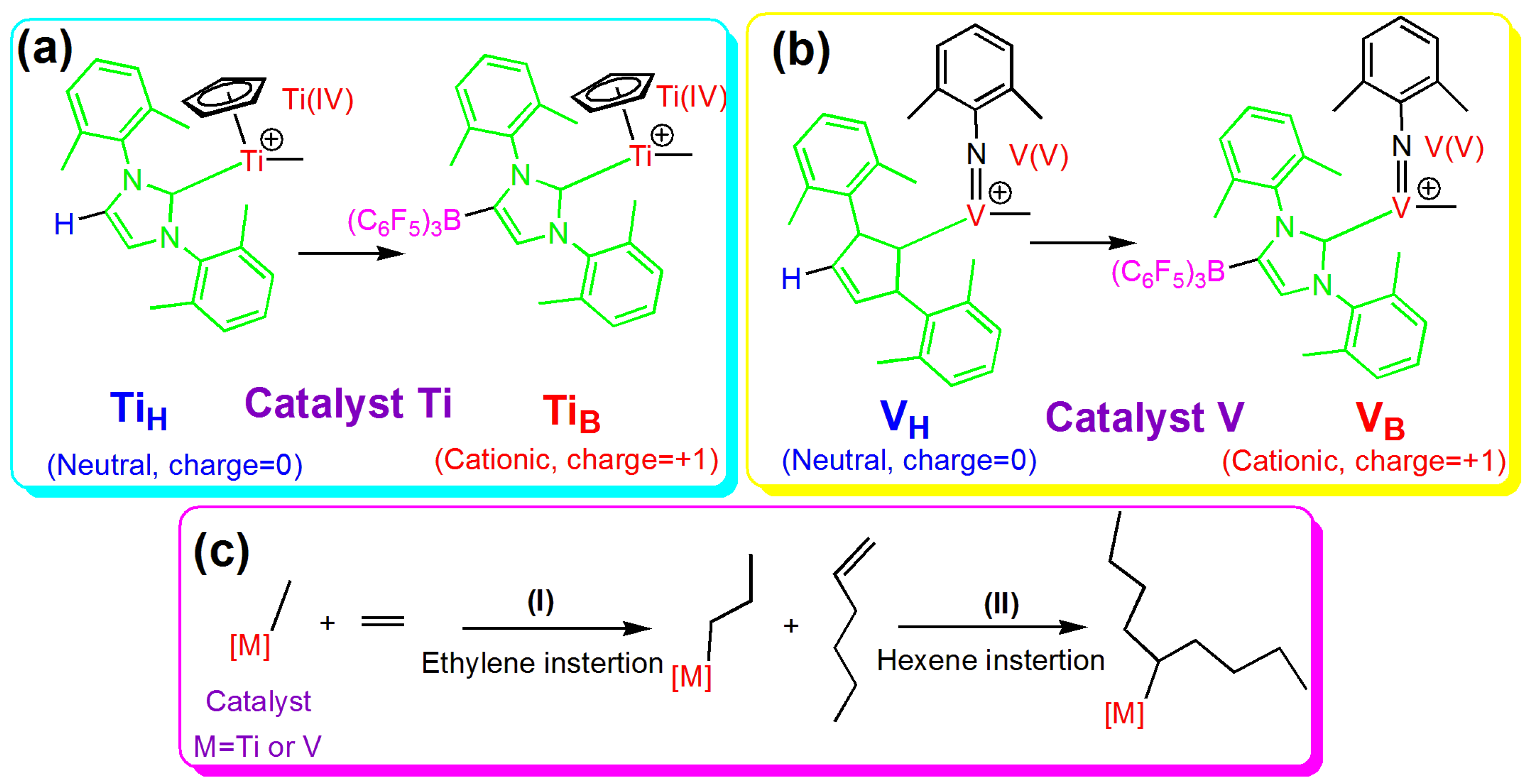
3.1. Ethylene Insertion into Titanium (TiH or TiB) Catalysts
Figure 1 depicts the pathway of ethylene insertion to titanium (
TiH or
TiB) catalysts. The optimized structures and IRI analysis of key structures are shown in
Figure 2.
Table 1 presents the WBIs and natural charges (Q
NBO) for certain key bonds and atoms. Evidently, the ethylene insertion to titanium (
TiH or
TiB) catalysts initially formed an intermediate (
TiIMH or
TiIMB), followed by a four-member ring transition state (
TiTSH or
TiTSB), ultimately generating the final product (
TiPRH or
TiPRB).
TiH and
TiB are half-titanocene catalysts containing N-heterocyclic carbene as a ligand, with the difference that in
TiB, one hydrogen of the N-heterocyclic carbene is substituted by B(C
6F
5)
3 as the remote coordinating ligand.
In TiH catalysts, ethylene initially bonded to Ti, thereby forming intermediate TiIMH. Owing to the coordination effect, the bond distance between Ti and the N-heterocyclic carbene (NHC) carbon atom (TiH–2CH, 2.271 Å) in the TiIMH structure was longer than that of TiH (2.174 Å) by 0.097 Å. Additionally, the 3CH–4CH (1.435 Å) bond was longer than that in the C2H4 molecule (1.330 Å) by 0.105 Å. The TiH–3CH and TiH–4CH bond lengths were 2.186 and 2.137 Å, respectively, with large WBIs of TiH–3CH (0.646) and TiH–4CH (0.689). Compared with reactants (TiH + C2H4), the energy of TiIMH was lower by 19.4 kcal/mol, thus indicating that TiIMH is highly stable. After TiIMH, the reaction reached a four-member ring transition state, TiTSH, with the TiH–3CH bond (2.160 Å) being shorter than that in TiIMH(2.186 Å) by 0.026 Å. The WBIs of TiH–3CH (0.572) and TiH–4CH (0.275) in TiTSH were lower than those in TiIMH. Following TiTSH, the product of ethylene insertion, TiPRH formed. The 1CH–4CH (1.537 Å) and TiH–3CH (2.138 Å) bonds in TiPRH were shorter than those in TiTSH by 0.545 and 0.022 Å, respectively. Conversely, the 3CH–4CH bond (1.538 Å) in TiPRH was longer than that in TiTSH by 0.105 Å.
During the reaction process, the WBIs of TiH–1CH (from 0.634 to 0.094) and 3CH–4CH (from 2.039 to 1.012) bonds diminished from the reactant (TiH + C2H4) to the product TiPRH. This decrease indicates the breakage of the TiH–1CH bond and the transition of the 3CH–4CH bond from a double to single bond. Meanwhile, the WBI gradually increased from 0.009 to 1.011 for the 1CH–4CH bond, thus indicating the formation of the 1CH–4CH bond. Compared with the reactant (TiH + C2H4), the energy of TiTSH was higher by 27.6 kcal/mol, whereas that of TiPRH was lower by 4.6 kcal/mol. The intermediate TiIMH was highly stable, and its energy was lower than that of the product TiPRH by 14.8 kcal/mol. Consequently, the reaction was prone to halt at the intermediate stage, thus rendering difficulty in yielding the product.
In TiB catalysts, one hydrogen of the N-heterocyclic carbene was substituted with B(C6F5)3 as the remote coordinating ligand. The QNBO value of the TiB atom (1.487 e) in the TiB catalyst was higher than that of the TiH atom (1.206 e) in the TiH catalyst. The process of ethylene insertion into TiB catalysts was similar to that of the TiH catalyst. The free energies demonstrate that ethylene insertion into the TiB catalyst is thermodynamically and kinetically preferred over the TiH catalyst. Compared with the reactant (TiB + C2H4), the energy barrier for TiTSB was 9.7 kcal/mol, which is significantly lower than that for the TiH catalyst (27.6 kcal/mol). Additionally, the reaction was exothermic by 8.7 kcal/mol, which is larger than the 4.6 kcal/mol for the TiH catalyst. Unlike the highly stable TiIMH, the energy of intermediate TiIMB was slightly higher than that of the reactant by 2.7 kcal/mol. Further, the WBIs of TiB–3CB (0.270) and TiB–4CB (0.156) in TiIMB were significantly lower than those (0.646 and 0.689) in TiIMH, thus indicating that the reaction proceeded smoothly to yield the product.
Bickelhaupt’s activation strain analysis is frequently employed to gain insight into the origin of the various contributions [
57,
58,
59]. This analysis decomposes the electronic activation energy ΔE
≠ of the transition state into the distortion energy (E
TS−dist) and interaction energy (E
TS−int) between two reaction fragments (
A:
C2H4 and
B:
Ti catalyst). Herein, this method was applied to analyze two transition states (
TiTSH and
TiTSB). As shown in
Scheme 2I, the distortion energies were calculated as E
TS-dist(A) = E
TS−A − E
A and E
TS−dist(B) = E
TS−B − E
B, where E
A (or E
B) and E
TS−A (or E
TS−B) are the energies of A (or B) in reactant ground state and transition state geometries, respectively. The interaction energies between A and B in the transition state were calculated by E
TS−int = ΔE
≠ − E
TS−dist + BE(AB), where E
TS−dist = E
TS−dist(A) + E
TS−dist(B), BE(AB) is the binding energy between A and B in the intermediate complex AB, and ΔE
≠ is the electronic activation energy for ethylene insertion reaction. When considering the intermediate complex AB as the reference, the electronic activation energy ΔE
≠ = E
TS−dist(eff) + E
TS−int, so E
TS−dist(eff)= E
TS−dist − BE(AB) represent the effective distortion energy.
We also applied the same approach to decompose the two coordination energies of intermediates (
TiIMH and
TiIMB) into distortion energy (E
IM−dist) and interaction energy (E
IM−int) between two reaction fragments (A:
C2H4 and B:
Ti catalyst). As shown in
Scheme 2II, the distortion energies were calculated as E
IM−dist(A) = E
IM−A − E
A and E
IM−dist(B) = E
IM-B − E
B, where E
A (or E
B) and E
IM−A (or E
IM−B) are the energies of A (or B) in reactant ground state and intermediate geometries, respectively. The interaction energies between A and B in the intermediates were calculated by E
IM−int = ΔE − E
IM−dist, where E
IM−dist = E
IM−dist(A) + E
IM−dist(B), and ΔE is the binding energy between A and B in the intermediate complex AB. The results are presented in
Table 2.
The E
IM−dist and E
TS−dist(eff) values of
TiIMH and
TiTSH were 30.2 and 75.8 kcal/mol, respectively, which are much higher than those of
TiIMB (9.6 kcal/mol) and
TiTSB (36.1 kcal/mol), respectively. This indicates that
TiIMH and
TiTSH undergo a more significant configuration change compared to reactants. This is consistent with the IRI analysis.
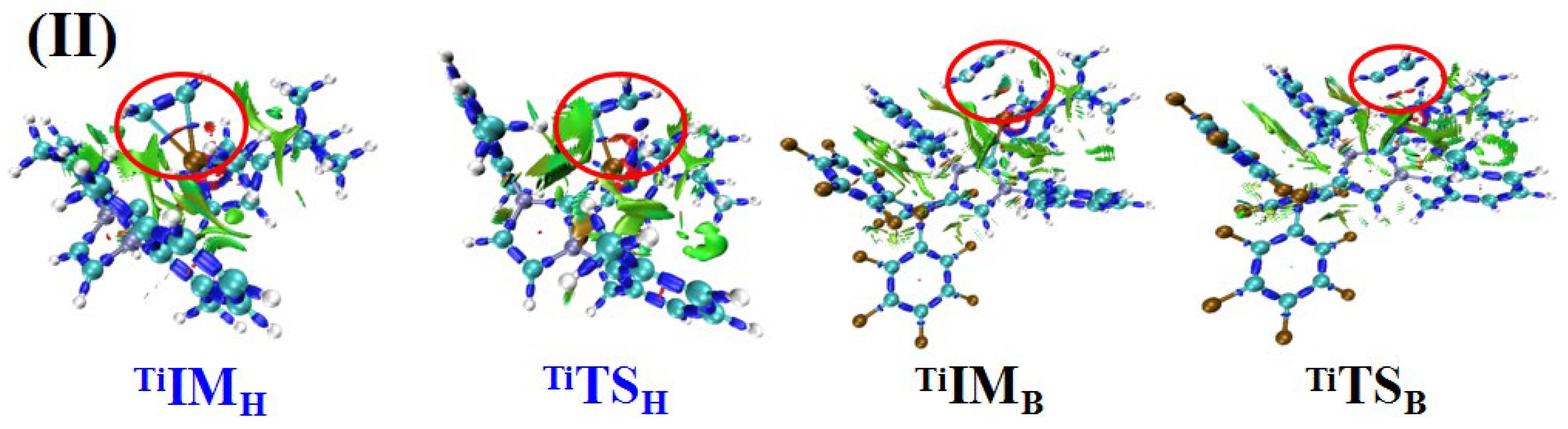
Figure 2II shows the IRI analysis of
TiIMH, TiTSH,
TiIMB, and
TiTSB [
48]. Evidently, the interactions between the two reaction fragments (
C2H4 and
Ti catalyst) in
TiIMH and
TiTSH were stronger than those in
TiIMB and
TiTSB.
The EIM−int of TiIMH (−65.3 kcal/mol) was much lower than that of TiIMB (−20.6 kcal/mol). This indicates that TiIMH is more stable, and the reaction for the TiH catalyst is prone to halt at the intermediate stage, whereas that for the TiB catalyst proceeds smoothly to yield the product. The ETS-int of TiTSH (−64.2 kcal/mol) was lower than that of TiTSB (−41.7 kcal/mol) by 22.5 kcal/mol. However, the ETS−dist(eff) (75.8 kcal/mol) of TiTSH was much higher than that (36.1 kcal/mol) of TiTSB by 39.7 kcal/mol. This explains why the energy barrier of TiTSB is lower than TiTSH, thus rendering the TiB catalyst more efficient in catalyzing ethylene polymerization.
3.2. 1-Hexene Insertion into Titanium (TiH or TiB) Catalysts
Following ethylene insertion into the
TiH or
TiB catalysts, the 1-hexene insertion reaction can be accomplished through 1,2 insertion or 2,1 insertion reactions. The 1-hexene 1,2 insertion and 2,1 insertion into the
TiH or
TiB catalysts involve an intermediate, followed by a four-member ring transition state, which yields the final product.
Figure 3 illustrates the reaction pathways of 1-hexene insertion into Ti catalysts, namely
TiH21 (blue),
TiH12 (green),
TiB21 (black), and
TiB12 (yellow).
Figure 4 shows the optimized structures of the 1-hexene insertion into Ti catalysts. The WBIs and Q
NBO involved in 1-hexene insertion into the
TiH and
TiB catalysts are presented in
Table 3.
For TiH catalysts, two pathways exist for 1-hexene insertion reactions: 1,2 insertion (TiH12) or 2,1 insertion (TiH21). The free energies indicated that the TiH21 pathway is thermodynamically and kinetically preferred over the TiH12 pathway. Compared with the reactant (TiPRH + Hex), the free energy barrier of TiTSH21 was 26.3 kcal/mol, which is lower than the 30.0 kcal/mol barrier for the TiTSH12 catalyst. Furthermore, the reaction for the TiH21 pathway was exothermic by 11.7 kcal/mol, which is higher than the 7.5 kcal/mol for the TiH12 pathway.
In the 1-hexene insertion TiH21 pathway, 1-hexene initially bonded to Ti, thereby forming the intermediate TiIMH21. Owing to the coordination effect, the 6CH–7CH bond (1.457 Å) was significantly longer than that in the 1-hexene (Hex) molecule (1.332 Å) by 0.125 Å. The TiH–6CH and TiH–7CH bond were 2.129 and 2.148 Å in length, respectively, with large WBIs of TiH–6CH (0.693) and TiH–7CH (0.677). Compared with reactants (TiPRH + Hex), the free energy of TiIMH21 was lower by 16.3 kcal/mol, thus indicating its stability. After TiIMH21, the reaction reached a four-member ring transition state, TiTSH21, with the TiH–7CH (2.266 Å) being longer than that in TiIMH21 (2.148 Å) by 0.118 Å. The WBIs of TiH–6CH (0.192) and TiH–7CH (0.393) in TiTSH21 were lower than those in TiIMH21. Following TiTSH21, the product of 1-hexene insertion, TiPRH21, formed. Compared with the TiTSH21 configuration, the 3CH–6CH (1.535 Å) and TiH–7CH (2.123 Å) bonds in TiPRH21 were shorter, whereas the 6CH–7CH bond (1.537 Å) in TiPRH21 was longer.
During the reaction process, the WBIs of the TiH–3CH (from 0.627 to 0.100) and 6CH–7CH bonds (from 1.980 to 0.999) decreased from the reactant (TiPRH + Hex) to the product TiPRH21. This decrease indicates the breakage of the TiH–3CH bond and the transition of the 6CH–7CH bond from a double to single bond. Meanwhile, the WBI for the 3CH–6CH bond gradually increased from 0.012 to 1.000, thus indicating the formation of the 3CH–6CH bond. Similar to the ethylene insertion reaction, the 1-hexene insertion intermediate TiIMH21 was stable, with its energy being equal to that of the product TiPRH21.
For TiB catalysts, 1,2 insertion (TiB12) or 2,1 insertion (TiB21) pathways exist for 1-hexene insertion reactions. The free energy results revealed that the TiB21 pathway is thermodynamically preferred over the TiB12 pathway. Compared with the reactant (TiPRB + Hex), the free energy barrier of TiTSB21 was 12.4 kcal/mol, which is higher than the 6.5 kcal/mol for the TiTSB12 catalyst. Additionally, the reaction for the TiB21 pathway was exothermic by 14.6 kcal/mol, significantly larger than the 9.8 kcal/mol for the TiB12 pathway. The energies of both transition states (TiTSB21 and TiTSB12) were not high, thus allowing them to be easily crossed during the reaction. Consequently, the product (TiPRB21) with lower product energy TiPRB21 became the main reaction product.
Comparison between TiH and TiB catalysts in the 1-hexene insertion reaction revealed that both catalysts followed the dominant 2,1 insertion pathway (TiH21 and TiB21). The free energy barrier for TiB21 was 12.4 kcal/mol, significantly lower than 26.3 kcal/mol for TiH21. Unlike the highly stable TiIMH21, the energy of the intermediate TiIMB21 was 13.3 kcal/mol higher than that of the product TiPRB21. The WBIs of the TiB–6CB (0.341) and TiB–7CB (0.120) in TiIMB21 were considerably lower than those in TiIMH21 (0.693 and 0.677). Therefore, the 1-hexene insertion reaction for TiB21 was easier to perform.
The comparison of the entire ethylene and 1-hexene insertion reaction process for
TiH and
TiB catalysts is shown in
Figure 5. In the ethylene and 1-hexene insertion into
TiH catalysts, the energy of the transition state
TiTSH was the highest at 27.6 kcal/mol, whereas that of the final product
TiPRH21 was −16.3 kcal/mol. However, the energy of the intermediate
TiIMH was −19.4 kcal/mol, lower than that of the final product
TiPRH21 (−16.3 kcal/mol)
, thus indicating that the intermediate is highly stable. Therefore, the reaction was prone to halt at the intermediate rather than proceed to yield the product. Conversely, for the entire ethylene and 1-hexene insertion into
TiB catalysts, compared with the reactant, the energy of the transition state
TiTSB was 9.7 kcal/mol, considerably lower than that of
TiTSH (27.6 kcal/mol). The energy of the final product
TiPRB21 was −23.3 kcal/mol, significantly lower than that of
TiPRH21 (−16.3 kcal/mol). Consequently, the entire ethylene and 1-hexene insertion reaction proceeded smoothly to reach the final product for the
TiB catalyst.
3.3. Ethylene and 1-Hexene Insertion into V Catalysts
Figure 6 shows the reaction pathways for ethylene and 1-hexene insertion into vanadium (
VH or
VB) catalysts. The key optimized structures and IRI analysis of important structures are shown in
Figure 7. The WBIs and Q
NBO involved in the ethylene and 1-hexene insertion into
VH or
VB catalysts are presented in
Table 4. Similar to the reaction pathway catalyzed by the Ti catalyst, the ethylene and 1-hexene insertion into
VH or
VB catalysts involved the formation of an intermediate, followed by a four-member ring transition state, thereby resulting in the final product. Both the
VH and
VB catalysts contain N-heterocyclic carbene as a ligand, with the difference that in the
VB catalyst, one hydrogen of the N-heterocyclic carbene is substituted with B(C
6F
5)
3 as the remote coordinating ligand. The Q
NBO value for the V
B atom (0.767 e) in the
VB catalyst was larger than that for the V
H atom (0.435 e) in the
VH catalyst.
For ethylene insertion into vanadium (VH and VB) catalysts, the energy results indicate that ethylene insertion into the VB catalyst is thermodynamically and kinetically preferred over the VH catalyst. Compared with the reactant, the energy barrier for VB ethylene insertion was 2.9 kcal/mol, significantly lower than the 13.1 kcal/mol for VH ethylene insertion. The reaction was exothermic by 20.7 kcal/mol for the VB catalyst, larger than the 7.9 kcal/mol for the VH catalyst. The intermediate VIMH was highly stable, with its energy being lower than that of the product VPRH by 12.6 kcal/mol. Therefore, the reaction was prone to halt at the intermediate stage, thus rendering difficulty in yielding the product. However, the energy of the intermediate VIMB was higher than that of the product VPRB by 10.8 kcal/mol. Additionally, the WBIs of VB–3CB (0.384) and VB–4CB (0.200) in VIMB were significantly lower than those in VIMH (0.621 and 0.712). Consequently, the reaction proceeded smoothly to yield the product for the VB ethylene insertion.
The results for the decomposition of activation energies of the transition states (
VTSH and
VTSB) and coordination energies of intermediates (
VIMH and
VIMB) into the distort energies and the interaction energies are presented in
Table 5.
The E
IM−dist and E
TS−dist(eff) values of
VIMH and
VTSH were 15.4 and 72.7 kcal/mol, respectively, higher than those of
VIMB (5.4 kcal/mol) and
VTSB (48.5 kcal/mol), respectively. This indicates that
VIMH and
VTSH underwent a more significant configuration change compared with the reactants, which is consistent with the IRI analysis.
Figure 7II shows the IRI analysis of
VIMH, VTSH,
VIMB, and
VTSB [
48]. The interactions between the two reaction fragments (
C2H4 and
V catalyst) in
VIMH and
VTSH were stronger than those in
VIMB and
VTSB.
The E
IM−int of
VIMH (−49.5 kcal/mol) was considerably lower than that of
VIMB (−28.0 kcal/mol). This suggests that the intermediate
VIMH was more stable, and the ethylene insertion reaction for the
VH catalyst was prone to halt at the intermediate. Conversely, for the
VB catalyst, the ethylene insertion reaction proceeded smoothly to yield the product. The E
TS−int of
VTSH (−73.7 kcal/mol) was lower than that of
vTSB (−59.5 kcal/mol) by 14.2 kcal/mol. However, the E
TS−dist(eff) (72.7 kcal/mol) of
vTSH was significantly larger than that of
vTSB (48.5 kcal/mol) by 24.2 kcal/mol. This explains why the energy barrier of
VTSB was lower than that of
VTSH, thus rendering the
VB catalyst more efficient in catalyzing ethylene polymerization. After ethylene insertion into
VH and
VB catalysts, the subsequent 1-hexene insertion reaction occurred through 1,2 insertion or 2,1 insertion reactions. The 1-hexene 1,2 insertion and 2,1 insertion into
VH and
VB catalysts involve an intermediate, followed by a four-member ring transition state, leading to the final product. For clarity, this study focused on the optimal reaction pathways (
VH12 and
VB21), and the detailed reaction pathways are shown in
Figures S1 and S2.
VH12 and
VB21 are the dominant 1-hexene insertion reaction pathways for
VH and
VB catalysts, respectively.
The energy results indicate that the
VB21 pathway is preferred over the
VH12 pathway. As shown in
Figure 6, the energy of the transition state
VTSB21 was lower than that of
VTSH12 by 13.9 kcal/mol. Moreover, the energy of product
VPRB21 was lower than that of
VPRH12 by 12.5 kcal/mol. Furthermore, the energy of
VIMH12 was lower than that of the corresponding product
VPRH12 by 8.0 kcal/mol, thus indicating its high stability. Therefore, the reaction was prone to halt at the intermediate rather than proceed to yield the product. However, the energy of intermediate
VIMB21 was higher than that of the product
VPRB21 by 11.5 kcal/mol. The WBIs of V
B–
6C
B (0.136) and V
B–
7C
B (0.257) in
VIMB21 were significantly lower than those in
VIMH12 (0.660 and 0.601). Thus, the reaction proceeded smoothly to yield the product for
VB 1-hexene insertion.
In the insertion of ethylene and 1-hexene into VH catalysts, the transition state energy (VTSH) was 13.1 kcal/mol, whereas that of the final product energy VPRH12 was −15.0 kcal/mol. However, the energies of intermediates VIMH and VIMH12 were −20.5 and −23.0 kcal/mol, respectively, lower than that of the final product VPRH12 (−15.0 kcal/mol). This indicates that the intermediate stages were highly stable, thus rendering the reaction prone to stopping at the intermediate stages and difficult to yield the final product. Conversely, for the ethylene and 1-hexene insertion into VB catalysts, the transition state energy VTSB was significantly lower at 2.9 kcal/mol compared with VTSH (13.1 kcal/mol). The energy of the final product VPRB21 was −27.5 kcal/mol, substantially lower than that of VPRH12 (−15.0 kcal/mol). Consequently, the ethylene and 1-hexene insertion reaction proceeded smoothly and yielded the final product when using the VB catalyst.
Further comparison between
TiB and
VB catalysts revealed that the energy of transition state
VTSB of the ethylene and 1-hexene insertion into
VB catalysts was 2.9 kcal/mol, which is lower than that of
TiTSB (9.7 kcal/mol). Additionally, the energy of the final product
VPRB21 was −27.5 kcal/mol, which is lower than that of
TiPRB21 (−23.3 kcal/mol). These results indicate that the
VB catalyst has higher reaction activity than the
TiB catalyst, which is in agreement with the experimental results. Further, the catalytic activity reached 4590 kg-PE/mol-Ti·h and 11,000 kg-PE/mol-V·h for ethylene polymerization [
24,
25].
To thoroughly investigate the reaction, both the GRI and ELF analyses were conducted on key molecules (
Table 6 and
Figure 8). The
VB and
TiB catalysts exhibited the largest electrophilicity (
ω) values, 9.449 and 7.478, respectively, thus signifying their electrophilic nature. By contrast,
VH and
TiH exhibited substantial global nucleophilicity (
NNu) values of 5.650 and 6.356, thus indicating that they are nucleophilic. The
C2H4 and
Hex molecules displayed moderate
NNu values of 1.863 and 2.361, thus suggesting a certain level of nucleophilicity. Thus,
VB and
TiB catalysts can facilitate olefin polymerization reactions, whereas
VH and
TiH do not. The
ω value of
VB (9.449) was slightly higher than that of
TiB (7.478); thus,
VB exhibited greater electrophilicity and reactivity, which is in agreement with the experimental results [
24,
25].
Figure 8 presents the ELF analysis for key molecules, including
TiH,
TiB,
VH, and
VB. The ELF analysis, which is typically used to analyze nucleophilic attack sites, was employed herein to obtain the ELF isosurface maps for these molecules (
Figure 8). A comparison of the ELF isosurface maps for
TiH and
VH with those of
TiB and
VB revealed that the ELF values in the atomic regions of
TiB and
VB were lower. This suggests that
TiB and
VB atoms are more susceptible to nucleophilic attacks. These observations align with the conclusions drawn from previous discussions.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}