, F.; Moritz, S.; Lindstrom, T.; Ankerfors, M.; Gray, D.; Dorris, A. Nanocelluloses: A new family of nature-based materials. Angew. Chem. Int. Ed. 2011, 50, 5438. [Google Scholar] [CrossRef] [PubMed]
- Auad, M.L.; Contos, V.S.; Nutt, S.; Aranguren, M.I.; Marcovich, N.E. Characterization of nanocellulose-reinforced shape memory polyurethanes. Polym. Int. 2008, 57, 651. [Google Scholar] [CrossRef]
- Vatansever, E.; Arslan, D.; Sarul, D.S.; Kahraman, Y.; Gunes, G.; Durmus, A.; Nofar, M. Development of CNC-reinforced PBAT nanocomposites with reduced percolation threshold: A comparative study on the preparation method. J. Mater. Sci. 2020, 55, 15523–15537. [Google Scholar] [CrossRef]
- Kamal, M.R.; Khoshkava, V. Effect of cellulose nanocrystals (CNC) on rheological and mechanical properties and crystallization behavior of PLA/CNC nanocomposites. Carbohydr. Polym. 2015, 123, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Khoshkava, V.; Kamal, M.R. Effect of cellulose nanocrystals (CNC) particle morphology on dispersion and rheological and mechanical properties of polypropylene/CNC nanocomposites. ACS Appl. Mater. Interfaces 2014, 6, 8146–8157. [Google Scholar] [CrossRef]
- Ishida, H.; Kaji, H.; Horii, F. Solid-State 13C NMR Analyses of the Structure and Chain Conformation of Thermotropic Liquid Crystalline Polyurethane Crystallized from the Melt through the Liquid Crystalline State. Macromolecules 1997, 30, 5799–5803. [Google Scholar] [CrossRef]
- Wang, H.L.; Kao, H.M.; Digar, M.; Wen, T.C. FTIR and Solid State 13C NMR Studies on the Interaction of Lithium Cations with Polyether Poly(urethane urea). Macromolecules 2001, 34, 529–537. [Google Scholar] [CrossRef]
- Hong, Y.; Miyoshi, T. Solid-State NMR Characterization of Polymer Chain Structure and Dynamics in Polymer Crystals. In Encyclopedia of Polymers and Composites; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Cheng, H. NMR Spectroscopy of Polymers: Innovative Strategies for Complex Macromolecules; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2011. [Google Scholar] [CrossRef]
- Stropnik, C.; Germic, L.; Zerjal, B. Morphology Variety and Formation Mechanisms of Polymeric Membranes Prepared by Wet Phase Inversion. J. Appl. Polym. Sci. 1996, 61, 1821–1830. [Google Scholar] [CrossRef]
- Mulder, M.; Mulder, J. Basic Principles of Membrane Technology; Springer Science & Business Media: Berlin, Germany, 1996. [Google Scholar]
- Sirvio, J.A.; Honkaniemi, S.; Visanko, M.; Liimatainen, H. Composite films of poly (vinyl alcohol) and bifunctional cross-linking cellulose nanocrystals. ACS Appl. Mater. Interfaces 2015, 7, 19691. [Google Scholar] [CrossRef] [PubMed]
- Strathman, H.; Koch, K. The formation mechanism of phase inversion membranes. Desalination 1997, 21, 241. [Google Scholar] [CrossRef]
- Cahn, J.W. Phase separation by spinodal decomposition in isotropic systems. J. Chem. Phys. 1965, 42, 93. [Google Scholar] [CrossRef]
- Ruiz, M.M.; Cavaillé, J.Y.; Dufresne, A.; Graillat, C.; Gérard, J.F. New waterborne epoxy coatings based on cellulose nanofillers. Macromol. Symp. 2001, 169, 211–222. [Google Scholar] [CrossRef]
- Bottino, A.; Capannelli, G.; Imperato, A.; Munari, S. UF of hydrosoluble polymers—Effect of operating conditions on the performance of the membrane. J. Membr. Sci. 1984, 21, 247. [Google Scholar] [CrossRef]
- Singh, R.; Hankins, N. (Eds.) Emerging Membrane Technology for Sustainable Water Treatment; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar] [CrossRef]
- Pappa, T.; Refetoff, S. Thyroid Hormone Transport Proteins: Thyroxine-Binding Globulin, Transthyretin, and Albumin; Elsevier: Amsterdam, The Netherlands, 2017; pp. 483–490. [Google Scholar] [CrossRef]
- Svedberg, T.; Pedersen, K.O. The Ultracentrifuge; Oxford University Press: Oxford, UK, 1940. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Name |
|---|---|
| PU | Poly(ester-urethane-urea), neat polyurethane |
| NC 1 | Polyurethane containing 1 % wt CNC |
| NC 2 | Polyurethane containing 2 % wt CNC |
| Sample | Tg (°C) | Melting Temperature | Enthalpy Fusion (ΔHm) J/g |
|---|---|---|---|
| PU | −53 | - | - |
| NC 1 | −53 | 45 | 28 |
| NC 2 | −53 | 43 | 7.8 |
| Sample | Young’s Modulus (MPa) | Yield Point (Mpa) | Deformation at Break (%) | Toughness (J/m3) |
|---|---|---|---|---|
| PU | 10 ± 0.8 | - | 760 ± 62 | 10 ± 1 |
| NC 1 | 220 ± 12 | 9.35 ± 4 | 411 ± 29 | 33.2 ± 7 |
| NC 2 | 83 ± 6 | 4 ± 0.36 | 470 ± 4 | 18 ± 1.2 |
| Pure Water Flux L m−2 h−1 | |||
|---|---|---|---|
| Pressure Transmembrane ΔP | |||
| Sample | 40.03 kPa | 98.06 kPa | 196.13 kPa |
| PU | 3 | 5 | 46 |
| NC 1 | 192 | 318 | 625 |
| NC 2 | 95 | 244 | 285 |
| Sample | Rm (kPa/L m−2 h−1) |
|---|---|
| PU | 0.19 |
| NC 1 | 3.34 |
| NC 2 | 1.72 |
| Sample | Water Content % | Porosity % | SR % |
|---|---|---|---|
| PU | 74 ± 1 | 52 ± 6 | 83 ± 5 |
| NC 1 | 66 ± 5 | 65 ± 13 | 29 ± 9 |
| NC 2 | 70 ± 2 | 58 ± 2 | 42 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antolín-Cerón, V.-H.; González-López, F.-J.; Astudillo-Sánchez, P.D.; Barrera-Rivera, K.-A.; Martínez-Richa, A. High-Performance Polyurethane Nanocomposite Membranes Containing Cellulose Nanocrystals for Protein Separation. Polymers 2022, 14, 831. https://doi.org/10.3390/polym14040831
Antolín-Cerón V-H, González-López F-J, Astudillo-Sánchez PD, Barrera-Rivera K-A, Martínez-Richa A. High-Performance Polyurethane Nanocomposite Membranes Containing Cellulose Nanocrystals for Protein Separation. Polymers. 2022; 14(4):831. https://doi.org/10.3390/polym14040831
Chicago/Turabian StyleAntolín-Cerón, Víctor-Hugo, Francisco-Jesús González-López, Pablo Daniel Astudillo-Sánchez, Karla-Alejandra Barrera-Rivera, and Antonio Martínez-Richa. 2022. "High-Performance Polyurethane Nanocomposite Membranes Containing Cellulose Nanocrystals for Protein Separation" Polymers 14, no. 4: 831. https://doi.org/10.3390/polym14040831
APA StyleAntolín-Cerón, V.-H., González-López, F.-J., Astudillo-Sánchez, P. D., Barrera-Rivera, K.-A., & Martínez-Richa, A. (2022). High-Performance Polyurethane Nanocomposite Membranes Containing Cellulose Nanocrystals for Protein Separation. Polymers, 14(4), 831. https://doi.org/10.3390/polym14040831