1. Introduction
The arrangement of the reactive center in a ligand has been the basis for many advances in inorganic as well as organometallic chemistry. Accordingly, significant time and effort have gone into designing and synthesizing ancillary ligands, besides studying the properties, reactivities, and geometries of the resulting complexes. In some cases, these efforts have resulted in new applications in stoichiometric chemistry, catalysis, and materials science [
1,
2,
3]. A large number of structural studies for systems with phosphinimide ligands have been published in this area. These chelating and anionic ligands (R
3PN) are easily obtained from neutral phosphinimine precursors, prepared utilizing the simple and long–established Staudinger reaction [
4,
5,
6]. Iminophosphorane compounds, also known as phosphoranimines, phosphinimines, or phosphazenes, were detected in 1919; they are organic composites with the general structure R
3P=NR. Iminophosphoranes contain nitrogen alongside phosphorus atoms, which coordinate to transition metals using the nitrogen atom’s lone pair of electrons. Multidentate ligands are produced by adding extra donor sites into the iminophosphorane ligand, and they are gaining popularity in both coordination chemistry and catalysis [
7,
8,
9]. Iminophosphoranes are resonance hybrids of the two recognized forms A and B, and have a highly polarized P=N bond (
Scheme 1). They can also integrate transition metals through the sp
2–hybridized nitrogen atom’s lone pair, resulting in stable complexes (C in
Scheme 1) [
10]. Iminophosphoranes, which are primarily s–donor ligands with only minimal p–acceptor characteristics, have a limited inherent coordinating capability since they can be easily substituted by other ligands.
Polydentate mixed ligands, such as those found in the generic metal complex structures shown in
Scheme 2 (D–G), can stabilize a wider range of metal ions than their monodentate R
3P = NR counterparts due to the addition of further donor sites to the iminophosphoranes. The reactivity of the metal center can be modified as needed by selecting and adjusting these new donor sites, as well as the appropriate linkers. It is regarded as a critical component in achieving high levels of activity and selectivity [
7].
Several procedures for the production of iminophosphoranes are recognized; however, the most dominant and extensively used are the Staudinger as well as Kirsanov reaction (
Scheme 3). A phosphine (PR
3) is used as a starting material in both of them. The Staudinger reaction involves direct oxidation with an organic azide, whereas the Kirsanov reaction involves initial bromination and consecutive reaction of the resulting phosphine dibromide compound with a primary amine in the presence of a base [
11,
12,
13].
There are various methods for the synthesis of phosphazenes, such as the free radical process, thermal ring opening polymerization, substitution polymerization and cationic or anionic living polymerization, and solution polymerization method [
14,
15,
16,
17]. Phosphazenes have highly tunable chemical as well as physical properties that count on the substituents attached to the phosphorus atom. Therefore, they are employed in an enormous range of fields, such as rechargeable batteries [
18], liquid crystals [
19], membranes [
20] and lubricants [
21], anticancer agents [
22], flame retardants [
23], antibacterial reagents [
24], biological materials [
25], synthetic bones [
26], photosensitive substrates [
27], thermally stable macromolecules [
28], a high limiting oxygen index (LOI) [
29], coordination metal supports and reagents for organometallic chemistry [
30], silicon-containing compounds [
31], and catalysts [
32].
Uranium is a chemical element with the symbol U, which belongs to the actinides. Due to its strategic significance in the energy sector, uranium became the most valuable heavy metal [
32,
33,
34,
35]. In nature, the Earth’s crust contains an average of 4.0 mg/L uranium [
36]; however, it deposits in different types of rocks. It is a hexavalent state in the secondary minerals, such as kasolite, uranophane, autunite, etc., while it has a tetravalent oxidation state in the form of primary minerals, e.g., coffinite, pitchblende, uraninite, etc. Firstly, uranium is leached from its ore in an acidic or alkaline process, and extracted from the resulting leach liquor using an ion exchanger or a solvent extraction technique [
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47]. Solvent extraction (SX) is a widely used technique for the recovery and separation of base metals and strategic metals in hydrometallurgy. Solvent extraction efficacy is subject to several factors including the type of separation apparatus (pulsed columns, mixer–settler, etc.), the type of feed solution, the applied flowsheets, the chemical composition of the organic solvent, the flow rates of both aqueous and organic phases in the extractions along with elution steps, etc. In the SX method, the extractant plays an essential role. It should have tremendous solubility in the organic solvent. Nevertheless, it must have an extremely low aqueous phase solubility. The solvent ought to be nonvolatile, industrially serviceable, nontoxic, nonflammable, and affordable for the element extraction process [
48,
49]. Previous studies have used several extractants for uranium extraction by solvent extraction. The derivatives of organophosphorus compounds have been employed since 1900 in uranium extraction processes. Compounds such as Cyanex 302, Cyanex 272, TBP, DEHPA, D2EHPA/TOPO mixtures, Primene JM–T/Alamine–336 mixture [
50], DDPA, HDPA, CMPO, DNPPA, and DBBP, which are used to extract uranium from different matrices, were studied [
51,
52,
53,
54,
55,
56]. Several ligands with high selectivity are employed for uptake of uranium ions, comprising organic crown ethers [
57], calixarenes [
58], and Schiff bases. The long-chain amines have proven to be outstanding extractants for a large number of anionic metal complexes applied widely in uranium removal, such as Adogen–383, TOA, and Aliquate–336 [
59,
60,
61].
Amides are deemed one of the outstanding as well as vital organic functional groups in pharmaceuticals, agrochemicals, polymers, and naturally occurring molecules. In addition, carboxamides have considerable value in coordination, medicinal, and organic chemistry. They can be obtained via the amidation process where a condensation reaction occurs between carboxylic acid and amine. One-pot synthesis of the chelating carboxamides using different catalysts were proposed. For instance, pyridine–2,6–dicarboxylic acid bis–(3–hydroxy phenyl) amide (Pydca) along with tetra–kis (2–ethylhexyl) pyridine–2,6–dicarboxamide (EHPyCA) were successfully synthesized and utilized for removal of thorium as well as uranium from leach liquors of ore samples in Egypt [
62,
63,
64].
In this study, a new synthetic N–hydroxy–N–trioctyl iminophosphorane (HTIP) chelating ligand was synthesized using an effective alternative technique compared to the traditional Staudinger and Kirsanov methods and employed for uranium extraction from acidic solution. Both the removal and elution factors were optimized. Furthermore, the study dealt with the equilibrium, kinetic, and thermodynamic characteristics of uranium extraction from G. Gattar leach fluid, North Eastern Desert of Egypt.
2. Materials and Methods
2.1. Apparatus
The acidity and alkalinity of solutions were detected using a digital pH meter (VSTAR10 series, Thermo Scientific™, Waltham, MA, USA) with an error of ±0.1. An analytical balance (AUW220D Series, Shimadzu, Kyoto, Japan) with standard deviation of 0.05 mg was utilized to measure all samples. A Vibromatic-384 shaker was employed to mix the contents in separating funnels. The crystal structure of materials was examined using X-ray diffraction (XRD) technique (D8 Discover Family, Bruker, Billerica, MA, USA). Quantitative analysis of U(VI) was executed with a double beam spectrometer (T80 UV/Vis, PG Instrument, Leicestershire, UK) using the arsenazo (III) indicator and 650 nm wavelength against an appropriate standard solution [
65]. Furthermore, oxidometric titration against ammonium metavanadate and sodium diphenyl amine sulfonate as an indicator using automatic titrator [
66,
67] (SCOTT Instrument, GmbH, DE, Sialkot, Pakistan) was also employed to confirm the concentration of U(VI) ions. An ICP-OES spectrometer (OPTIMA 5300 DV, PerkinElmer, Waltham, MA, USA) was applied to specify the concentration of uranium and metal ions of G. Gattar leachate. A Reichert Thermovar was used to determine the melting point. The elemental analysis of uranium concentrate product and HTIP ligand were recorded using EDX (JSM–7900F, Jeol, Tokyo, Japan). An FTIR spectrophotometer (IRPrestige–21, Shimadzu, Kyoto, Japan) was employed to record the IR spectra using KBr disc. The
1H and
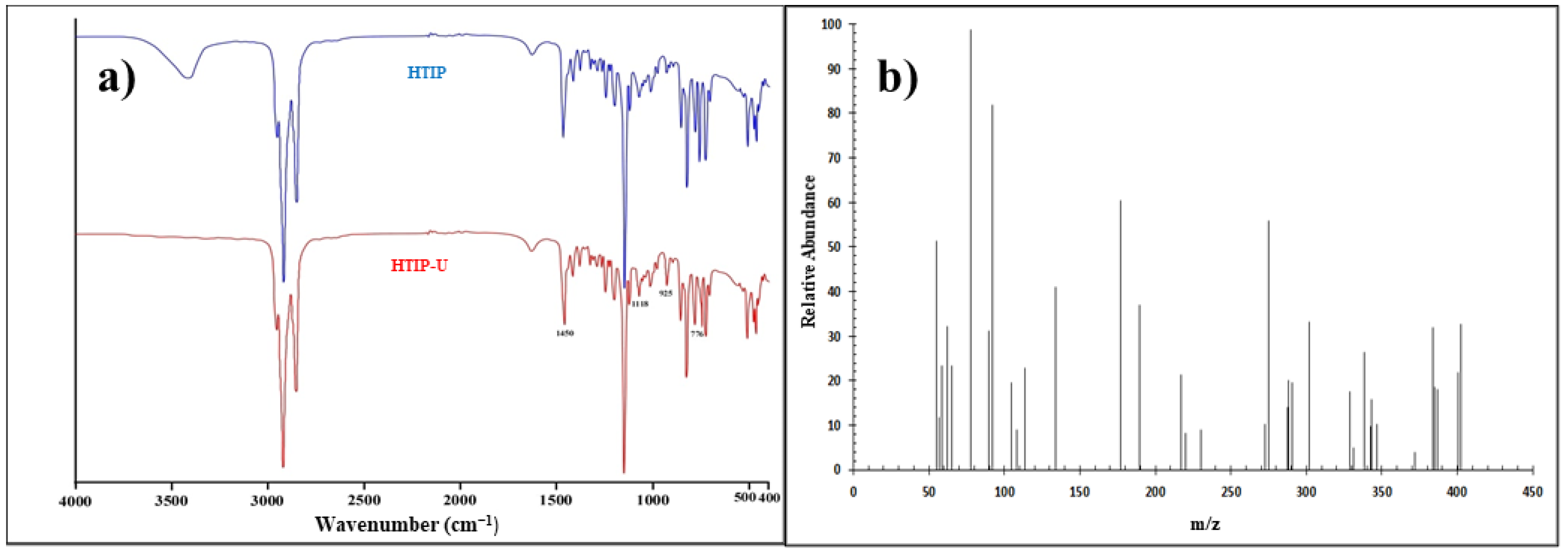
13C–NMR spectra were obtained at 500 MHz using an NMR spectrometer (Bruker Avance TM 500, Bruker, Billerica, MA, USA). The coupling constant (J) was measured in Hertz (Hz), while the chemical shift (δ) was measured in ppm. A mass spectrometer (Finnigan SSQ 7000 spectrometer, Thermo Finnigan, San Jose, CA, USA) was used for the molecular formula. The laboratories of National Research Center (NRC), Cairo, Egypt performed the FTIR, GC–MS, 1H, and 13C-NMR analyses.
2.2. Reagents
All of the reagents were made with analytical grade chemicals. HCl, H2SO4, NaOH, and HNO3 were purchased from POCH S.A., Gliwice, Poland. Trioctylphosphine oxide (TOPO) and N–hydroxylamine hydrochloride were obtained from Thermo Fisher Scientific–Acros Organics Inc., Geel, Belgium. Ammonium metavanadate, sodium nitrite, FeSO4.7H2O, AlCl3, and urea were supplied by Scharlau Chemie. S.A., Barcelona, Spain. Uranyl acetate dihydrate, arsenazo III, and sodium diphenyl amine sulfonate were obtained from Merck, Darmstadt, Germany. Furthermore, methanol, DMF, and ethyl acetate were purchased from Fluka, Gillingham, UK. All reactions were performed utilizing flame-dried glassware. Thin paper chromatography (PC) was used to observe the reaction’s development. Ethanol plus ethyl acetate (50:50 v/v) was adopted as an eluent. A UV lamp was used to visualize spots on the PC plates (250 nm).
2.3. Synthesis of N–Hydroxy–N–Trioctyl Iminophosphorane (HTIP) Chelating Ligand
Two primary procedures were used to produce N–Hydroxy–N–trioctyl iminophosphorane (HTIP). The first stage in neutralization was to mix 0.1 mole of NaOH (5.0 g, over the stoichiometric quantity) with 0.1 mole of NH2OH.HCl (7.0 g) in 50.0 mL of DMF as diluent. For 2.0 h, the mixture was refluxed at 50 °C. The vital goal of the neutralization phase was to make NH2OH more nucleophilic. The second swelling process started with 0.1 mole trioctylphosphine oxide (TOPO, 38.6 g) and 0.1 mole (13.3 g) AlCl3 hard Lewis acid in 50.0 mL DMF in a condenser for 2.0 h at 50 °C. Finally, the two additions were added to each other and allowed to condense for 6.0 h at 100 °C. The reaction was monitored using paper chromatography (PC) sheets and a solvent mixture of ethanol plus ethyl acetate 50:50 v/v. A UV lamp was used to detect the spots. The resulting HTIP appeared as a crystalline white to pale yellow solid with a density of ≈0.943 g/cm3. After completion of the reaction, the product was obtained by washing it several times with deionized water to remove any leftover DMF and AlCl3. The residue was washed, and the recrystallization procedure was performed with an ethanol/DMF mixture.
2.4. Preparation of U(VI) Standard Stock Solution
A 1000 mg/L (4.2 × 10−3 mol/L) U(VI) standard stock solution was prepared by dissolving 1.872 g of UO2(CH3COOH)2.2H2O in deionized water that had been acidified with 5.0 mL concentrated HNO3 to avoid hydrolysis in a 1000 mL volumetric flask. In addition, numerous standard stock solutions of 1000 mg/L of probable different ions during U(VI) extraction by HTIP/CHCl3 chelating ligand were generated by dissolving proper amounts of their salts in 1000 mL deionized water.
2.5. Extraction and Stripping Procedures
The pH value, shaking time, initial uranium(VI) conc., HTIP conc., temperature, and different ions were all tuned to optimize U(VI) ion extraction from synthetic solution by HTIP/CHCl
3. In these experiments, 25.0 mL of a 100 mg/L (4.2 × 10
−4 mol/L) synthetic U(VI) ions solution was mechanically shaken for a predefined period of time with 25.0 mL of varied concentrations of HTIP/CHCl
3. Both the extraction distribution ratio
D and stripping ratio
D′ were calculated using the following Equation (1) [
68]:
where
Co and
Ca (mg/L) correspond to the concentration of U(VI) in organic and aqueous phase, respectively. Moreover, the distribution coefficient (
Kd) and extraction percentage (
E%) were calculated using Equations (2) and (3), respectively:
where
Ci and
Ce (mg/L) symbolise for the initial and equilibrium concentration of U(VI) ions, respectively.
Vo and
Va (mL) represent organic and aqueous phase volumes, respectively. Nonetheless, the stripping procedures were performed by shaking different volumes of the loaded organic solvent with the eluent (2.0 mol H
2SO
4) for 10 min at ambient temperature. After equilibration, the two layers were entirely separated, and the U(VI) ion concentration was measured. The stripping percentage (
S%) can be expressed using the next Equation (4):
2.6. Production of G. Gattar Granite Leach Solution
Uranium–rich ore sample was collected from G. Gattar granite, North Eastern Desert, Egypt. Percolation leaching technique was applied; H2SO4 was used as a leachant. The leaching parameters were optimized at 75.0 g/L of H2SO4, −100 mesh particle size for 4.0 h leaching time, and 1:1 S/L phase ratio at room temperature. ICP-OES and colorimetric analysis were used to detect the chemical composition of the G. Gattar ore sample and its leachate.
4. Conclusions
A novel synthetic chelating agent, N–hydroxy–N–trioctyl–iminophosphorane (HTIP), was synthesized using an effective substitutional technique compared to the conventional methods and was utilized to uptake U(VI) ions from leach liquor of G. Gattar, North Eastern Desert, Egypt. Characterization was performed successfully using numerous analytical techniques, including 1H–NMR, 13C–NMR, FTIR, EDX, and GC–MS analyses. The uranium sequestration procedures were optimized by mixing 25.0 mL of U(VI) ion solution containing 0.42 × 10−4 mol/L with 0.99 × 10−3 mol/L HTIP/CHCl3 at pH 3.0, 1:1 A/O phase ratio, at 25 °C for 30 min. The utmost retention capacity of HTIP/CHCl3 was 247.5 mg/g. From the stoichiometric calculations, approximately 1.5 hydrogen atoms were released during the extraction at pH 3.0, and 4.0 moles of HTIP ligand were responsible for chelation of 1 mole of uranyl ions. The kinetic modeling data were well-suited to the pseudo first-order model. Furthermore, thermodynamic study demonstrated a negative ΔS° value, indicating that the uptake process is less disordered. Moreover, the rise in ΔG° value pointed to the spontaneousness and possibility of removing U(VI) ions at low temperatures. The elution of uranium loaded on HTIP was achieved using 2.0 M of H2SO4 as uranyl sulfate with 99.0% efficiency. Lastly, uranium concentrate (Na2U2O7, Y.C) with a purity of 93.24% was obtained by adding 30.0% NaOH to the elution and adjusting the pH to 7.0–8.0 with continuous stirring for 2 h.


 ,
, 


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}