1. Introduction
Polyurethane (PU) has been widely applied in many industries as a versatile material due to its relatively easy manufacture, low cost, the accessible source of their raw materials and because they are typically used as glues, fibers, coatings and hoses among other applications. Recently, PUs have been used in medical and environmental applications such as in the removal of dyes and organic solvents and as filters for macromolecules [
1]. Many researchers have used PU for the preparation of membranes because of its high flux capability, high salt rejection properties and high hydrophilicity [
2,
3]. However, PU lacks enough good mechanical and thermal properties by itself, and porosity is in general not adequate for certain applications in membrane science.
Polymer nanocomposites reinforced with a low fraction of nanofillers have received great attention due to the fascinating properties that they present, and could compete with those of the most advanced materials available in the market [
4,
5]. Cellulose nanocrystals (CNCs) are highly crystalline rod-like nanomaterials isolated from cellulose fibers by acid hydrolysis [
6]. CNCs have been incorporated into a wide variety of polymer matrices as reinforcing fillers, due to their intrinsic properties such as nanoscale dimensions, high surface area, unique morphology, low density, high specific strength and Young’s modulus, as well as a very low coefficient of thermal expansion [
7,
8,
9].
Some of the most important characteristics that dialysis membranes must have are: (i) good biocompatibility; (ii) affordable cost; and (iii) appropriate morphology, from which PUs are one important choice. Among the materials with these characteristics, polyurethanes (PU) stand out as good candidates for these applications. Another particular feature of these materials is the hydrophilicity enhancement when they are reinforced with CNCs, allowing also for the elimination of macrovoids during membrane preparation [
10,
11]. Additionally, Bai et al. [
12] reported that the poly(vinylidene fluoride) (PVDF) membrane containing low amounts of CNC (from 0 to 0.25 wt%) shows increased water flux, mechanical strength and thermostability. Due to their singular hydrophilicity, CNCs are considered as attractive to reinforce membranes, as they improve water permeability and the capability to remove organic substances, metal ions, dyes and microbes [
11]. The wettability of membranes can then be improved and their biocompatibility enhanced by adding CNC, instead of using other approaches such as the chemical modification of polymer. This procedure also increased the diffusive transport properties of solute through the membrane. CNC has been proven to act as a pore forming agent, which is important in attaining appreciable porosity and pore size of the membrane [
13].
The molecular weights of proteins, which are believed to cause various chronic side reactions in dialysis patients, are in a range of 10–55 kDa. In particular, it is well known that the accumulation of 2-microglobulin (2-MG, 11,800 Da) in the body causes amyloidosis [
14]. In hemodialysis separation, low- and middle-molecular-weight uremic toxins such as urea, uric acid, creatinin and 2-microglobulin have to be removed from blood. However, proteins such as serum albumin (66 kDa) should be retained. Therefore, the desirable kidney dialysis membranes must have a suitable pore size distribution which can selectively act for the blood components previously mentioned. Additionally, higher mechanical strength can be reached when the membranes possess a sponge-like asymmetric structure bearing high transmembrane pressures and preventing membrane rupturing and leakage, which might happen in the membrane structure with macrovoids and after continuous use [
13].
In a previous work, we reported the synthesis and characterization of a biodegradable membranes filled with functionalized carbon nanotubes and showed evidence that nanofiller provides sites for hydrogen bonding interactions in the matrix which influence the mechanical properties and the porous morphology of the membrane [
15]. In this work, filtration of egg albumin was attempted in order to explore the properties of and interaction between PU and CNC for hemodialysis membranes. The optimal experimental conditions to obtain a high-performance hemodialysis membrane for protein separation are reported.
2. Materials and Methods
2.1. Materials
Poly(1,4 butylene adipate) (Diexter G 4400-57) with an hydroxyl equivalent weight of 984 was supplied by Coim USA Inc. (West Deptford, NJ, USA), isophorone diisocyanate (IPDI) from Watane. Hexamethylene diamine, Tin (II) 2-ethyl hexanoate and N,N dimethyl formamide (DMF) were acquired from Aldrich (Toluca, México). Dichloroethane and absolute ethanol were purchased from Merck (Naucalpan, México), ethylenediamine from Spectrum (Jersey City, NJ, USA), NaH2SO4 from Honeywell Fluka (Charlotte, NC, USA), chloroform 99.8% from Karal México, and Na2HSO4 and egg albumin from Meyer (Mexico City, México). All reagents were dried before being used.
2.2. Synthesis of Cellulose Nanocrystal
CNCs were prepared by acid hydrolysis of commercial cellulose microcrystals (MCCs), following a method reported in the literature [
16,
17,
18]. First, 20 g of MCC and 175 mL of sulphuric acid solution (64% (
w/w)) were mixed in a 250 mL three-neck round-bottom flask and homogenized with a mechanical stirrer. Hydrolysis was carried out at 45 °C for 30 min. The obtained product was diluted in 4 L of deionized water to stop the hydrolysis reaction. Next, to remove the acid excess, the suspension was centrifuged and 1 L of the CNC suspension was obtained. Suspension was dialyzed for 5 days for neutralization. To purify the suspension, ion exchange resin (Dowex Marathon MR-3 hydrogen and hydroxide form) was added and stirred for 24 h and then removed by filtration. The pH of the CNC suspension was adjusted to around 9.0 adding dropwise upon stirring 1.0% NaOH aqueous solution [
17,
18,
19]. The CNC suspension was sonicated in order to obtain a stable suspension of the nanocrystals, which was stored in a fridge at 3 °C to avoid bacterial growth.
2.3. Synthesis of PU and Their Nanocomposites
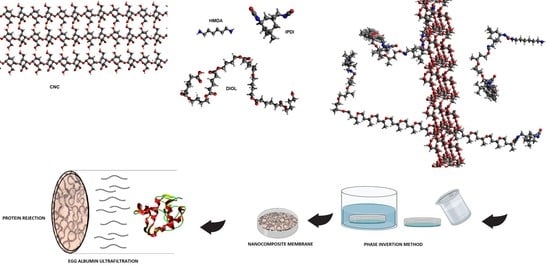
Dry polyester diol and isophorone diisocyanate (OH:NCO mol ratio 1:2) (2.5 g:0.57 g, 2.58 meq:5.16 meq) were charged into a round-bottom flask in which a solution of 1,2 dichloroethane (20 mL) containing stannous 2-ethyl hexanoate (18 μL, 0.055 mmol) was previously placed. The mixture was stirred for 3 h at 80 °C and then the chain extender (HMDA, 0.15 g, 2.58 meq) was added dropwise. The reaction mixture was kept at the same temperature for 3 h. For nanocomposite synthesis (see
Scheme 1), the specific amount of CNC was added to the initial solution of polyester diol and IPDI in 1,2 dichloroethane, and this mixture was sonicated for 15 min. Then the catalyst was added, and the reaction mixture was heated at 80 °C under stirring for 3 h. HMDA was added dropwise and the reaction was maintained for 3 h at 80 °C. The product was poured over a leveled aluminum mold at room temperature for 24 h. Evaporation of the solvent was carried out until a film was obtained. The film was released and dried in vacuum at room temperature for 12 h.
2.4. Membrane Preparation
Membranes were prepared following the phase inversion procedure reported in the literature [
20,
21,
22]. A 20% wt polymer or nanocomposite solution using
N,N-dimethylformamide was prepared. The solution was casted on a Petri dish until a thin layer was formed at room temperature. A distilled water container was used separately as a gelation bath at room temperature. The petri dish was immersed in the bath and kept underwater during the night in order to ensure efficient mass transfer. The formed membrane was washed with distilled water and dried at room temperature for 48 h. The thickness of the membranes varied from 0.10 to 0.25 mm.
2.5. Microfiltration
An acrylic cylindrical dead-end microfiltration cell with a 1 L capacity was used to test membrane performance. The cell consists of two nylon caps and threaded rod flanges, in which an “O” ring rubber and a stainless-steel grid were inserted to hold the membrane. This microfiltration cell was connected to an air compressor with a pressure control valve and gauge through a feed reservoir.
Prepared membranes were cut into the size needed for fixing it up in the microfiltration cell of 20.2 cm
2 area. The membranes were subjected to pure water flux studies at transmembrane pressures of 40, 98 and 198 kPa. Flux was measured under steady-state flow. The water flux was measured every 10 min. The pure water flux was determined using Equation (1) [
23,
24]:
where
Q is the quantity of permeate collected (L),
Jw is the water flux (L m
−2 h
−1), Δ
t is the sampling time (h), and
A is the membrane area (m
2). Measurements were carried out in triplicate.
To determine the hydraulic resistance of the membrane (
Rm), the pure water flux of the membranes was measured at transmembrane pressures (Δ
P) of 49, 98 and 196 kPa.
Rm was evaluated from the slope of
Jw versus Δ
P plot [
25]:
2.6. Water Content and Porosity
The water content fraction (
ε) and the porosity (
P) of the membranes were calculated using Equations (3) [
26] and (4) [
27].
where
A is the membrane surface (m
2),
h is the membrane thickness (m),
ε is the percentage of water content,
P is the membrane porosity percentage,
Wwet is the wet sample weight (g),
Wdry is the dry sample weight (g),
ρ is the density,
A is the membrane area (cm
2) and
h is the membrane thickness (mm).
2.7. Protein Rejection
After mounting the membrane in the ultrafiltration cell, the chamber was filled with protein solution and immediately pressurized to the desired level (149 kPa) and maintained at a constant level throughout the run. Albumin egg protein was dissolved (0.1%) in a phosphate buffer (0.05 M, pH 7.2) and used as standard solution. The permeate was collected over measured time intervals in graduated tubes and then analyzed for protein content by UV-Vis spectrophotometry (Hash-1000) at λ
max 450 nm after adding biuret reagent protein. Separation was calculated from the concentrations of feed and permeate using Equation (5) [
28]:
where %
SR is the % solute rejection, and
CP and
CF are concentrations of permeate and feed, respectively.
2.8. Differential Scanning Calorimetry (DSC)
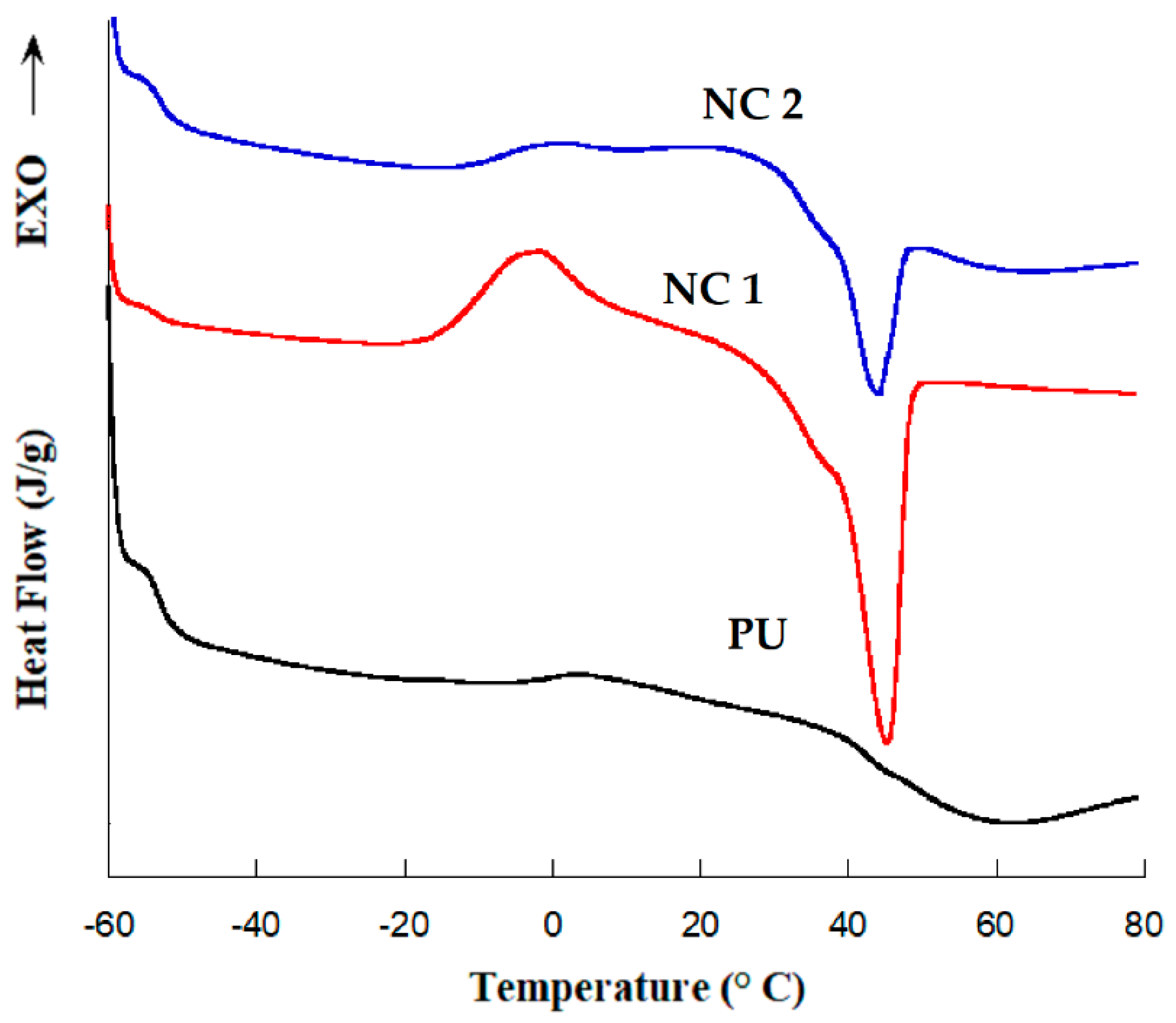
DSC was used to study the thermal properties of the prepared nanocomposites. The measurements were carried out on a Mettler Toledo DSC model DSC822e previously calibrated with indium. All tests were performed under a nitrogen atmosphere. Thermograms were recorded by heating the samples from −90 to 80 °C at 10 °C/min; only the second scan was reported. Sample weights ranged between 5 and 10 mg. The glass transition temperature (Tg) of the polymer matrix was evaluated by the inflexion point criteria and the melting enthalpy was calculated from the area under the endothermic peak.
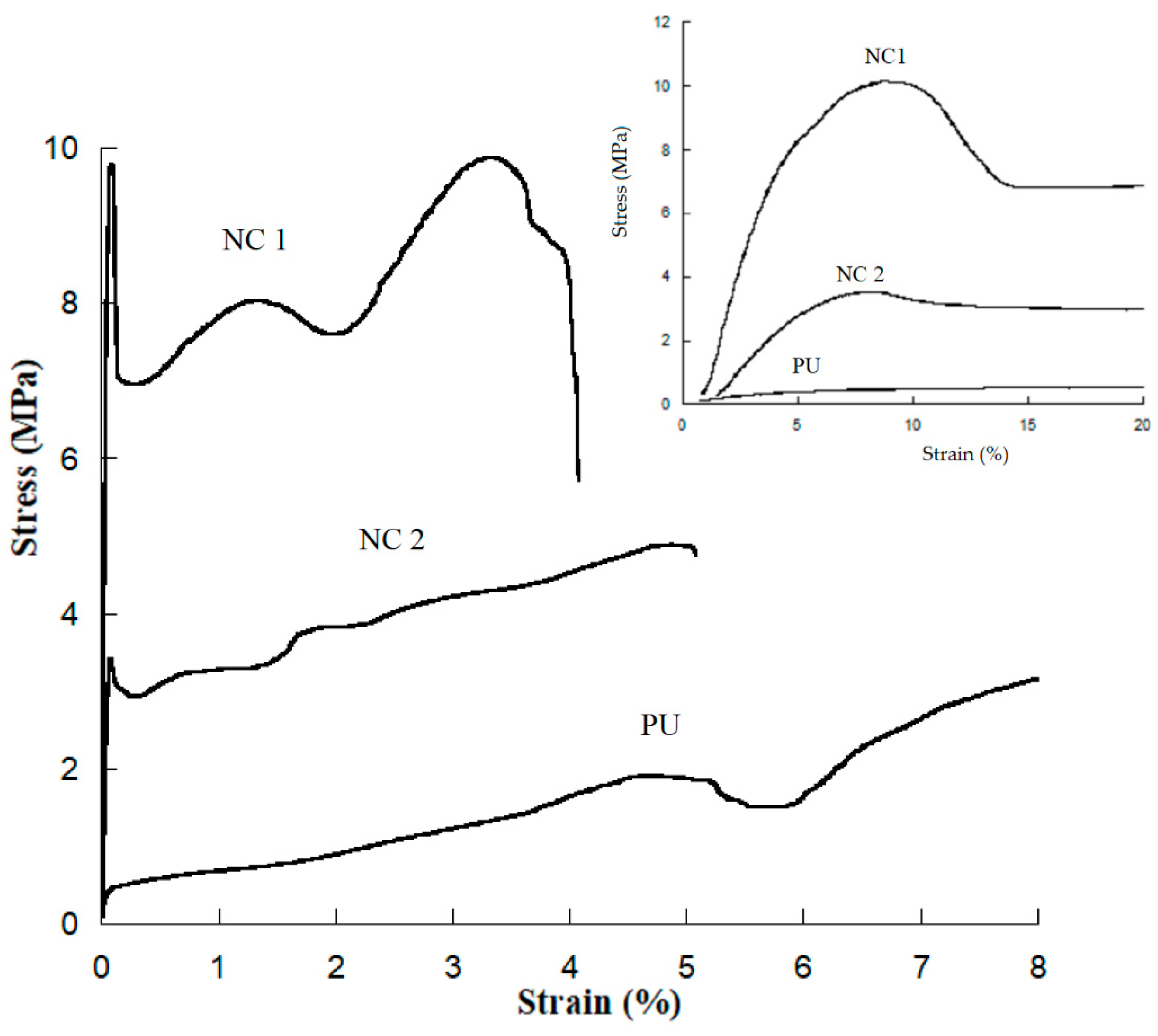
2.9. Mechanical Test
Tensile stress–strain tests were performed at a deformation rate of 50 mm/min in a United machine model SFM-10. The tested samples had a rectangular prism shape (55 × 12 × 0.5 mm) according to the ASTM D882 standard test method. Five samples were tested for each polymer composition.
2.10. Infrared Spectroscopy (FTIR)
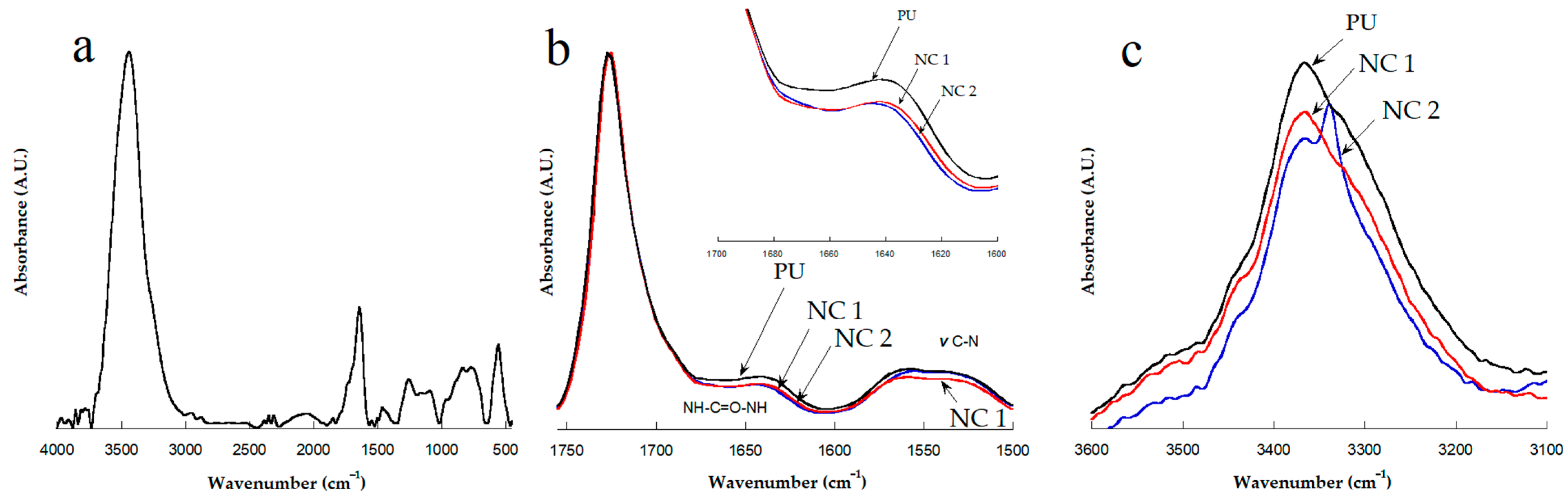
FTIR spectra of the CNC, PU and nanocomposites were obtained on an Alpha II spectrophotometer from Bruker, using an attenuated total reflection (ATR) unit. Each spectrum is an average of four scans with a resolution of 2 cm−1.
2.11. Optical Microscopy
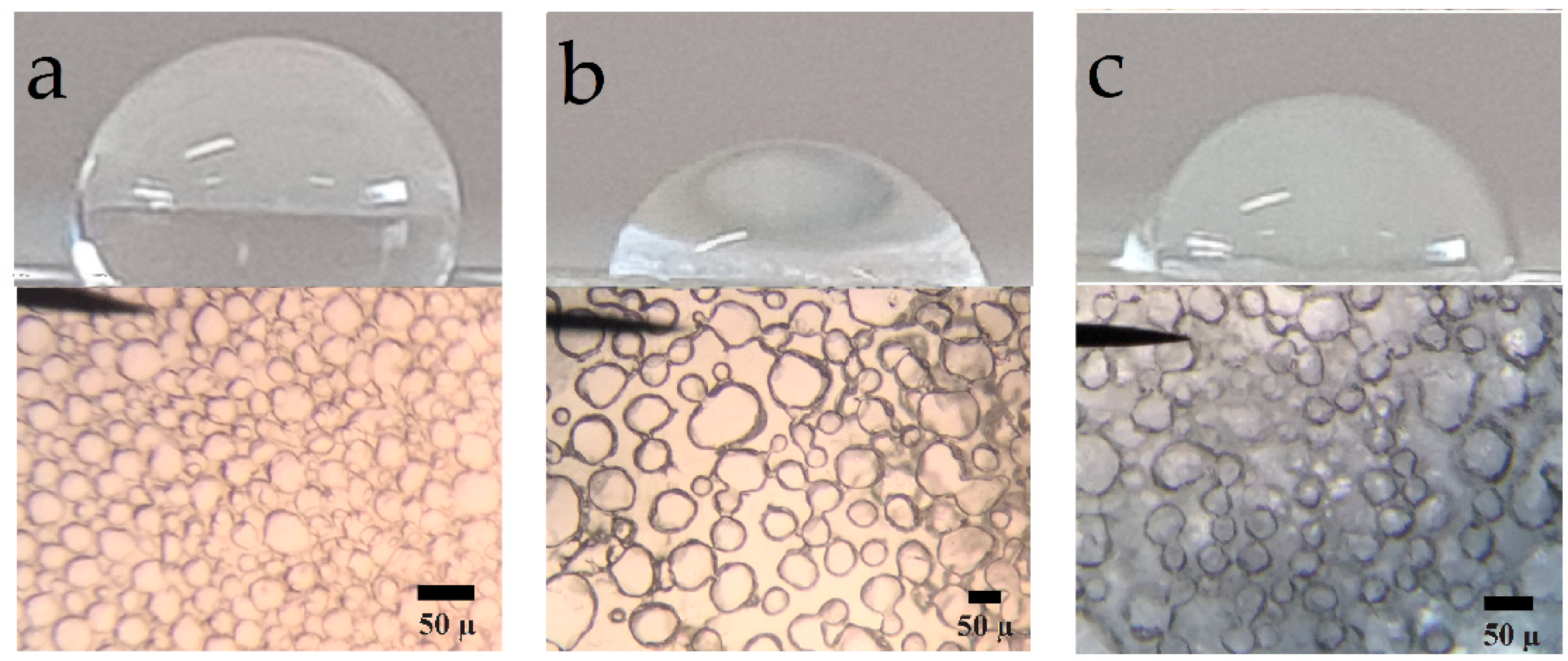
A homemade dual slide test-cell configuration was used to explore two-dimension morphology. A spacer formed by three coverslips packed was placed between the two slides and the assembly was held together with a commercial plastic clamp. This device was submerged into a coagulation bath in order to isolate the casting solution between the two slides. The microscopy visualization studies of cover morphology and size porous developed during the dry-cast process of the PU/DMF/H2O system were performed using an Amscope optical microscope. Images were recorded with the aid of a cell phone camera.
2.12. Scanning Electronic Microscopy (SEM)
A cross-section morphology of the membranes was examined with a field emission scanning electron microscope (FE-SEM), model MIRA 3LU of Tescan (Brno, Czech Republic) with an accelerating voltage of 15 kV. Samples were frozen in liquid nitrogen, broken, mounted into the sample holder and gilded for 20 s.
2.13. Water Contact Angle (WCA) Measurement
For the water contact angle measurement, a drop of water was placed on the surface of the membrane film with a pipette, and the contact angle between the water drop and film was measured.
2.14. Carbon-13 Solid-State NMR
Solid-state carbon-13 NMR spectra were recorded under proton decoupling on a Bruker Avance 400 operating at 100.613 MHz for 13C. A Bruker probe equipped with 4 mm rotors was used. CP-MAS spectra were obtained under Hartmann–Hahn matching conditions and a spinning rate of 6.0 kHz. A contact time of 2.5 ms and a repetition time of 4 s were used. The measurements were made using spin-lock power in radiofrequency units of 60 kHz and typically 4000 transients were recorded. Chemical shifts were externally referenced to tetramethylsilane using adamantane.
4. Conclusions
Polyurethane nanocomposite membranes were effectively synthesized and tested for aqueous separation of egg albumin protein by microfiltration. CNC plays an important role in controlling pore size and flux. We find that nanocomposite pore formation occurs via a spinodal decomposition mechanism, whereas for neat PU the nucleation and growth mechanism operates. The formation of a co-continuous structure is attributed to the growing of the polymer-poor phase by coalescence after spinodal decomposition, as well as the flux of nonsolvent into the polymer-poor phase due to the presence of dispersed CNC. There is enough evidence that the CNCs are hosted in a SS, as evidenced by: (i) the increase of Young’s Modulus (governed by crystals of diol) in the linear zone of the strain–stress curve, (ii) the melting endotherms recorded by DSC, and (iii) narrower peaks observed for carbons of polyester diol moiety observed in the Carbon-13 CP-MAS NMR spectra of NC 1 sample. Results of tensile strength and elastic modulus showed that 1 wt% of CNC is the optimal concentration to improve the mechanical properties of PU. Improvement in mechanical properties is indeed significant by the presence of the CNC. Pure water flux had a wide range of values, which provides the versatility needed for uses in biomedicine. Protein separation on the prepared materials is suitable for kidney dialysis, since NC 2 and neat PU can remove proteins under the molecular size of the serum albumin protein. NC 1 retains less protein than the other membranes. This behavior is mainly attributed to (i) a decrease in the degree of phase separation between hard and soft segments and (ii) intermolecular interactions (bidentate hydrogen bonds) among urea groups, as detected by infrared spectroscopy. These facts lead to a change in the shear thinning of polymer solution and also to the hydrophilicity of CNC, which supports the mass transfer in the phase inversion process, leading to higher porosity in the membrane.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}