
Semi-Continuous Heterophase Polymerization to Synthesize Poly(methacrylic acid)-Based Nanocomposites for Drug Delivery

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis and Purification of Synthesized CNTs
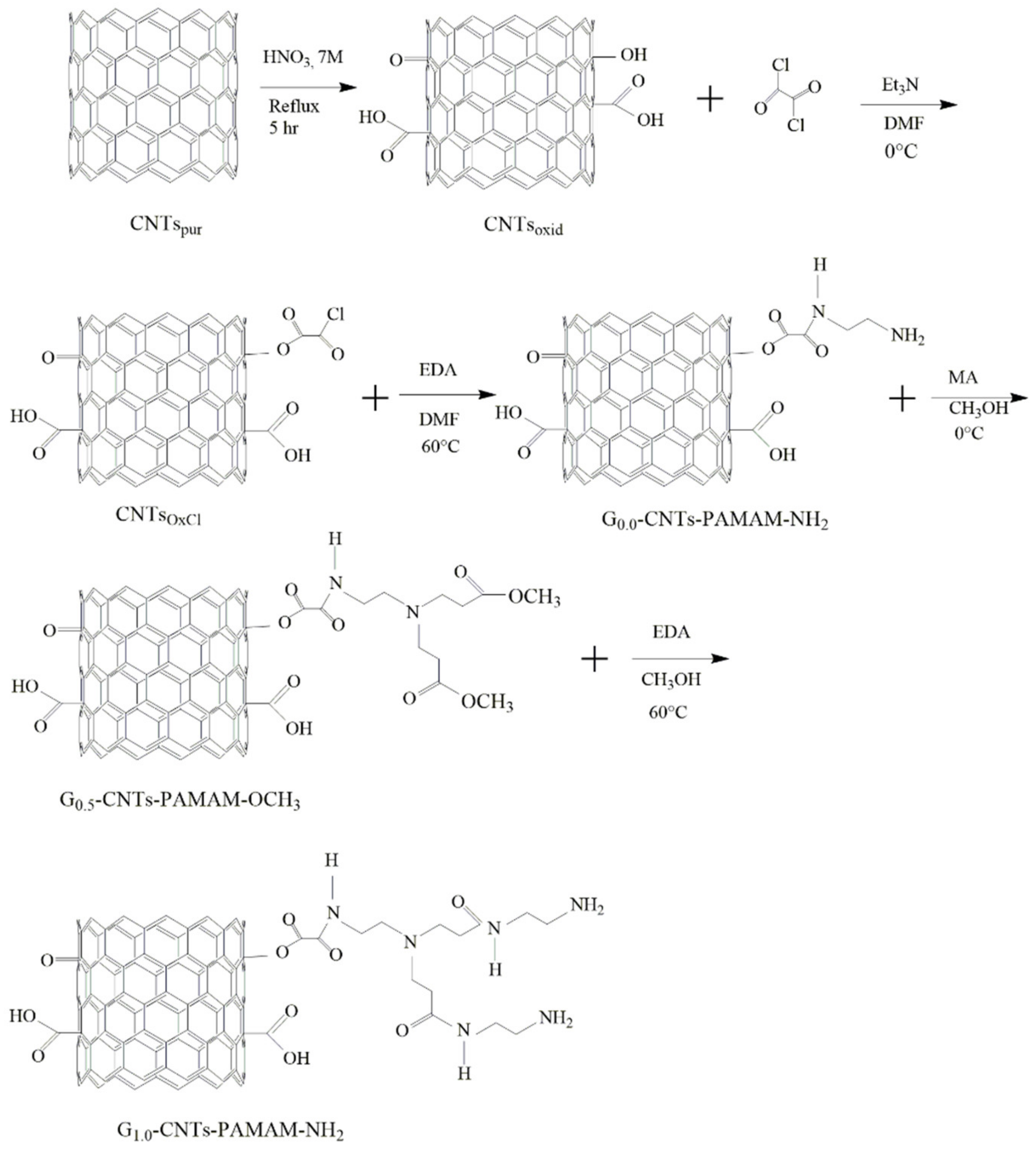
2.3. Functionalization of CNTs
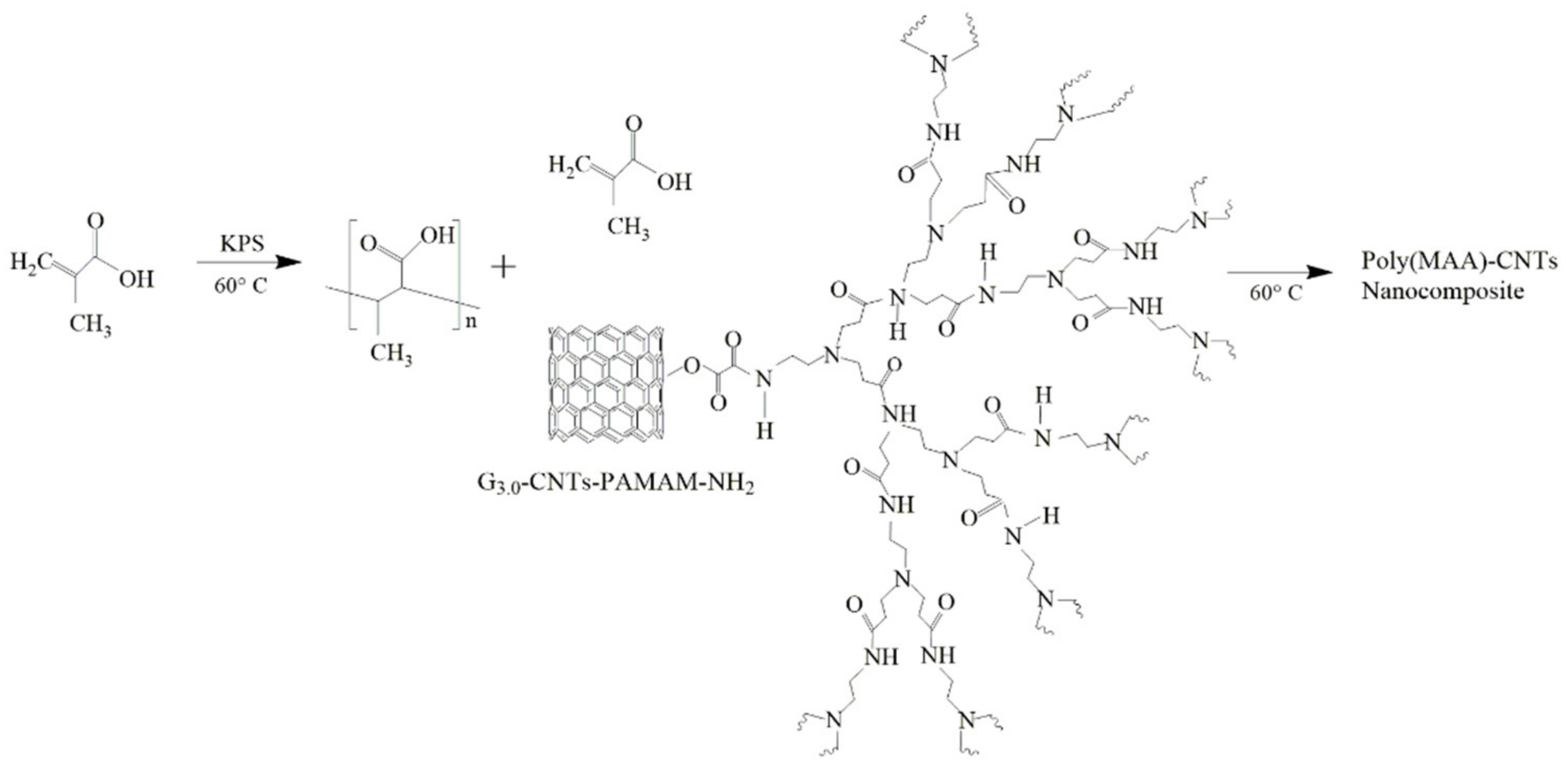
2.4. Synthesis of Poly(methacrylic acid) and Their Poly(MAA)–CNTs Nanocomposites by SHP
2.5. Characterization Techniques
3. Results and Discussion
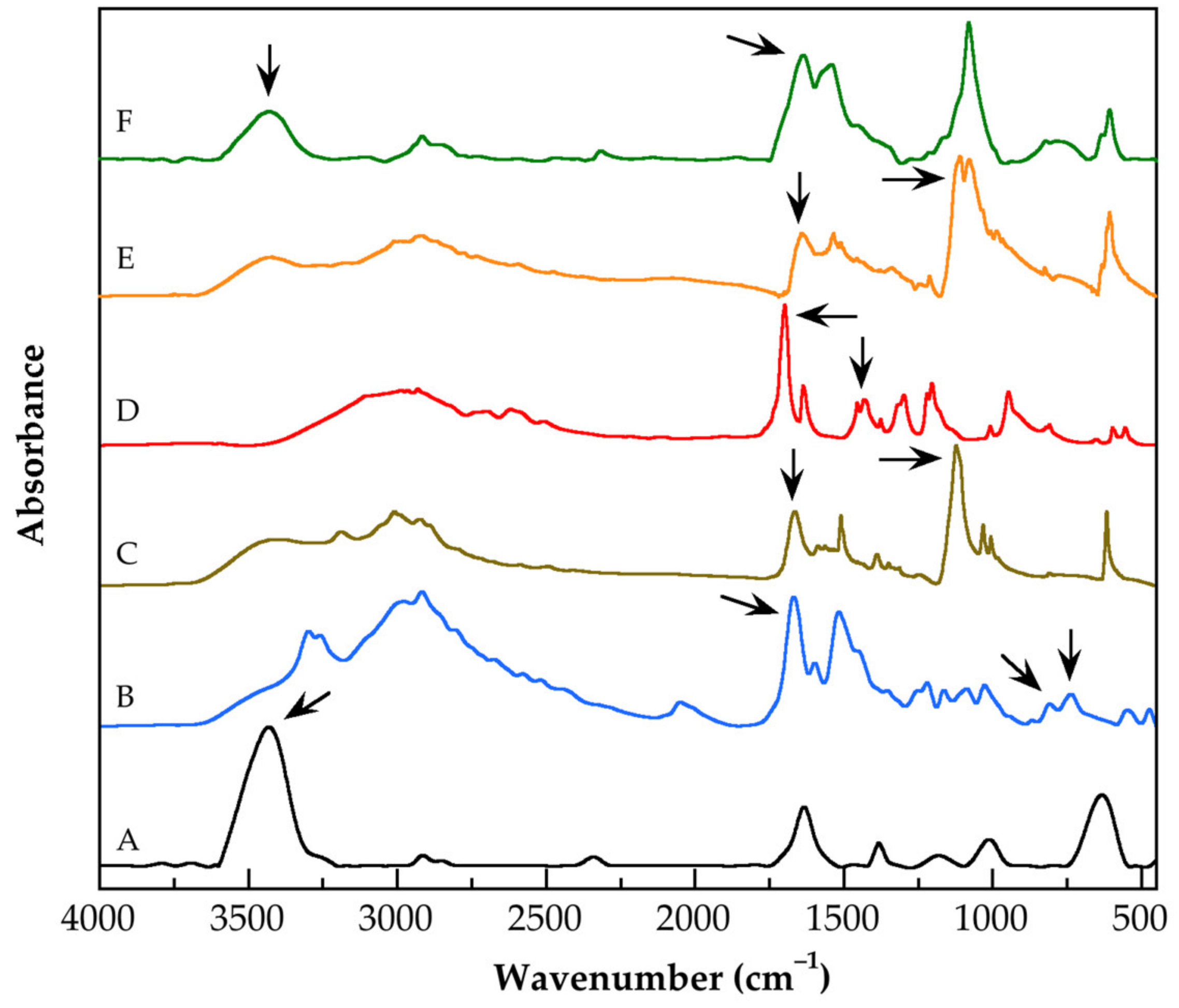
3.1. Characterization by FT-IR Spectroscopy of the Functionalized Carbon Nanotubes
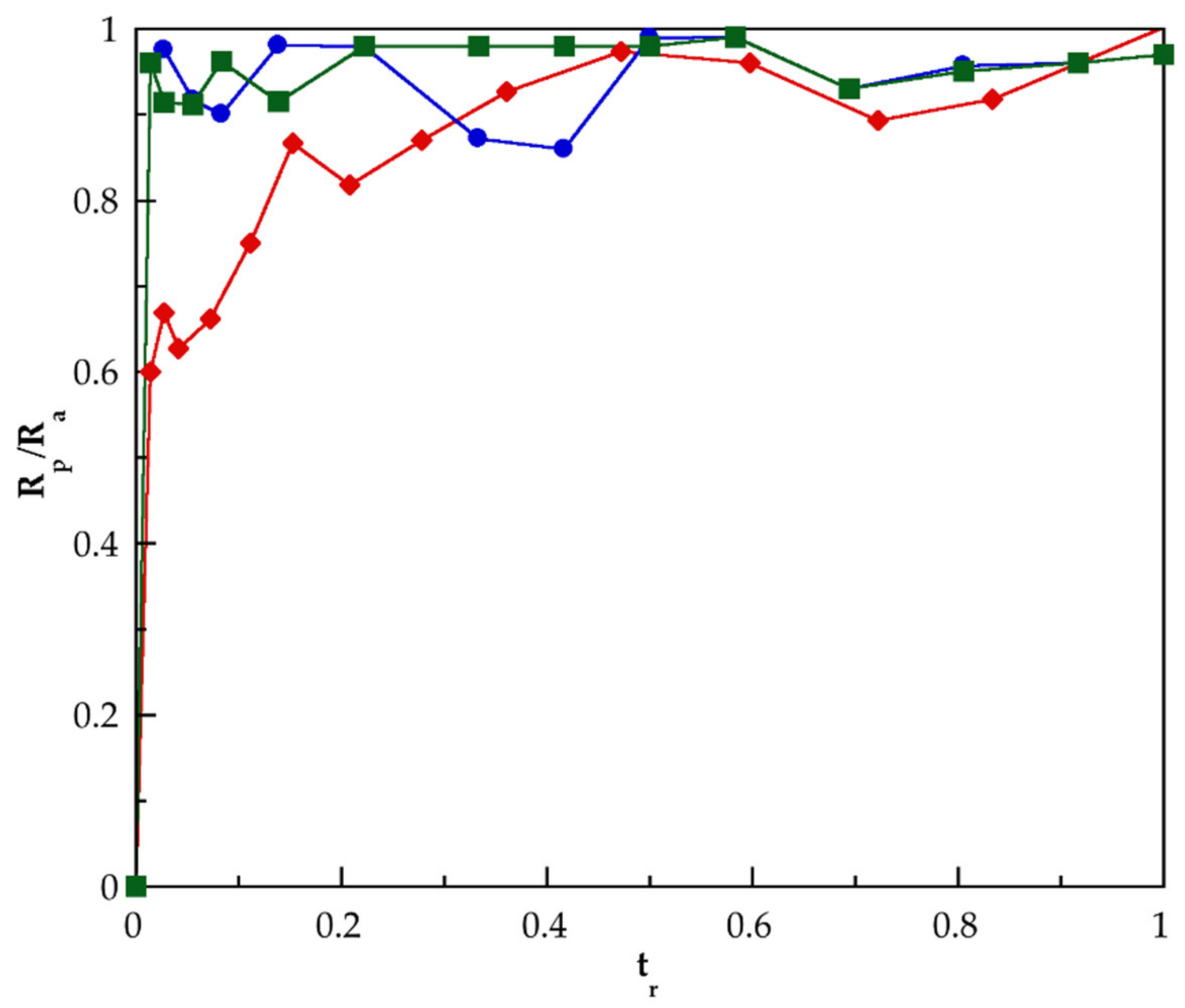
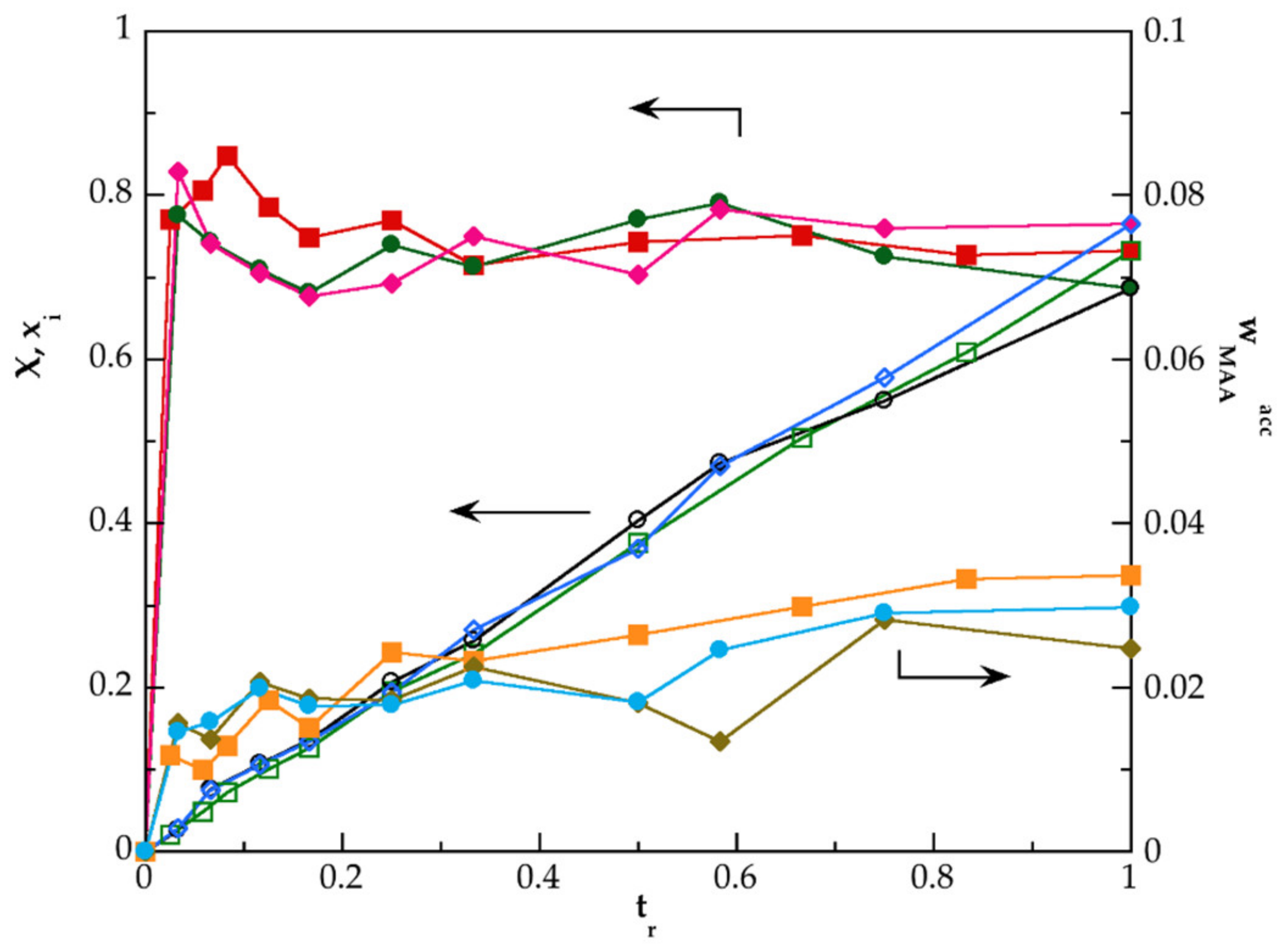
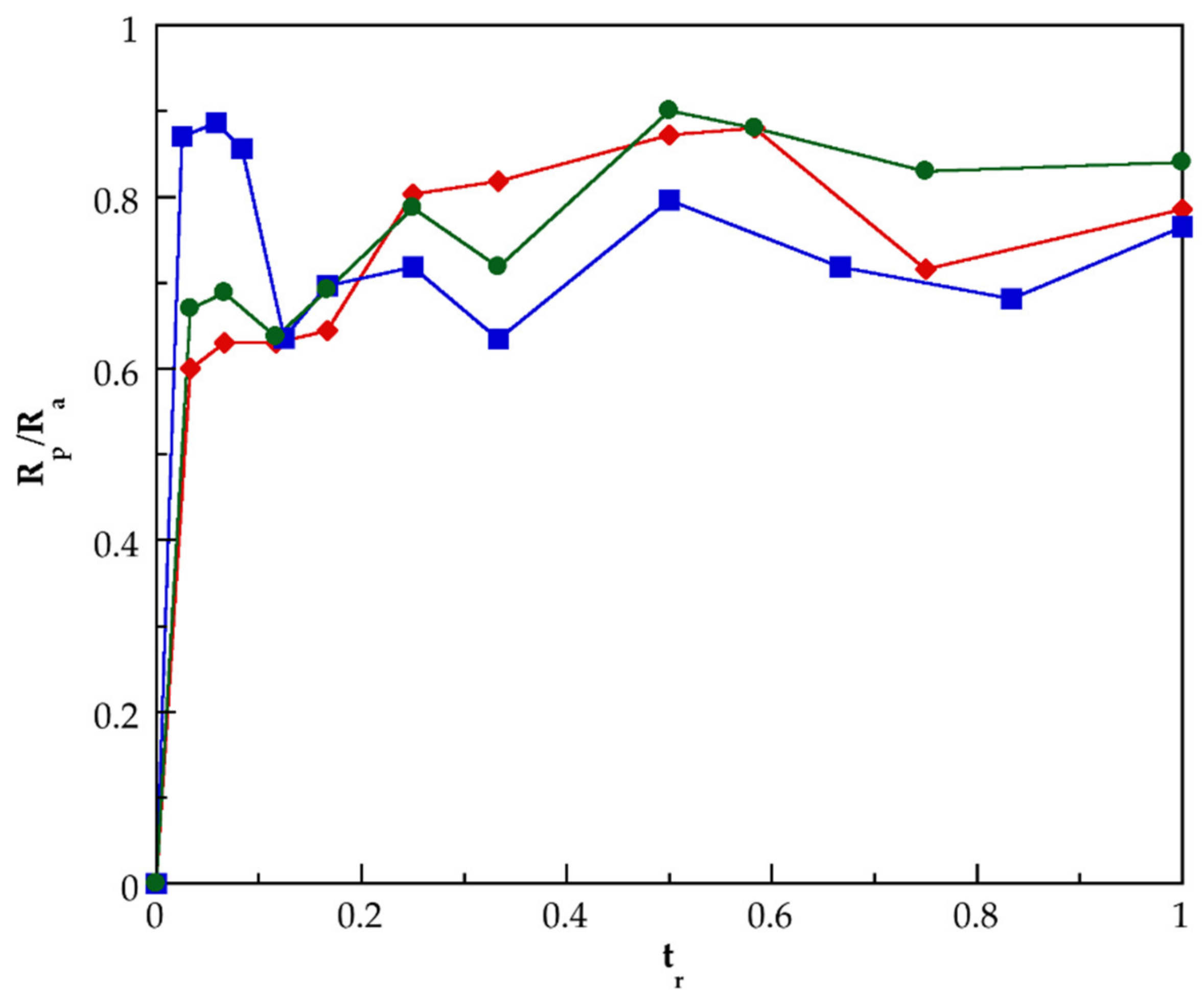
3.2. Kinetic of Polymerization of PMAA and of Their Nanocomposites Synthesized by SHP
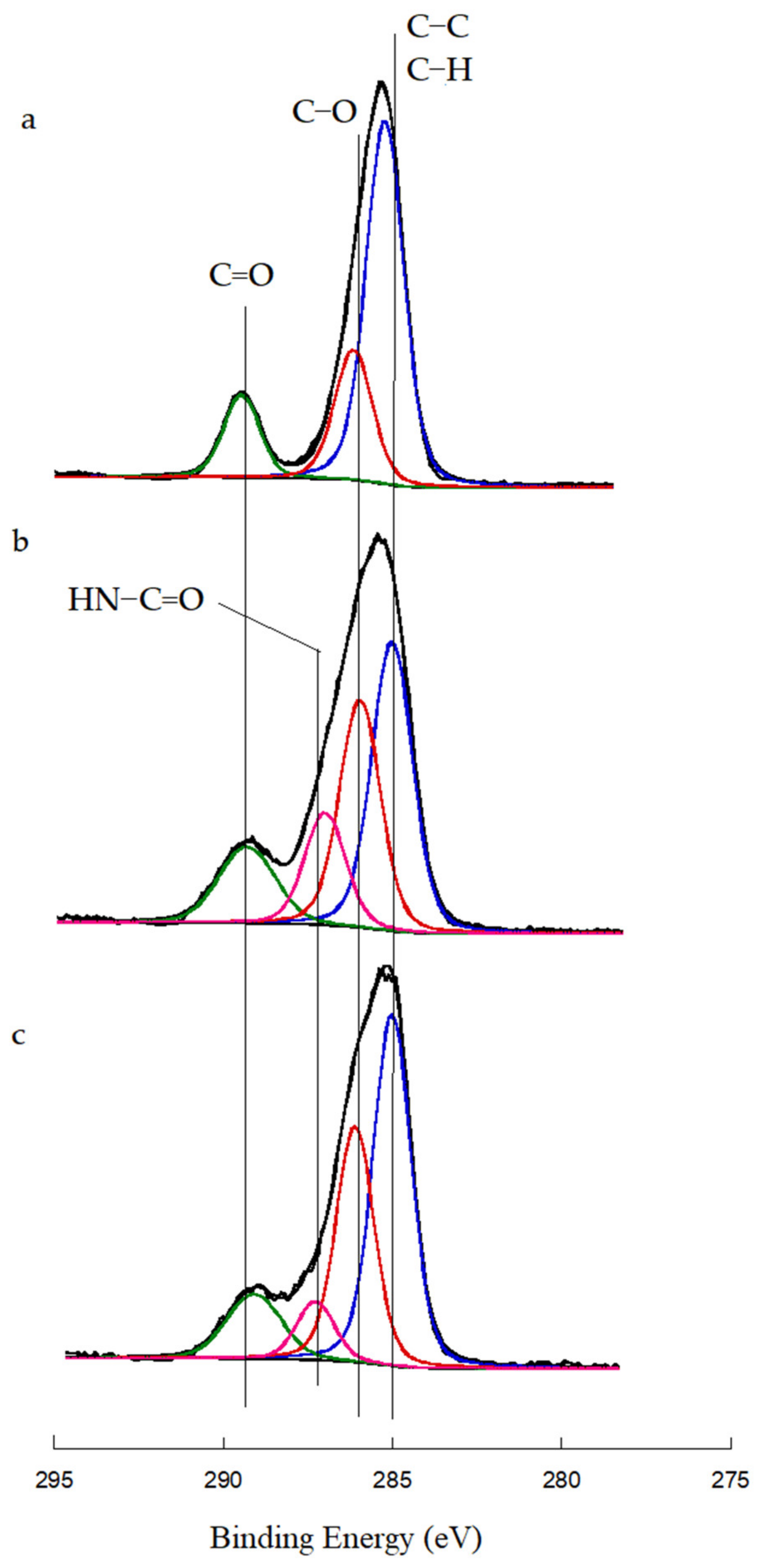
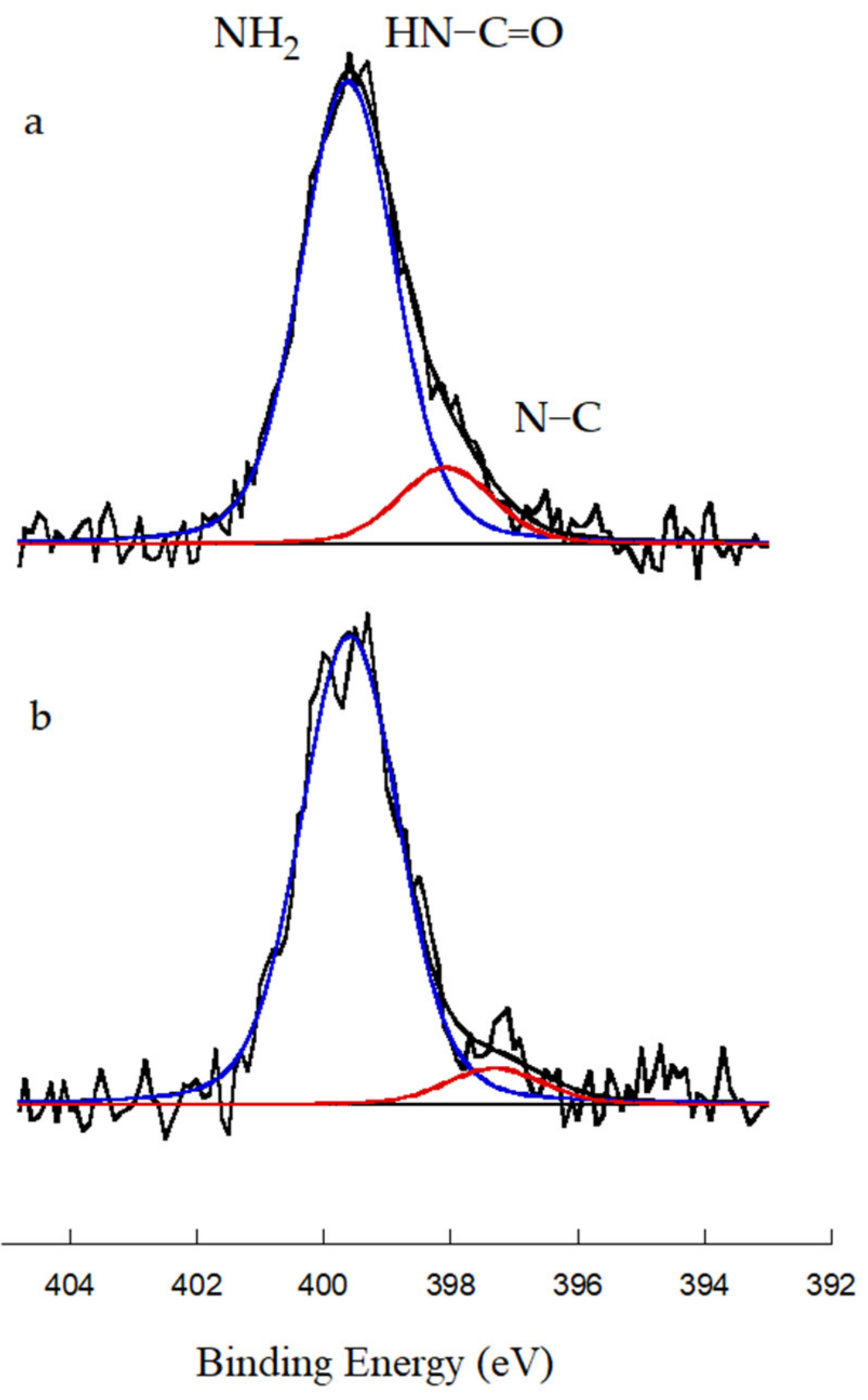
3.3. Evaluation of the Grafting of Functionalized CNTs Onto the Polymeric Matrix of the Prepared Nanocomposites
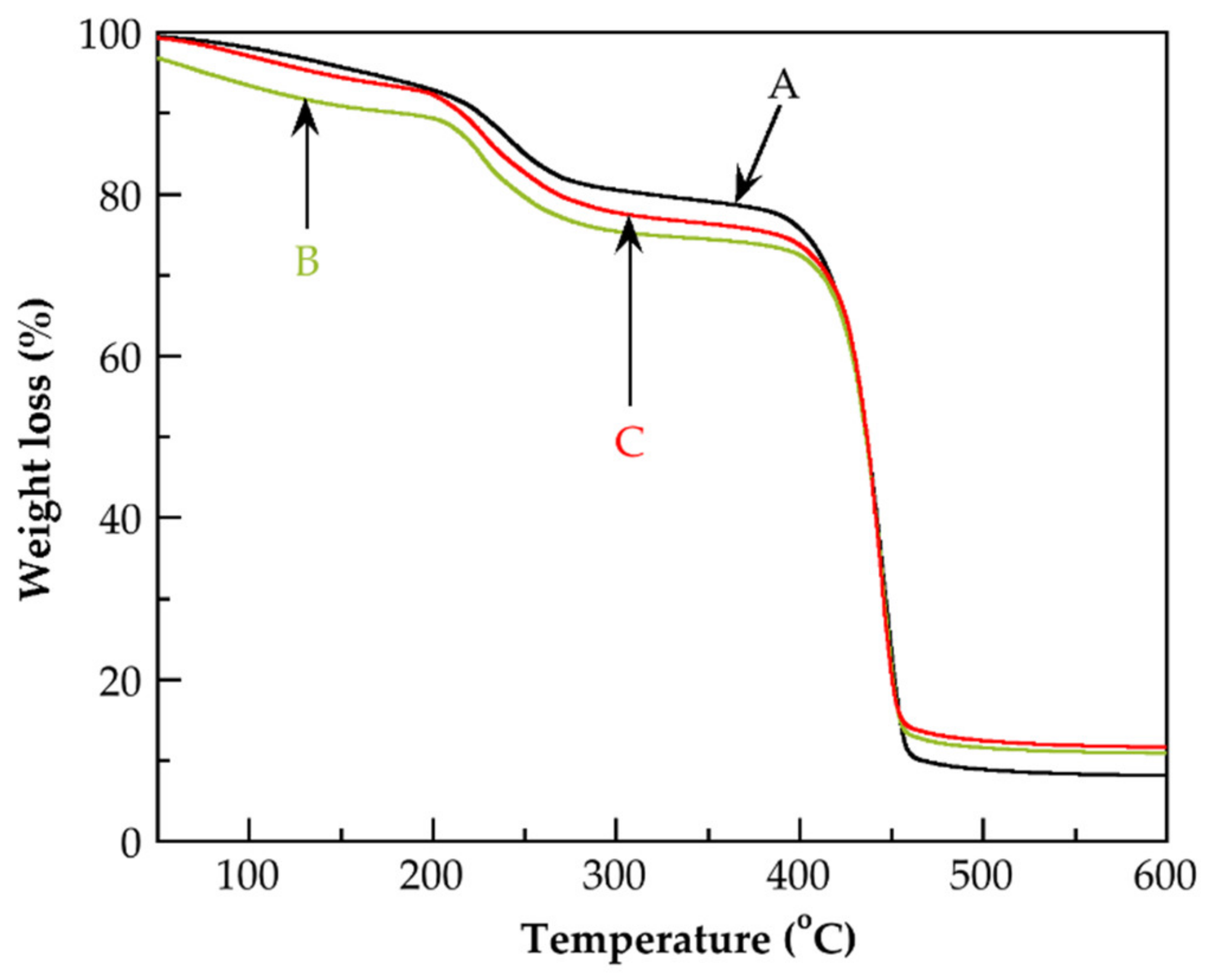
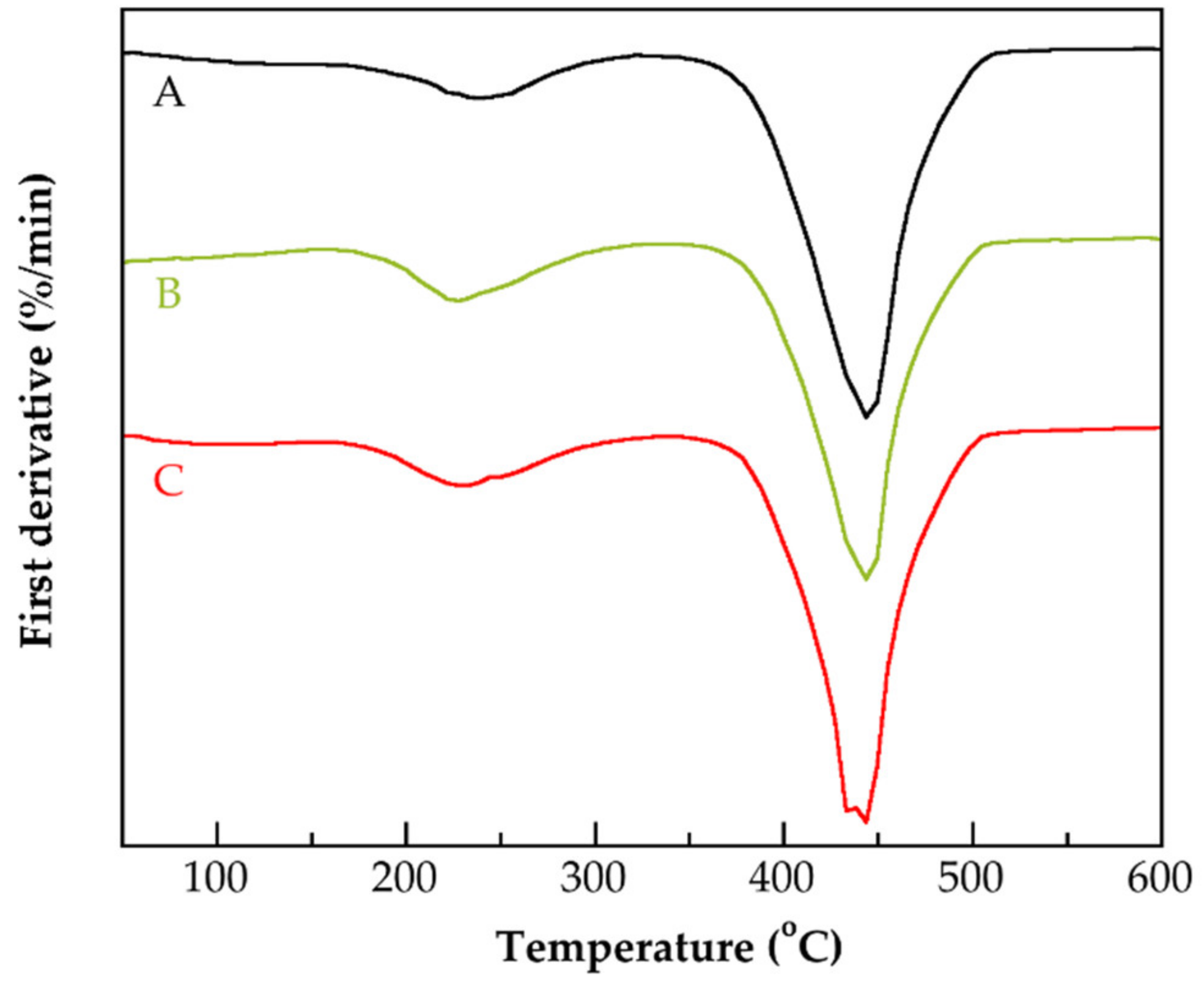
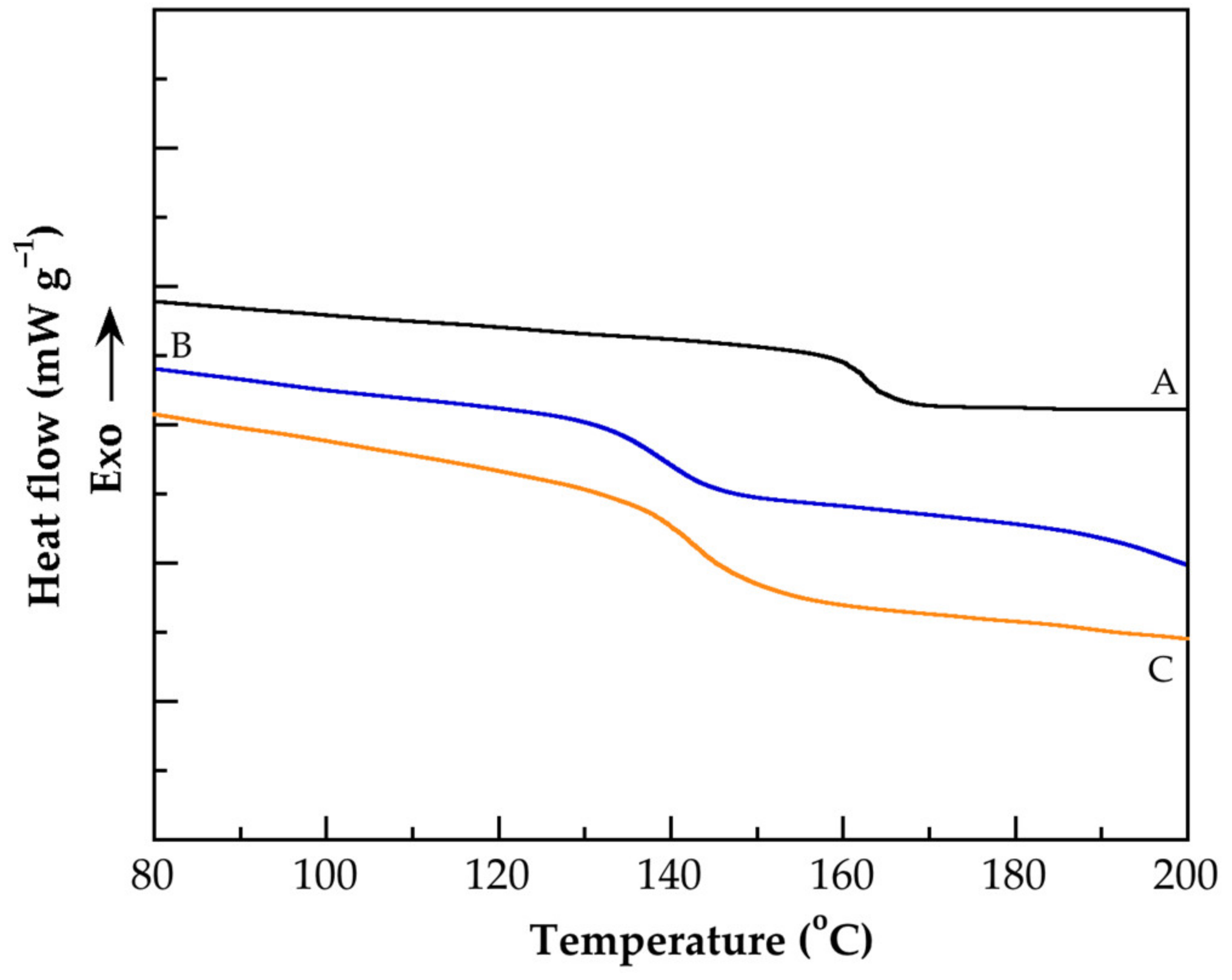
3.4. Thermal Characterization
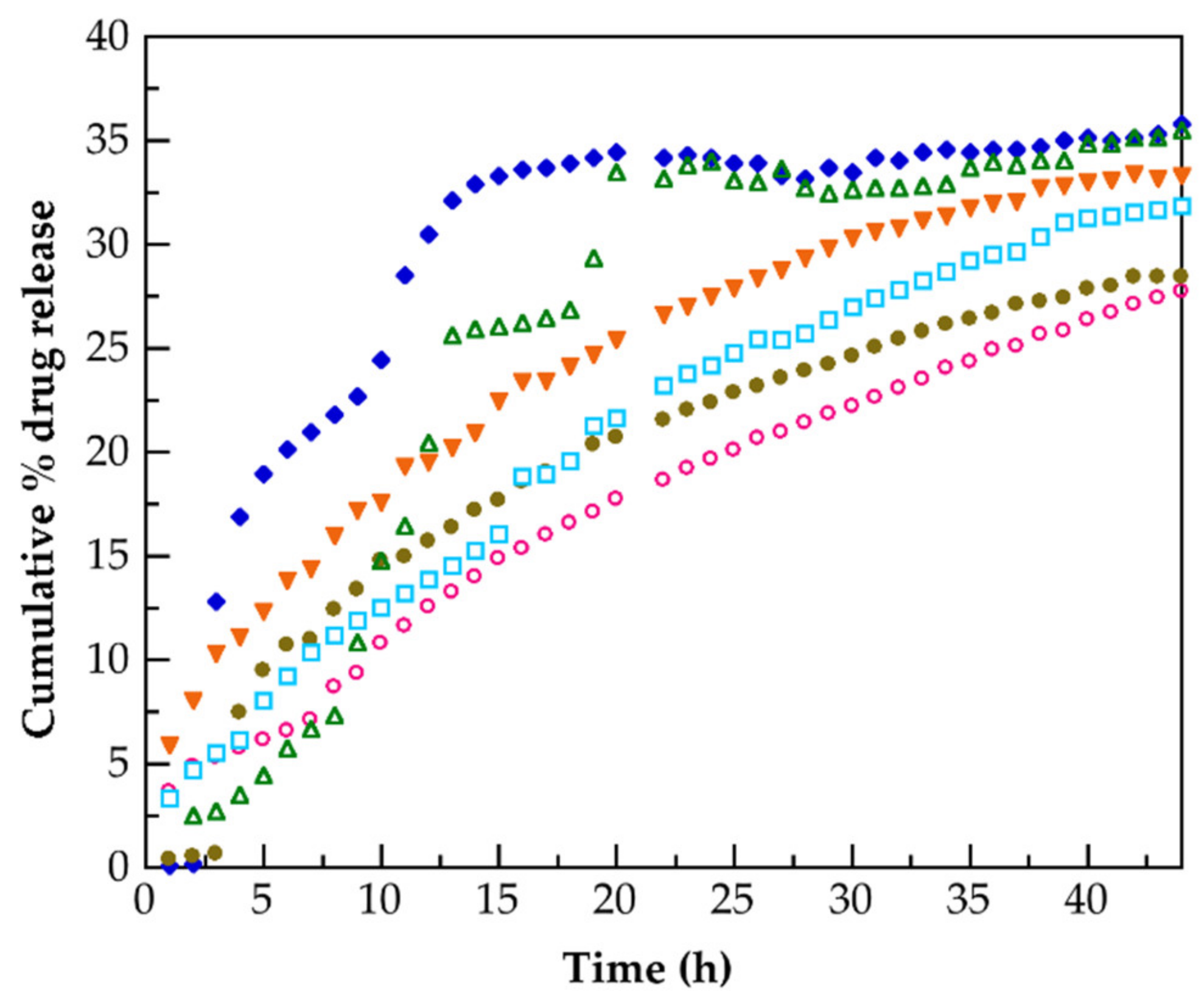
3.5. Release of Hydrocortisone from PMAA and of Their Nanocomposites
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, J.-Y.; Han, S.-W.; Huh, W.-S.; Tan, L.-S.; Baek, J.-B. In situ grafting of carboxylic acid-terminated hyperbranched poly(ether-ketone) to the surface of carbon nanotubes. Polymer 2007, 48, 4034–4040. [Google Scholar] [CrossRef]
- Pandey, G.; Thostenson, E.T. Carbon nanotube-based multifunctional polymer nanocomposites. Polym. Rev. 2012, 52, 355–416. [Google Scholar] [CrossRef]
- Ma, P.-C.; Siddiqui, N.A.; Marom, G.; Kim, J.-K. Dispersion and functionalization of carbon nanotubes for polymer-based Nanocomposites: A review. Compos. Part A 2010, 41, 1345–1367. [Google Scholar] [CrossRef]
- Wang, H.; Shao, Y.; Mei, S.; Lu, Y.; Zhang, M.; Sun, J.; Matyjaszewski, K.; Antonietti, M.; Yuan, J. Polymer-derived heteroatom-doped porous carbon materials. Chem. Rev. 2020, 120, 9363–9419. [Google Scholar] [CrossRef]
- Li, T.; Lee, J.-H.; Wang, R.; Kang, Y.T. Enhancement of heat transfer for thermal energy storage application using stearic acid nanocomposite with multi-walled carbon nanotubes. Energy 2013, 55, 752–761. [Google Scholar] [CrossRef]
- Jang, D.; Park, J.-E.; Kim, Y.-K. Evaluation of (CNT@CIP)-embedded magneto-resistive sensor based on carbon nanotube and carbonyl iron powder polymer composites. Polymers 2022, 14, 542. [Google Scholar] [CrossRef]
- Feldman, D. Polymers a polymer nanocomposites for cancer therapy. Appl. Sci. 2019, 9, 3899. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yang, W.-W.; Xu, D.-G. Stimuli-responsive nanoscale drug delivery systems for cancer therapy. J. Drug Target. 2019, 27, 423–433. [Google Scholar] [CrossRef]
- Peppas, N.A. Devices based on ontelligent biopolymers for oral protein delivery. Int. J. Pharm. 2004, 277, 11–17. [Google Scholar] [CrossRef]
- Zhang, J.; Peppas, N.A. Synthesis and characterization pf pH- and temperatura-sensitive poly(methacrylic acid)/poly(N-isopropylacrylamide) interpenetrating polymeric networks. Macromolecules 2000, 33, 102–107. [Google Scholar] [CrossRef]
- Kalay, S.; Stetsyshyn, Y.; Donchak, V.; Harhay, K.; Lishchynskyi, O.; Ohar, H.; Panchenko, Y.; Voronov, S.; Culha, M. pH-controlled fluorescence switching in water-dispersed polymer brushes grafted to modified boron nitride nanotubes for cellular imaging. Beilstein J. Nanotechnol. 2019, 10, 2428–2439. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, B.; Wang, Q.; Shi, X.; Xiao, Z.; Lin, J.; Fang, X. Carbon nanotubes as molecular transporters for walled plant cells. Nano Lett. 2009, 9, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Hampel, S.; Kunze, D.; Haase, D.; Krämer, K.; Rauschenbach, M.; Ritschel, M.; Leonhardt, A.; Thomas, J.; Oswald, S.; Hoffmann, V.; et al. Carbon nanotubes filled with a chemotherapeutic agent: A nanocarrier mediates inhibition of tumor cell growth. Nanomedicine 2008, 3, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhang, X.; Lu, Q.; Fei, Z.; Dyson, P.J. Single walled carbon nanotubes as drug delivery vehicles: Targeting doxorubicin to tumors. Biomaterials 2012, 33, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, D.; Patnaik, S.; Sood, S.; Das, N. Carbon nanotubes: Evaluation of toxicity at biointerfaces. J. Pharm. Anal. 2019, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Sayes, C.M.; Liang, F.; Hudson, J.L.; Mendez, J.; Guo, W.; Beach, J.M.; Moore, V.C.; Doyle, C.D.; West, J.L.; Billups, W.E.; et al. Functionalized density dependence of single-walled carbon nanotubes cytotoxicity in vitro. Toxicol. Lett. 2006, 161, 135–142. [Google Scholar] [CrossRef]
- Francis, A.P.; Devasena, T. Toxicity of carbon nanotubes: A review. Toxicol. Ind. Health 2018, 34, 200–210. [Google Scholar] [CrossRef]
- Fisher, C.; Rider, A.E.; Han, Z.J.; Kumar, S.; Levchenko, I.; Ostrikov, K. Applications and nanotoxicity of carbon nanotubes and grapheme in biomedicine. J. Nanomater. 2012, 2012, 315185. [Google Scholar] [CrossRef] [Green Version]
- Wick, P.; Manser, P.; Limbach, L.K.; Dettlaff-Weglikowskab, U.; Krumeich, F.; Roth, S.; Stark, W.J.; Bruinink, A. The degree and kind of agglomeration affect carbon nanotube cytotoxicity. Toxicol. Lett. 2007, 168, 121–131. [Google Scholar] [CrossRef]
- Sadegh, H.; Shahryari-ghoshekandi, R. Functionalization of carbon nanotubes and its application in nanomediciene: A review. Nanomed. J. 2015, 2, 231–248. [Google Scholar]
- Kolanowska, A.; Kuziel, A.W.; Jedrysiak, R.G.; Krzywiecki, M.; Korczeniewski, E.; Wisniewski, M.; Terzyk, A.P.; Boncel, S. Ullmann reactions of carbon nanotubes-advantageous and unexplored functionalization toward tunable surface chemistry. Nanomaterials 2019, 9, 1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giofre, S.V.; Tiecco, M.; Celesti, C.; Patane, S.; Triolo, C.; Gulino, A.; Spitaleri, L.; Scalese, S.; Scuderi, M.; Iannazzo, D. Eco-friendly 1,3-dipolar cycloaddition reactions on graphene quantum dots in natural deep eutectic solvent. Nanomaterials 2020, 10, 2549. [Google Scholar] [CrossRef] [PubMed]
- Tuccitto, N.; Spitaleri, L.; Destri, G.L.; Pappalardo, A.; Gulino, A.; Sfrazzetto, G.T. Supramolecular sensing of a chemical warfare agents simulant by functionalized carbon nanoparticles. Molecules 2020, 25, 5731. [Google Scholar] [CrossRef] [PubMed]
- Mountrichas, G.; Pispas, S.; Tagmatarchis, N. Grafting-to approach for the functionalization of carbon nanotubes with polystyrene. Mater. Sci. Eng. B 2008, 152, 40–43. [Google Scholar] [CrossRef]
- Qin, S.; Qin, D.; Ford, W.T.; Resasco, D.E.; Herrera, J.E. Functionalization of single-walled carbon nanotubes with polystyrene via grafting to and grafting from methods. Macromolecules 2004, 37, 752–757. [Google Scholar] [CrossRef]
- Salami-Kalajahi, M.; Haddadi-Asl, V.; Behboodi-Sadabad, F.; Rahimi-Razin, S.; Roghani-Mamaqani, H. Properties of PMMA/carbon nanotubes nanocomposites prepared by “grafting through” method. Polym. Compos. 2012, 33, 215–224. [Google Scholar] [CrossRef]
- Donchak, V.; Stetsyshyn, Y.; Bratychak, M.; Broza, G.; Harhay, K.; Stepina, N.; Kostenko, M.; Voronov, S. Nanoarchitectonics at surfaces using mutifunctional initiators of surface-initiated radical polymerization for fabrication of the nanocomposite. Appl. Surf. Sci. Adv. 2021, 5, 100104. [Google Scholar] [CrossRef]
- D’Emanuele, A.; Attwood, D. Dendrimer-drug interactions. Adv. Drug Deliv. Rev. 2005, 57, 2147–2162. [Google Scholar] [CrossRef]
- Luo, D.; Haverstick, K.; Belcheva, N.; Han, E.; Saltzman, W.M. Poly(ethylene glycol)-conjugated PAMAM dendrimer for biocompatible, high-efficiency DNA delivery. Macromolecules 2002, 35, 3456–3462. [Google Scholar] [CrossRef]
- Lu, X.; Imae, T. Dendrimer-mediated synthesis of water-dispersable carbon-nanotube-supported oxide nanoparticles. J. Phys. Chem. C 2007, 111, 8459–8462. [Google Scholar] [CrossRef]
- Shi, X.; Wang, S.H.; Shen, M.; Antwerp, M.E.; Chen, X.; Li, C.; Petersen, E.J.; Huang, Q.; Weber, W.J., Jr.; Baker, J.R., Jr. Multifunctional dendrimer-modified multiwalled carbon nanotubes: Synthesis, characterization, and in vitro cancer cell targeting and imaging. Biomacromolecules 2009, 10, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bryantsev, V.S.; Diallo, M.S.; Goddard, W.A., III. PAMAM Dendrimers undergo pH responsive conformational changes without swelling. J. Am. Chem. Soc. 2009, 131, 2798–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maingi, V.; Kumar, M.V.S.; Maiti, P.K. PAMAM Dendrimer-drug interactions: Effect of pH on the binding and release pattern. J. Phys. Chem. B 2012, 116, 4370–4376. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.-L.; Mai, Y.-W.; Zhou, X.-P. Dispersion and alignment of carbon nanotubes in polymer matrix: A review. Mater. Sci. Eng. R 2005, 49, 89–112. [Google Scholar] [CrossRef]
- Aguilar, J.; Rabelero, M.; Nuño-Donlucas, S.M.; Mendizábal, E.; Martínez-Richa, A.; López, R.G.; Arellano, M.; Puig, J.E. Narrow size-distribution poly(methyl methacrylate) nanoparticles made by semicontinuous heterophase polymerization. J. Appl. Polym. Sci. 2011, 119, 1827–1834. [Google Scholar] [CrossRef]
- Silva-Jara, J.M.; Manríquez-González, R.; López-Dellamary, F.A.; Puig, J.E.; Nuño-Donlucas, S.M. Semi-continuous heterophase polymerization to synthesize nanocomposites of poly(acrylic acid)-functionalized carbon nanotubes. J. Macromol. Sci. Pure Appl. Chem. 2015, 52, 732–744. [Google Scholar] [CrossRef]
- Saade, H.; Barrera, C.; Espinoza, A.; López-Quintanilla, M.L.; Fernandez, S.; Lopez, R.G. Ultrafine nanoparticles of Ibuprofen-poly(methyl methacrylate) by a polymerization-loading method. Drug Deliv. Lett. 2013, 3, 54–60. [Google Scholar] [CrossRef]
- Pérez-García, M.G.; Rabelero, M.; Nuño-Donlucas, S.M.; Mendizábal, E.; Martínez-Richa, A.; López, R.G.; Arellano, M.; Puig, J.E. Semi-continuous heterophase polymerization of n-butyl methacrylate: Effect of monomer feeding rate. J. Macromol. Sci. Pure Appl. Chem. 2012, 49, 539–546. [Google Scholar] [CrossRef]
- Dorff, T.B.; Crawford, E.D. Management and challenges of corticosteroid therapy in men with metastatic castrate-resistant prostate cancer. Ann. Oncol. 2013, 24, 31–38. [Google Scholar] [CrossRef]
- Rúan-Esparza, L.; Soto, V.; Gómez-Salazar, S.; Rabelero, M.; Ávalos-Borja, M.; Luna-Bárcenas, G.; Prokhorov, E.; Nuño-Donlucas, S.M. Poly[ethylene-co-(acrylic acid)]-based nanocomposites: Thermal and mechanical properties and their structural characteristics studied by Raman spectroscopy. Polym. Compos. 2011, 32, 1181–1189. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Zhang, P. De novo growth of poly(amidoamine) dendrimers on the surface of multi-walled carbon nanotubes. J. Mater. Sci. 2014, 49, 3469–3477. [Google Scholar] [CrossRef]
- Xia, R.; Li, M.; Zhang, Y.; Qian, J.; Yuan, X. Surface modification of MWNTs with BA-MMA-GMA terpolymer by single-step grafting technique. J. Appl. Polym. Sci. 2011, 119, 282–289. [Google Scholar] [CrossRef]
- Dutta, A. Fourier transform infrared spectroscopy. In Spectroscopic Methods for Nanomaterials Characterization; Thomas, S., Thomas, R., Zachariah, A., Mishra, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 73–93. [Google Scholar]
- Gulino, A.; Papanikolaou, G.; Lanzafame, P.; Aaliti, A.; Primerano, P.; Spitaleri, L.; Triolo, C.; Dahrouch, Z.; Khaskhoussi, A.; Schiavo, S.L. Synthesis, characterization and photocatalytic behavior of SiO2@nitrized-TiO2 nanocomposites obtained by straightforward novel approach. ChemistryOpen 2021, 10, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Iglesias, M. Estudios Conformacionales en [Alfa]-Dicetonas. Ph.D. Thesis, Universidad Complutense de Madrid, Madrid, Spain, 1974. [Google Scholar]
- Coleman, M.M.; Skrovanek, D.J.; Hu, J.; Painter, P.C. Hydrogen bonding in polymer blends. 1. FTIR Studies of urethane-ether blends. Macromolecules 1988, 21, 59–65. [Google Scholar] [CrossRef]
- Martinez-Felipe, A.; Brebner, F.; Zaton, D.; Concellon, A.; Ahmadi, S.; Piñol, M.; Oriol, L. Molecular recognition via hydrogen bonding in supramolecular complexes: A Fourier transform infrared spectroscopy study. Molecules 2018, 23, 2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajjadi, S.; Brooks, B.W. Unseeded semibatch emulsion polymerization of butyl acrylate: Bimodal particle size distribution. J. Polym. Sci. A Polym. Chem. 2000, 38, 528–545. [Google Scholar] [CrossRef]
- Galano, A. Carbon nanotubes: Promising agents against free radicals. Nanoscale 2010, 2, 373–380. [Google Scholar] [CrossRef]
- Nymark, P.; Jensen, K.A.; Suhonen, S.; Kembouche, Y.; Vippola, M.; Kleinjans, J.; Catalán, J.; Norppa, H.; van Delft, J.; Briedé, J.J. Free radical scavenging and formation by multi-walled carbon nanotubes in cell free conditions and in human bronchial epithelial cells. Part. Fibre. Toxicol. 2014, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Fenoglio, I.; Tomatis, M.; Lison, D.; Muller, J.; Fonseca, A.; Nagy, J.N.; Fubini, B. Reactivity of carbon nanotubes: Free radical generation or scavenging activity? Free Radical Bio. Med. 2006, 40, 1227–1233. [Google Scholar] [CrossRef]
- Lucente-Schultz, R.M.; Moore, V.C.; Leonard, A.D.; Price, B.K.; Kosynkin, D.V.; Lu, M.; Partha, R.; Conyers, J.L.; Tour, J.M. Antioxidant single-walled carbon nanotubes. J. Am. Chem. Soc. 2009, 131, 3934–3941. [Google Scholar] [CrossRef]
- Amiri, A.; Memarpoor-Yazdi, M.; Shanbedi, M.; Eshghi, H. Influence of different amino acid groups on the free radical scavenging capability of multi walled carbon nanotubes. J. Biomed. Mater. Res. Part A 2013, 101, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Gulino, A. Structural and electronic characterization of self-assembled molecular nanoarchitectures by X-ray photoelectron spectroscopy. Anal. Bioanal. Chem. 2013, 405, 1479–1495. [Google Scholar] [CrossRef] [PubMed]
- Spitaleri, L.; Gangemi, C.M.A.; Purrello, R.; Nicotra, G.; Sfrazzeto, G.T.; Casella, G.; Casarin, M.; Gulino, A. Covalently conjugated gold-porphyrin nanostructures. Nanomaterials 2020, 10, 1644. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Gomez, A.; Bravo-Sanchez, M.; Ceballos-Sanchez, O.; Vazquez-Lepe, M.O. Practical methods for background subtraction in photoemission spectra. Surf. Interface Anal. 2014, 46, 897–905. [Google Scholar] [CrossRef]
- Ratner, B.D.; Castner, D.G. Electron spectroscopy for chemical analysis. In Surface Analysis: The Principal Techniques, 2nd ed.; Vickerman, J.C., Gilmore, I.S., Eds.; Wiley: Chichester, UK, 2009; pp. 53–79. [Google Scholar]
- Beamson, G.; Briggs, D. High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, 1st ed.; Wiley: Chichester, UK, 1992; pp. 110–117. [Google Scholar]
- Yan, X.; Xu, T.; Chen, G.; Yang, S.; Liu, H.; Xue, Q. Preparation and characterization of electrochemically deposited carbon nitride firms on silicon substrate. J. Phys. D Appl. Phys. 2004, 37, 907–913. [Google Scholar] [CrossRef]
- Graf, N.; Yegen, E.; Gross, T.; Lippitz, A.; Weigel, W.; Krakert, S.; Terfort, A.; Unger, W.E.S. XPS and NEXAFS studies of aliphatic and aromatic amine species in functionalized surfaces. Surf. Sci. 2009, 603, 2849–2860. [Google Scholar] [CrossRef]
- Pipping, F.; Sarghini, S.; Holländer, A.; Paulussen, S.; Terryn, H. TFAA chemical derivation and XPS. Analysis of OH and NHx polymers. Surf. Interface Anal. 2009, 41, 421–429. [Google Scholar] [CrossRef]
- Tamon, H.; Ishizaka, H.; Yamamoto, T.; Suzuki, T. Preparation of mesoporous carbon by freeze drying. Carbon 1999, 37, 2049–2055. [Google Scholar] [CrossRef]
- Schmiers, H.; Friebel, J.; Streubel, P.; Hesse, R.; Köpsel, R. Change of chemical bonding of nitrogen of polymeric N-heterocyclic compounds during pyrolysis. Carbon 1999, 37, 585–590. [Google Scholar] [CrossRef]
- Lahaye, J.; Nanse, G.; Bagreev, A.; Strelko, V. Prorous structure and surface chemistry of nitrogen containing carbons from polymers. Carbon 1999, 37, 585–590. [Google Scholar] [CrossRef]
- Genest, A.; Portinha, D.; Fleury, E.; Ganachaud, F. The aza-Michael reaction as an alternative strategy to generate advanced silicon-based (macro)molecules and materials. Prog. Polym. Sci. 2017, 72, 61–110. [Google Scholar] [CrossRef]
- Ozturk, O.; Black, T.J.; Perrine, K.; Pizzolato, K.; Williams, C.T.; Parsons, F.W.; Ratliff, J.S.; Gao, J.; Murphy, C.J.; Xie, H.; et al. Thermal decomposition of generation-4 polyamidoamine dendrimer films: Decomposition catalyzed by dendrimer-encapsulated Pt particles. Langmuir 2005, 21, 3998–4006. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.-C.; Lee, Y.-D.; Chin, W.-K. Thermal degradation of polymethacrylic acid. J. Polym. Sci. Part A Polym. Chem. 1992, 30, 2389–2397. [Google Scholar] [CrossRef]
- Schild, H.G. Thermal degradation of poly(methacrylic acid): Further studies applying TGA/FTIR. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 2403–2405. [Google Scholar] [CrossRef]
- Babal, A.S.; Gupta, R.; Singh, B.P.; Dhakate, S.R. Depression in glass transition temperature of multiwalled carbon nanotubes reinforced polycarbonate composites: Effect of functionalization. RSC Adv. 2015, 5, 43462–43472. [Google Scholar] [CrossRef] [Green Version]
- Khare, K.S.; Khare, R. Effect of carbon nanotube dispersion on glass transition in cross-linked epoxy-carbon nanotube nanocomposites: Role of interfacial interactions. J. Phys. Chem. B 2013, 117, 7444–7454. [Google Scholar] [CrossRef]
- Peeterbroeck, S.; Laoutid, F.; Swoboda, B.; Lopez-Cuesta, J.-M.; Moreau, N.; Nagy, J.B.; Alexandre, M.; Dubois, P. How carbon nanotube crushing can improve flame retardant behavior in polymer nanocomposites? Macromol. Rapid Commun. 2007, 28, 260–264. [Google Scholar] [CrossRef]
- Pötschke, P.; Bhattacharyya, A.R.; Janke, A. Carbon nanotube-filled polycarbonate composites produced by melt mixing and their use in blends with polyethylene. Carbon 2004, 42, 965–969. [Google Scholar] [CrossRef]
- Zhang, X.; Takegoshi, K.; Hikichi, K. Phase separation and thermal degradation of poly(vinyl alcohol)/poly(methacrylic acid) and poly(vinyl alcohol)/poly(acrylic acid) systems by 13C cp/masnmr. Polymer 1992, 33, 718–724. [Google Scholar] [CrossRef]
- Hadjiioannou, T.P.; Christian, G.D.; Koupparis, M.A. Quantitative Calculations in Pharmaceutical Practice and Research; VCH Publishers Inc.: New York, NY, USA, 1993. [Google Scholar]
- Bourne, D.W. Pharmacokinetics. In Modern Pharmaceutics, 4th ed.; Banker, G.S., Rhodes, C.T., Eds.; Marcel Dekker Inc.: New York, NY, USA, 2002; pp. 67–92. [Google Scholar]
- Higuchi, T. Mechanism of sustained action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Hixson, A.W.; Crowell, J.H. Dependence of reaction velocity upon surface and agitation: I-Theoretical consideration. Ind. Eng. Chem. 1931, 23, 923–931. [Google Scholar] [CrossRef]
- Shoaib, M.H.; Tazeen, J.; Merchant, H.A.; Yousuf, R.I. Evaluation of drug release kinetics from ibuprofen matrix tablets using HPMC. Pak. J. Pharm. Sci. 2006, 19, 119–124. [Google Scholar] [PubMed]
- Bruschi, M.L. Strategies to Modify the Drug Release from Pharmaceutical Systems, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 37–62. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Name | Content of MAA 1 (wt.%) | Content of G3.0-CNTs-PAMAM-NH2 2 (wt.%) | Rate of Addition of MAAor the Mixture of MAA/G3.0-CNTs-PAMAM-NH2 (g/min) |
|---|---|---|---|
| PMAA 1 | 4 | 0 | 0.1 |
| PMAA 2 | 8 | 0 | 0.1 |
| PMAA 3 | 4 | 0 | 0.2 |
| PMAA 4 | 8 | 0 | 0.2 |
| PMAA 5 | 4 | 0 | 0.3 |
| PMAA 6 | 8 | 0 | 0.3 |
| PMAA-G3.0-CNTs 1 | 4 | 0.5 | 0.1 |
| PMAA-G3.0-CNTs 2 | 4 | 1.0 | 0.1 |
| PMAA-G3.0-CNTs 3 | 8 | 0.5 | 0.1 |
| PMAA-G3.0-CNTs 4 | 8 | 1.0 | 0.1 |
| PMAA-G3.0-CNTs 5 | 4 | 0.5 | 0.2 |
| PMAA-G3.0-CNTs 6 | 4 | 1.0 | 0.2 |
| PMAA-G3.0-CNTs 7 | 8 | 0.5 | 0.2 |
| PMAA-G3.0-CNTs 8 | 8 | 1.0 | 0.2 |
| PMAA-G3.0-CNTs 9 | 4 | 0.5 | 0.3 |
| PMAA-G3.0-CNTs 10 | 4 | 1.0 | 0.3 |
| PMAA-G3.0-CNTs 11 | 8 | 0.5 | 0.3 |
| PMAA-G3.0-CNTs 12 | 8 | 1.0 | 0.3 |
| Name of Functionalized CNTs | Wavelength (cm−1) | Chemical Group |
|---|---|---|
| CNTsoxid | 3433 | O–H stretching |
| CNTsOxCl | 1668 808 737 | di-carbonyl stretching C–Cl stretching C–Cl stretching |
| G0.0-CNTs-PAMAM-NH2 | 1663 1123 | C=O stretching C–N stretching |
| G0.5-CNTs-PAMAM-OCH3 | 1699 1432 | C=O stretching C–H symmetric deformation |
| G3.0-CNTs-PAMAM-NH2 | 1641 1112 | C=O stretching C–N stretching |
| Mixture of G3.0-CNTs-PAMAM-NH2 and MAA | 1637 3431 | N–H bending N–H stretching |
| Identification Name | Tg (°C) |
|---|---|
| PMAA 1 | 155 |
| PMAA 2 | 163 |
| PMAA 3 | 163 |
| PMAA 4 | 165 |
| PMAA 5 | 179 |
| PMAA 6 | 159 |
| PMAA-G3.0-CNTs 1 | 148 |
| PMAA-G3.0-CNTs 2 | 149 |
| PMAA-G3.0-CNTs 3 | 140 |
| PMAA-G3.0-CNTs 4 | 142 |
| PMAA-G3.0-CNTs 5 | 151 |
| PMAA-G3.0-CNTs 6 | 157 |
| PMAA-G3.0-CNTs 7 | 152 |
| PMAA-G3.0-CNTs 8 | 150 |
| PMAA-G3.0-CNTs 9 | 155 |
| PMAA-G3.0-CNTs 10 | 149 |
| PMAA-G3.0-CNTs 11 | 153 |
| PMAA-G3.0-CNTs 12 | 149 |
| Tested Materials | Models | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Zero-Order | First-Order | Higuchi | Korsmeyer–Peppas | Hixson–Crowell | |||||||
| R2 | K0 (h−1) | R2 | KI (h−1) | R2 | KH (h–1/2) | R2 | n | KKP (h–n) | R2 | KHC (h–1/3) | |
| PMAA 1 | 0.85 | 0.37 | 0.91 | 0.011 | 0.97 | 2.65 | 0.93 | 0.29 | 5.80 | 0.43 | 0.039 |
| PMAA-G3.0-CNTs 1 | 0.79 | 0.44 | 0.82 | 0.010 | 0.96 | 3.22 | 0.99 | 0.29 | 7.43 | 0.40 | 0.040 |
| PMAA-G3.0-CNTs 2 | 0.61 | 0.47 | 0.43 | 0.012 | 0.85 | 3.66 | 0.88 | 0.41 | 6.01 | 0.48 | 0.021 |
| PMAA 2 | 0.80 | 0.40 | 0.71 | 0.012 | 0.97 | 2.91 | 0.99 | 0.36 | 5.15 | 0.77 | 0.021 |
| PMAA-G3.0-CNTs 3 | 0.88 | 0.45 | 0.89 | 0.012 | 0.98 | 3.13 | 0.96 | 0.33 | 5.78 | 0.47 | 0.042 |
| PMAA-G3.0-CNTs 4 | 0.78 | 0.53 | 0.71 | 0.014 | 0.93 | 3.81 | 0.91 | 0.41 | 5.37 | 0.48 | 0.046 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrade-Melecio, H.A.; Antolín-Cerón, V.H.; Alvarado-Mendoza, A.G.; Vázquez-Lepe, M.; Barrera-Rivera, K.A.; Martínez-Richa, A.; Nuño-Donlucas, S.M. Semi-Continuous Heterophase Polymerization to Synthesize Poly(methacrylic acid)-Based Nanocomposites for Drug Delivery. Polymers 2022, 14, 1195. https://doi.org/10.3390/polym14061195
Andrade-Melecio HA, Antolín-Cerón VH, Alvarado-Mendoza AG, Vázquez-Lepe M, Barrera-Rivera KA, Martínez-Richa A, Nuño-Donlucas SM. Semi-Continuous Heterophase Polymerization to Synthesize Poly(methacrylic acid)-Based Nanocomposites for Drug Delivery. Polymers. 2022; 14(6):1195. https://doi.org/10.3390/polym14061195
Chicago/Turabian StyleAndrade-Melecio, Hugo A., Víctor H. Antolín-Cerón, Abraham G. Alvarado-Mendoza, Milton Vázquez-Lepe, Karla A. Barrera-Rivera, Antonio Martínez-Richa, and Sergio M. Nuño-Donlucas. 2022. "Semi-Continuous Heterophase Polymerization to Synthesize Poly(methacrylic acid)-Based Nanocomposites for Drug Delivery" Polymers 14, no. 6: 1195. https://doi.org/10.3390/polym14061195