
High-Glass-Transition Polyesters Produced with Phthalic Anhydride and Epoxides by Ring-Opening Copolymerization (ROCOP)

 ,
,  , and
, and
Abstract
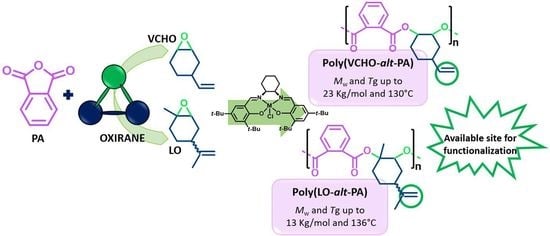
:
1. Introduction
2. Materials and Methods
2.1. Materials
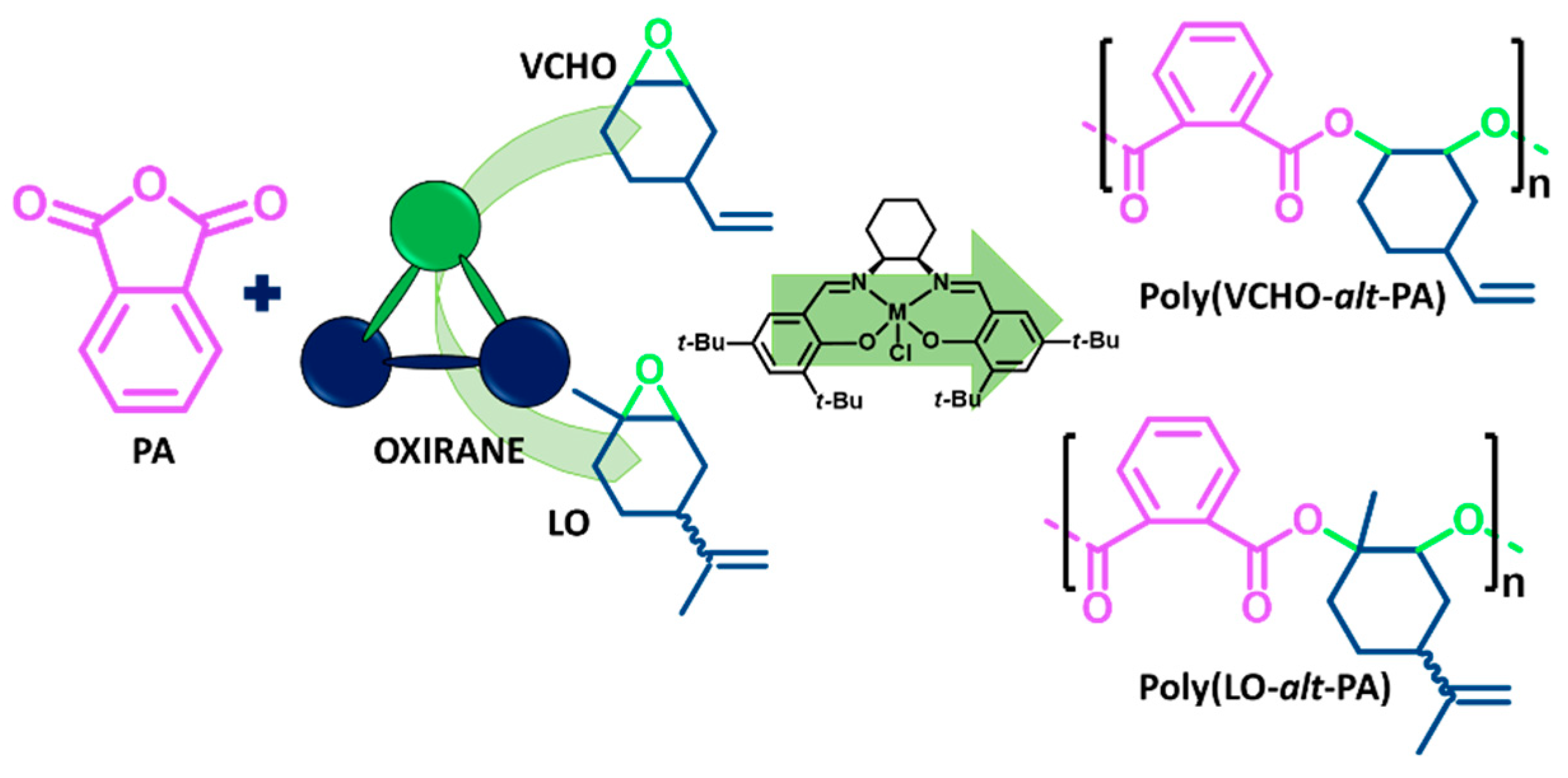
2.2. Polymerization in Solvent with Precontact Step
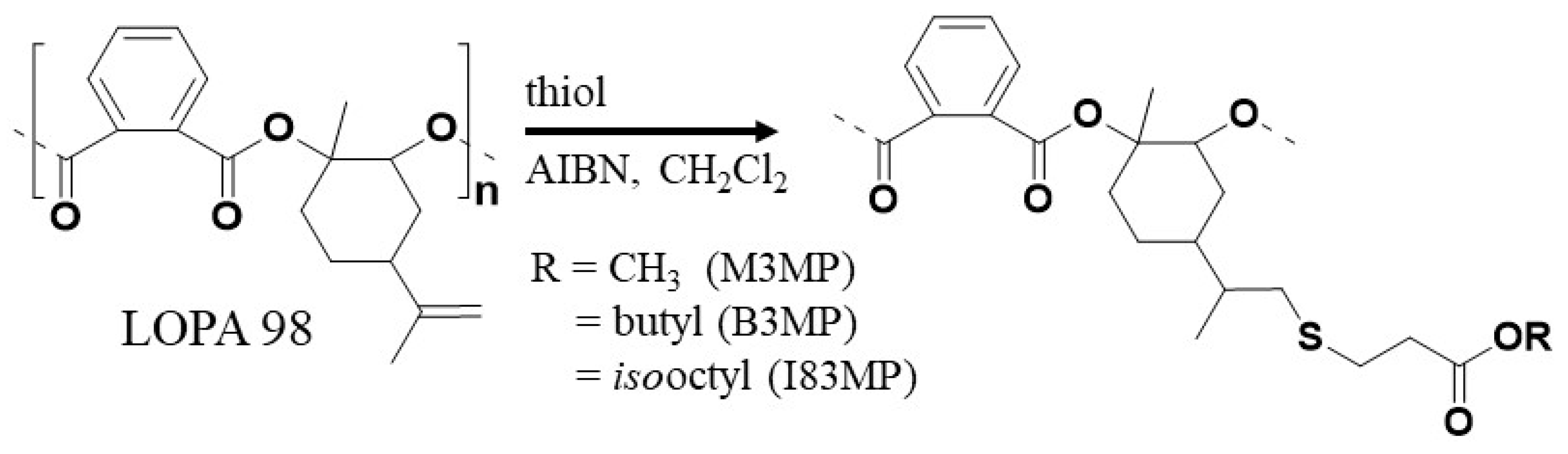
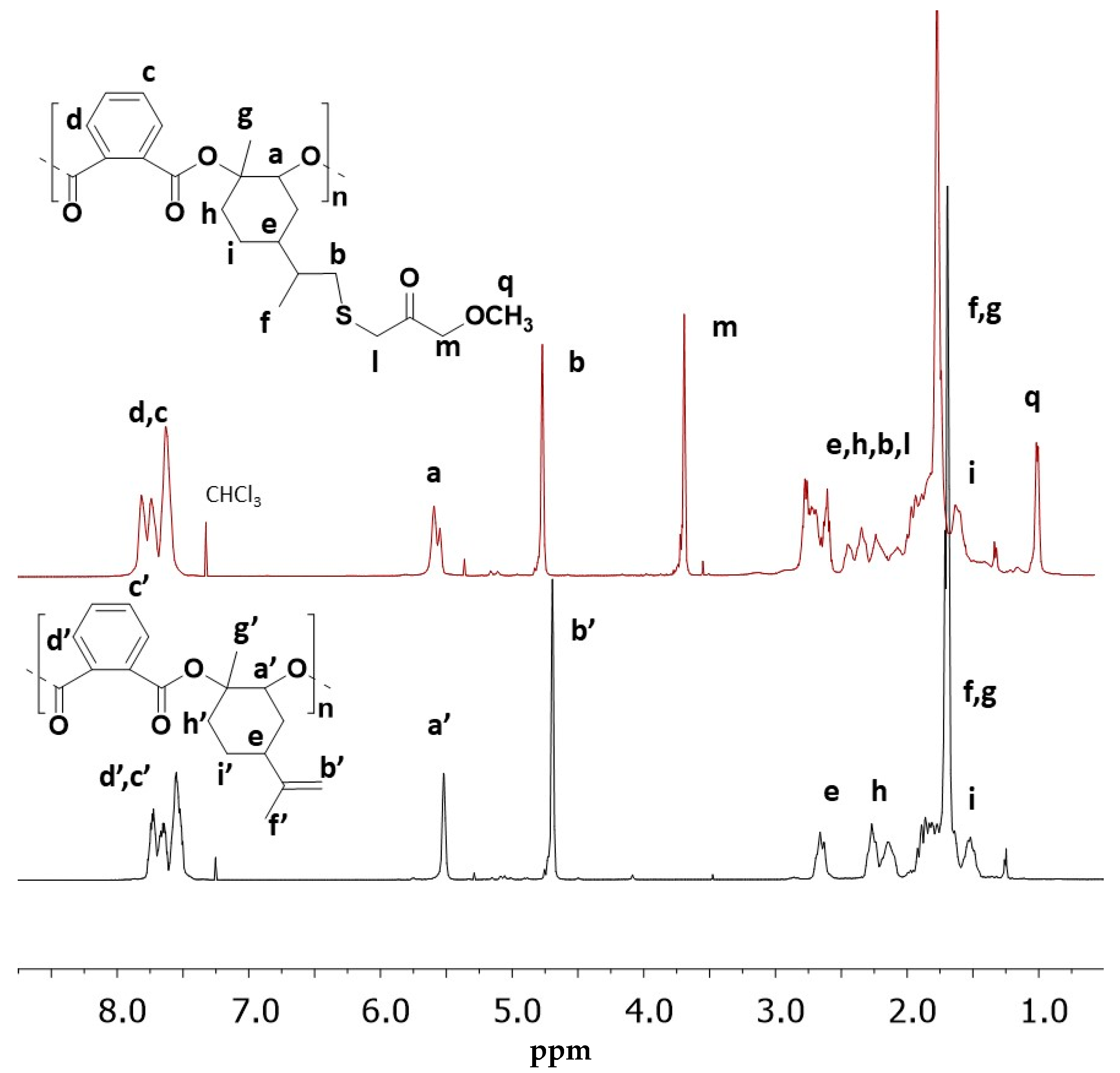
2.3. Preparation of LOPASH through Functionalization of Poly(LO-alt-PA) with Thiols
2.4. Methods
3. Results and Discussion
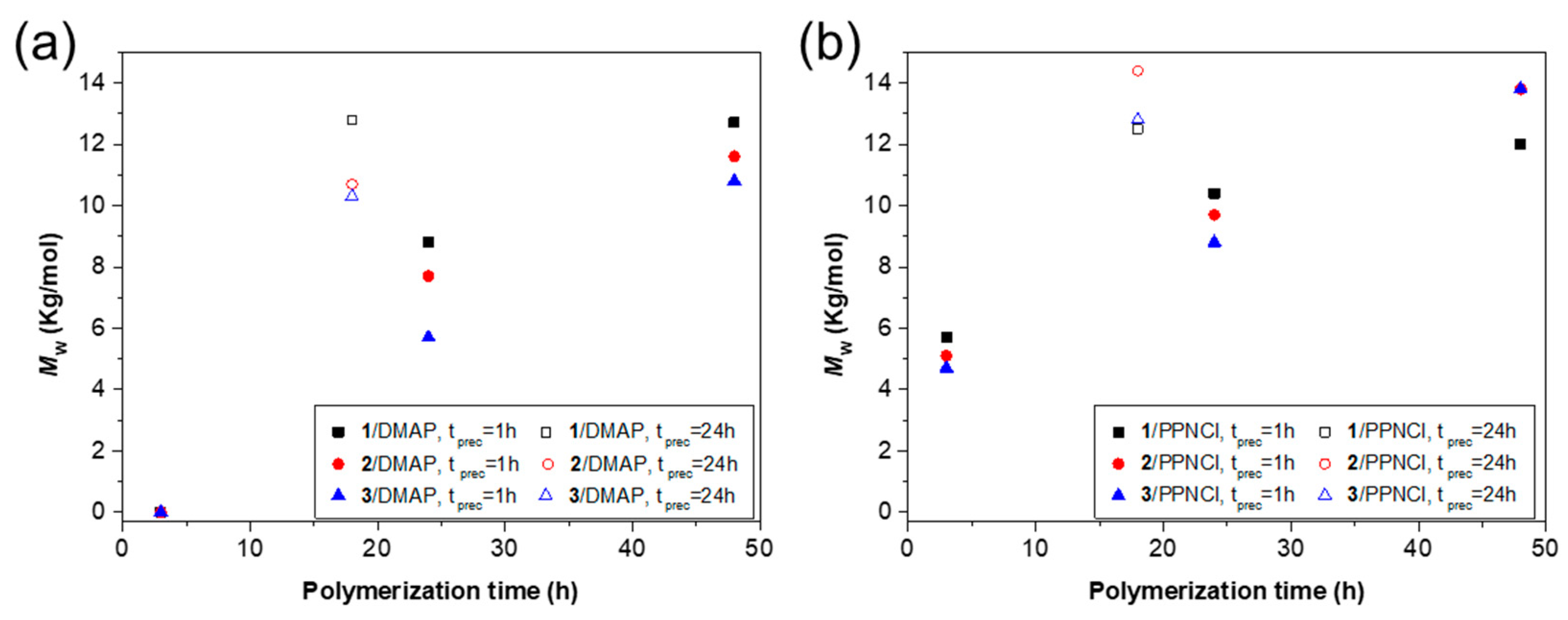
3.1. ROCOP of Limonene Oxide and Phthalic Anhydride
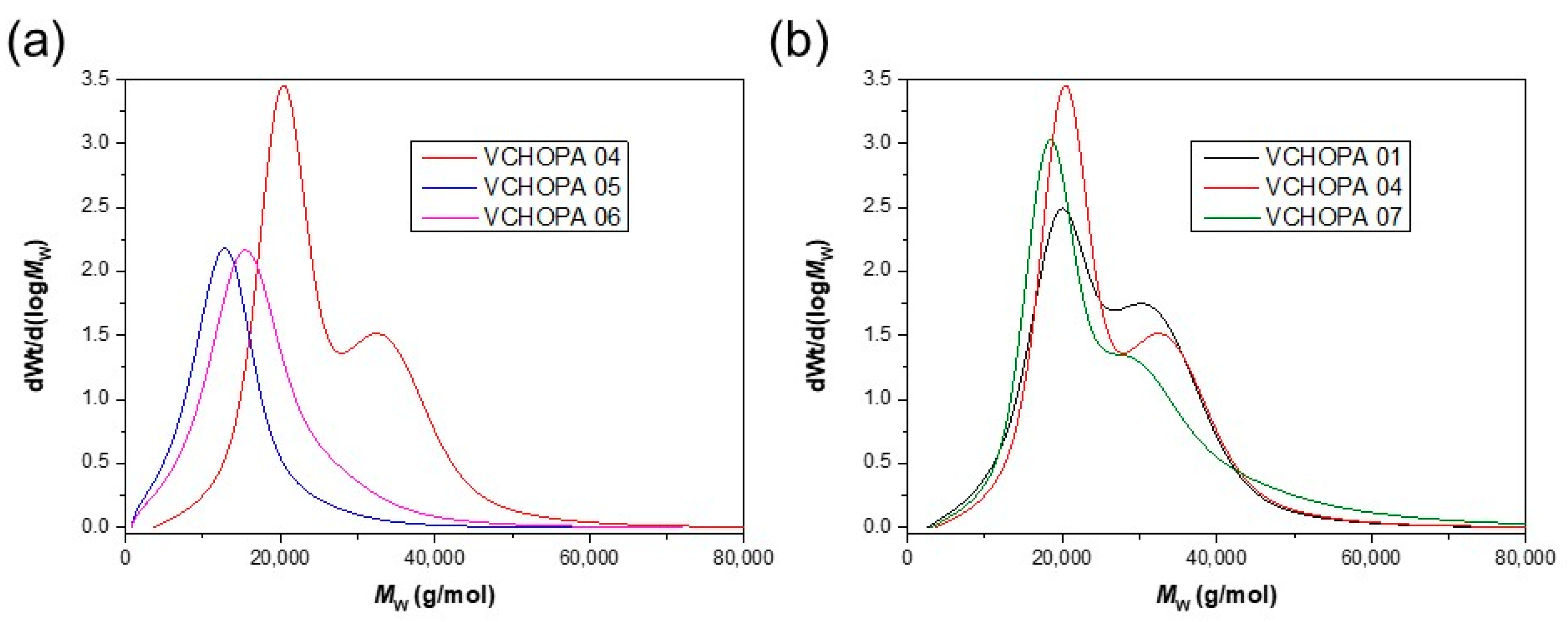
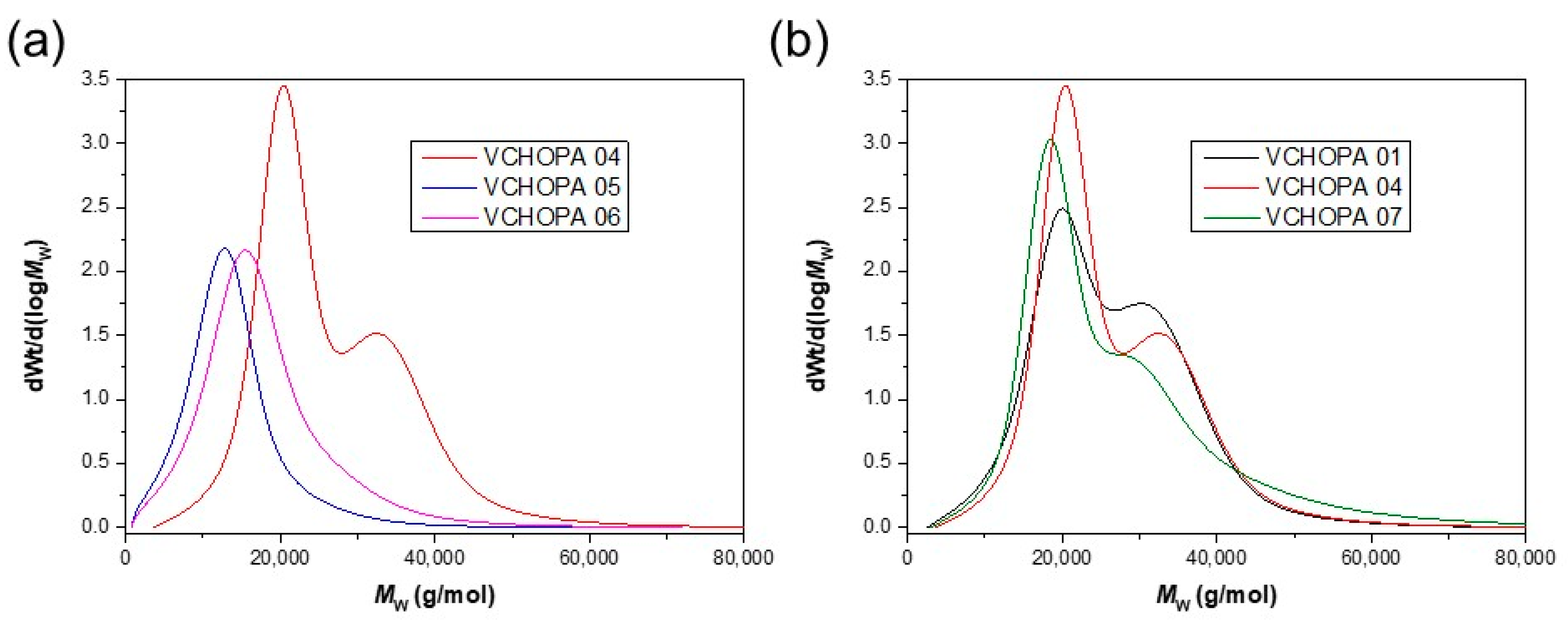
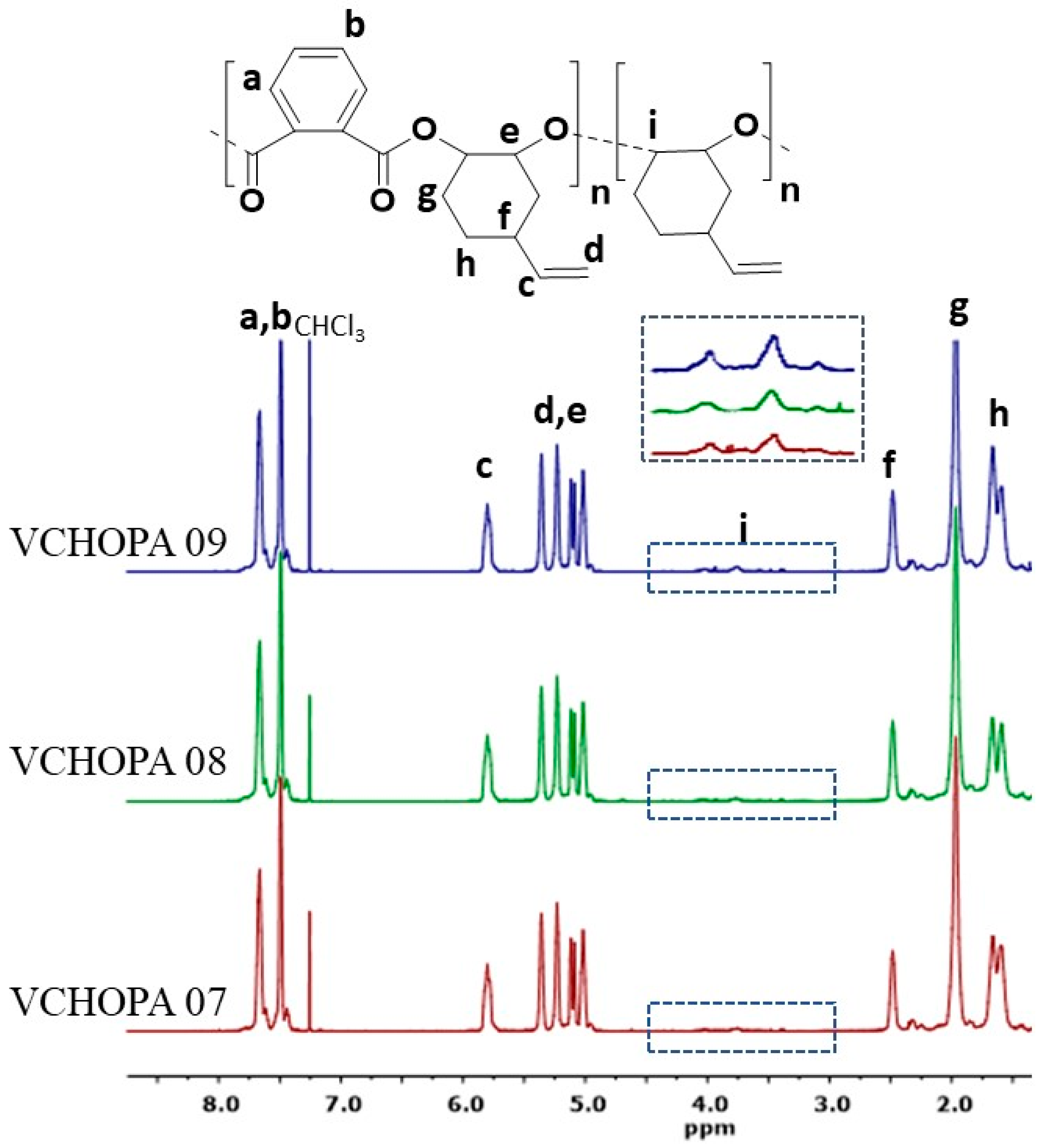
3.2. ROCOP of Vinylcyclohexene Oxide and Phthalic Anhydride
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, C.K.; Hillmyer, M.A. Polymers from Renewable Resources: A Perspective for a Special Issue of Polymer Reviews. Polym. Rev. 2008, 48, 1–10. [Google Scholar] [CrossRef]
- Ishimoto, K.; Arimoto, M.; Okuda, T.; Yamaguchi, S.; Aso, Y.; Ohara, H.; Kobayashi, S.; Ishii, M.; Morita, K.; Yamashita, H.; et al. Biobased Polymers: Synthesis of Graft Copolymers and Comb Polymers Using Lactic Acid Macromonomer and Properties of the Product Polymers. Biomacromolecules 2012, 13, 3757–3768. [Google Scholar] [CrossRef] [PubMed]
- Diment, W.T.; Rosetto, G.; Ezaz-Nikpay, N.; Kerrand, R.W.F.; Williams, C.K. A highly active, thermally robust iron(iii)/potassium(i) heterodinuclear catalyst for bio-derived epoxide/anhydride ring-opening copolymerizations. Green Chem. 2023, 25, 2262–2267. [Google Scholar] [CrossRef]
- Cywar, R.M.; Rorrer, N.A.; Hoyt, C.B.; Beckham, G.T.; Chen, E.Y.X. Bio-based polymers with performance-advantaged properties. Nat. Rev. Mater. 2022, 7, 83–103. [Google Scholar] [CrossRef]
- Payne, J.; Jones, M.D. The Chemical Recycling of Polyesters for a Circular Plastics Economy: Challenges and Emerging Opportunities. ChemSusChem 2021, 14, 4041–4070. [Google Scholar] [CrossRef] [PubMed]
- Hillmyer, M.A. The promise of plastics from plants. Science 2017, 358, 868–870. [Google Scholar] [CrossRef]
- Winnacker, M.; Rieger, B. Recent Progress in Sustainable Polymers Obtained from Cyclic Terpenes: Synthesis, Properties, and Application Potential. ChemSusChem 2015, 8, 2455–2471. [Google Scholar] [CrossRef]
- Chen, T.T.D.; Carrodeguas, L.P.; Sulley, G.S.; Gregory, G.L.; Williams, C.K. Bio-based and Degradable Block Polyester Pressure-Sensitive Adhesives. Angew. Chem. Int. Ed. 2020, 59, 23450–23455. [Google Scholar] [CrossRef]
- Della Monica, F.; Kleij, A.W. From terpenes to sustainable and functional polymers. Polym. Chem. 2020, 11, 5109–5127. [Google Scholar] [CrossRef]
- Brandolese, A.; Kleij, A.W. Catalyst Engineering Empowers the Creation of Biomass-Derived Polyesters and Polycarbonates. Acc. Chem. Res. 2022, 55, 1634–1645. [Google Scholar] [CrossRef]
- Longo, J.M.; Sanford, M.J.; Coates, G.W. Ring-opening copolymerization of epoxides and cyclic anhydrides with discrete metal complexes: Structure-properties relationships. Chem. Rev. 2016, 116, 15167–15197. [Google Scholar] [CrossRef]
- Quilter, H.C.; Hutchby, M.; Davidson, M.G.; Jones, M.D. Polymerisation of a terpene-derived lactone: A bio-based alternative to ε-caprolactone. Polym. Chem. 2017, 8, 833–837. [Google Scholar] [CrossRef] [Green Version]
- Martín, C.; Kleij, A.W. Terpolymers Derived from Limonene Oxide and Carbon Dioxide: Access to Cross-Linked Polycarbonates with Improved Thermal Properties. Macromolecules 2016, 49, 6285–6295. [Google Scholar] [CrossRef]
- Peña Carrodeguas, L.; González-Fabra, J.; Castro-Gómez, F.; Bo, C.; Kleij, A.W. AlIII-Catalysed Formation of Poly(limonene)carbonate: DFT Analysis of the Origin of Stereoregularity. Chem. Eur. J. 2015, 21, 6115–6122. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Qiao, L.; Qin, Y.; Zhao, X.; Chen, X.; Wang, X.; Wang, F. Synthesis and Stabilization of Novel Aliphatic Polycarbonate from Renewable Resource. Macromolecules 2009, 42, 9251–9254. [Google Scholar] [CrossRef]
- Xin, Y.; Uyama, H.J.J. Synthesis of new bio-based polycarbonates derived from terpene. J. Polym. Res. 2012, 19, 15. [Google Scholar] [CrossRef]
- Louisy, E.; Khodyrieva, V.; Olivero, S.; Michelet, V.; Mija, A. Use of Limonene Epoxides and Derivatives as Promising Monomers for Biobased Polymers. ChemPlusChem 2022, 87, e202200190. [Google Scholar] [CrossRef] [PubMed]
- Ciriminna, R.; Lomeli-Rodriguez, M.; Demma Carà, P.; Lopez-Sanchez, J.A.; Pagliaro, M. Limonene: A versatile chemical of the bioeconomy. Chem. Commun. 2014, 50, 15288–15296. [Google Scholar] [CrossRef] [PubMed]
- Ajellal, N.; Carpentier, J.F.; Guillaume, C.; Guillaume, S.M.; Helou, M.; Poirier, V.; Sarazin, Y.; Trifonov, A. Metal-catalyzed immortal ring-opening polymerization of lactones, lactides and cyclic carbonates. Dalton Trans. 2010, 39, 8363–8376. [Google Scholar] [CrossRef]
- Dijkstra, P.J.; Du, H.; Feijen, J. Single site catalysts for stereoselective ring-opening polymerization of lactides. Polym. Chem. 2011, 2, 520–527. [Google Scholar] [CrossRef]
- Zhang, X.; Fevre, M.; Gavin, O.J.; Waymouth, R.M. Catalysis as an Enabling Science for Sustainable Polymers. Chem. Rev. 2018, 118, 839–885. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Romain, C.; Meier, M.A.R.; Williams, C.K. Renewable Polycarbonates and Polyesters from 1,4-Cyclohexadiene. Green Chem. 2015, 17, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Proverbio, M.; Galotto Galotto, N.; Losio, S.; Tritto, I.; Boggioni, L. Influence of Co-catalysts and Polymerization Conditions on Properties of Poly(anhydride-alt-epoxide)s from ROCOP using Salen Complexes with Different Metals. Polymers 2019, 11, 1222. [Google Scholar] [CrossRef] [Green Version]
- Jeske, R.C.; Di Ciccio, A.M.; Coates, G.W. Alternating Copolymerization of Epoxides and Cyclic Anhydrides: An Improved Route to Aliphatic Polyesters. J. Am. Chem. Soc. 2007, 129, 11330–11331. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; de Montigny, F.; Thomas, C.M. Tandem synthesis of alternating polyesters from renewable resources. Nat. Commun. 2011, 2, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini Nejad, E.; Paoniasari, A.; van Melis, C.G.W.; Koning, C.E.; Duchateau, R. Catalytic Ring-Opening Copolymerization of Limonene Oxide and Phthalic Anhydride: Toward Partially Renewable Polyesters. Macromolecules 2013, 46, 631–637. [Google Scholar] [CrossRef]
- Peña Carrodeguas, L.; Martin, C.; Kleij, A.W. Semiaromatic polyesters derived from renewable terpene oxides with high glass transitions. Macromolecules 2017, 50, 5337–5345. [Google Scholar] [CrossRef]
- Isnard, F.; Lamberti, M.; Pellecchia, C.; Mazzeo, M. Ring-Opening Copolymerization of Epoxides with Cyclic Anhydrides Promoted by Bimetallic and Monometallic Phenoxy–Imine Aluminum complexes. ChemCatChem 2017, 9, 2972–2979. [Google Scholar] [CrossRef]
- Santulli, F.; D’Auria, I.; Boggioni, L.; Losio, S.; Proverbio, M.; Costabile, C.; Mazzeo, M. Bimetallic Aluminum Complexes Bearing Binaphthyl-Based Iminophenolate Ligands as Catalysts for the Synthesis of Polyesters. Organometallics 2020, 39, 1213–1220. [Google Scholar] [CrossRef]
- Bukowski, W.; Bukowska, A.; Sobota, A.; Pytel, M.; Bester, K. Copolymerization of Phthalic Anhydride with Epoxides Catalyzed by Amine-Bis(Phenolate) Chromium(III) Complexes. Polymers 2021, 13, 1785. [Google Scholar] [CrossRef]
- Si, G.; Zhang, L.; Han, B.; Duan, Z.; Li, B.; Dong, J.; Li, X.; Liu, B. Novel Chromium Complexes with a [OSSO]-Type Bis(Phenolato) Dianionic Ligand Mediate the Alternating Ring-Opening Copolymerization of Epoxides and Phthalic Anhydride. Polym. Chem. 2015, 6, 6372–6377. [Google Scholar] [CrossRef]
- Shaw, M.S.; Bates, M.R.; Jones, M.D.; Ward, B.D. Metallocene catalysts for the ring-opening copolymerisation of epoxides and cyclic anhydrides. Polym. Chem. 2022, 13, 3315–3324. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Q.; Li, L.; Urban, M.W. Recent Advances in Stimuli-Responsive Commodity Polymers. Macromol. Rapid Commun. 2021, 42, 2100054. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Poland, R.R.; Escobedo, C. Kinetic Studies of the Alternating Copolymerization of Cyclic Acid Anhydrides and Epoxides, and the Terpolymerization of Cyclic Acid Anhydrides, Epoxides, and CO2 Catalyzed by (salen)CrIIICl. Macromolecules 2012, 45, 2242–2248. [Google Scholar] [CrossRef]
- Hauenstein, O.; Agarwal, S.; Greiner, A. Bio-based polycarbonate as synthetic toolbox. Nat. Commun. 2016, 7, 11862. [Google Scholar] [CrossRef]
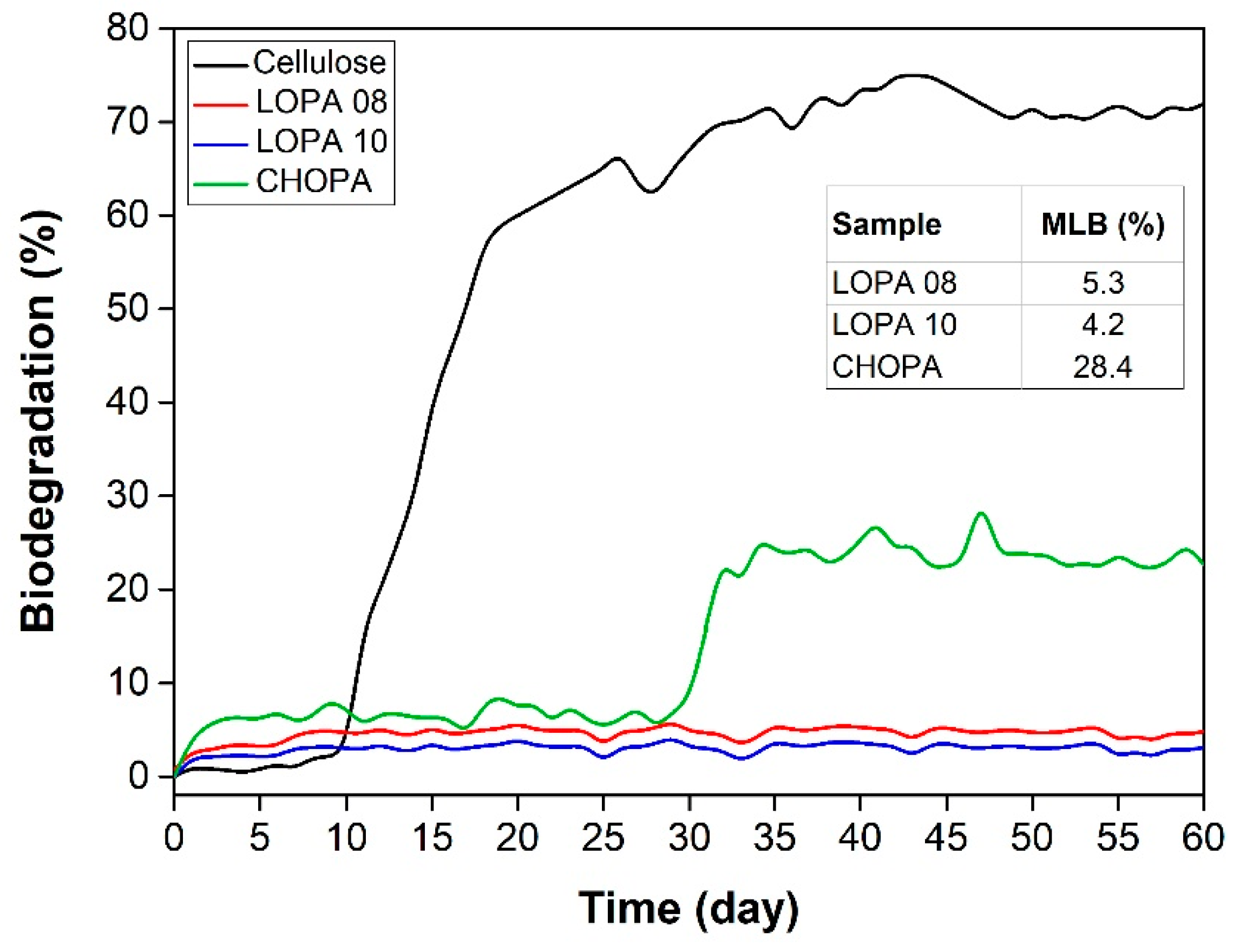
- ISO 14851; Determination of the Ultimate Aerobic Biodegradability of Plastic Materials in An Aqueous Medium—Method by Measuring the Oxygen Demand in A Closed Respirometer. ISO: Geneva, Switzerland, 1999.
- Penn, M.R.; Pauer, J.J.; Mihelcic, J.R. Biochemical Oxygen Demand. In Environmental and Ecological Chemistry; Sabljic, A., Ed.; EOLSS: Oxford, UK, 2009; Volume 2, pp. 278–297. [Google Scholar]
- Hosseini Nejad, E.; van Melis, C.G.W.; Vermeer, T.J.; Koning, C.E.; Duchateau, R. Alternating Ring-Opening Polymerization of Cyclohexene Oxide and Anhydrides: Effect of Catalyst, Cocatalyst, and Anhydride Structure. Macromolecules 2012, 45, 1770–1776. [Google Scholar] [CrossRef]
- Rapisarda, M.; Mistretta, M.C.; Scopelliti, M.; Leanza, M.; La Mantia, F.P.; Rizzarelli, P. Influence of Calcium Carbonate Nanoparticles on the Soil Burial Degradation of Polybutyleneadipate-Co-Butylenetherephthalate Films. Nanomaterials 2022, 12, 2275. [Google Scholar] [CrossRef]
- Bester, K.; Bukowska, A.; Myśliwiec, B.; Hus Tomczyk, D.; Urbaniak, P.; Bukowski, W. Alternating ring-opening copolymerization of phthalic anhydride with epoxydes catalysed by salophen chromium(III) complexes. An effect of substitution in salophen ligands. Polym. Chem. 2018, 9, 2147–2156. [Google Scholar] [CrossRef]
- Melchiors, M.S.; Vieira, T.Y.; Pereira, L.P.S.; Feuser, P.E.; Ferrão, V.; Machado, F.; Carciofi, B.A.M.; de Araujo, P.H.H.; de Oliveira, D.; Sayer, C. Copolymerization of limonene oxide and cyclic anhydrides catalyzed by ionic liquid BMI·Fe2Cl7, nanoparticles preparation, crosslinking, and cytoxicity studies. J. Pol. Res. 2022, 29, 336–348. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst/Cocatalyst | Precontact Time (h) | Polymerization Time (h) | Yield (%) | Mw (kg/mol) | Đ | Tg (° C) |
|---|---|---|---|---|---|---|---|
| LOPA 07 | 1/DMAP | 1 | 3 | / | / | / | / |
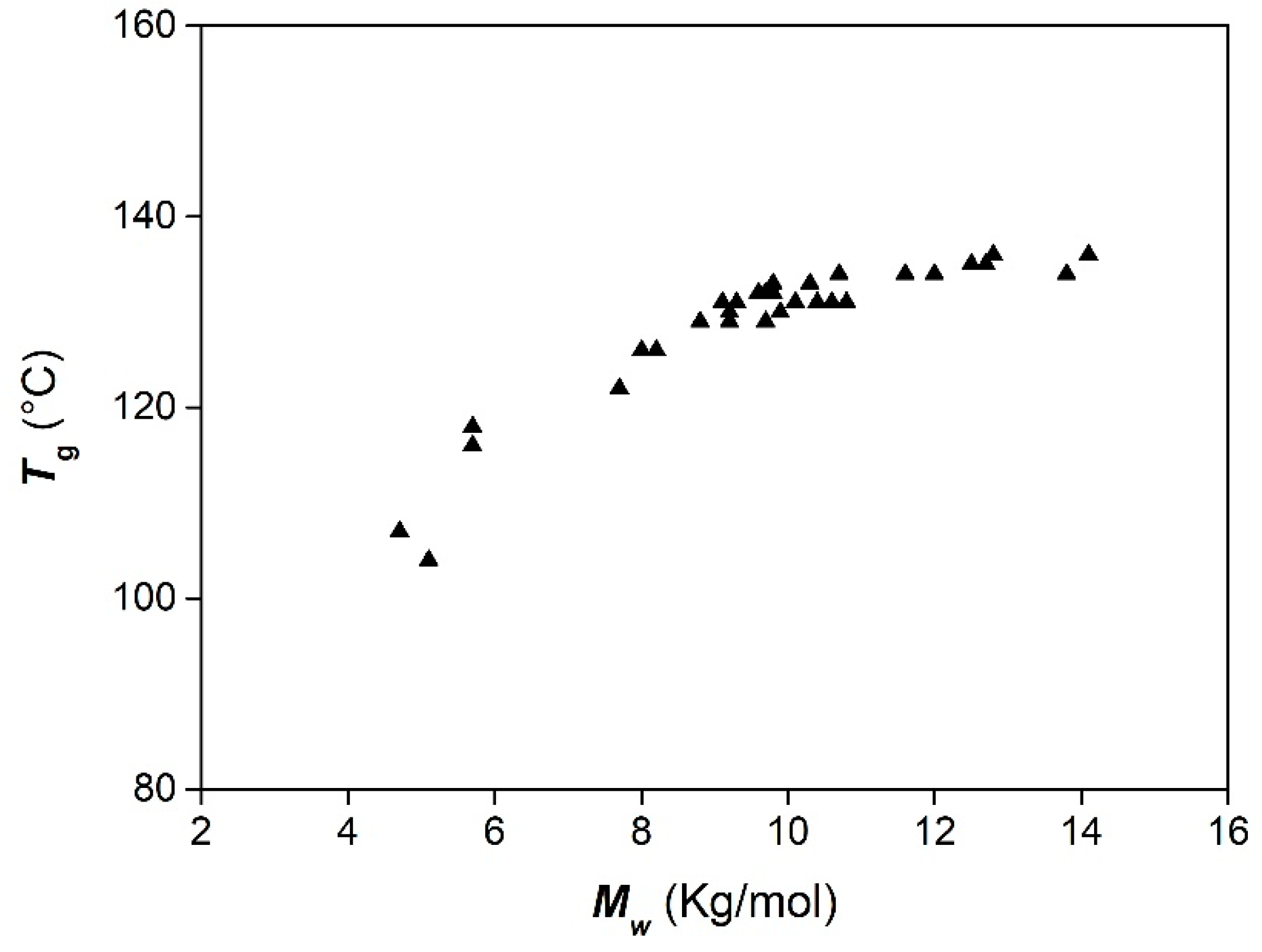
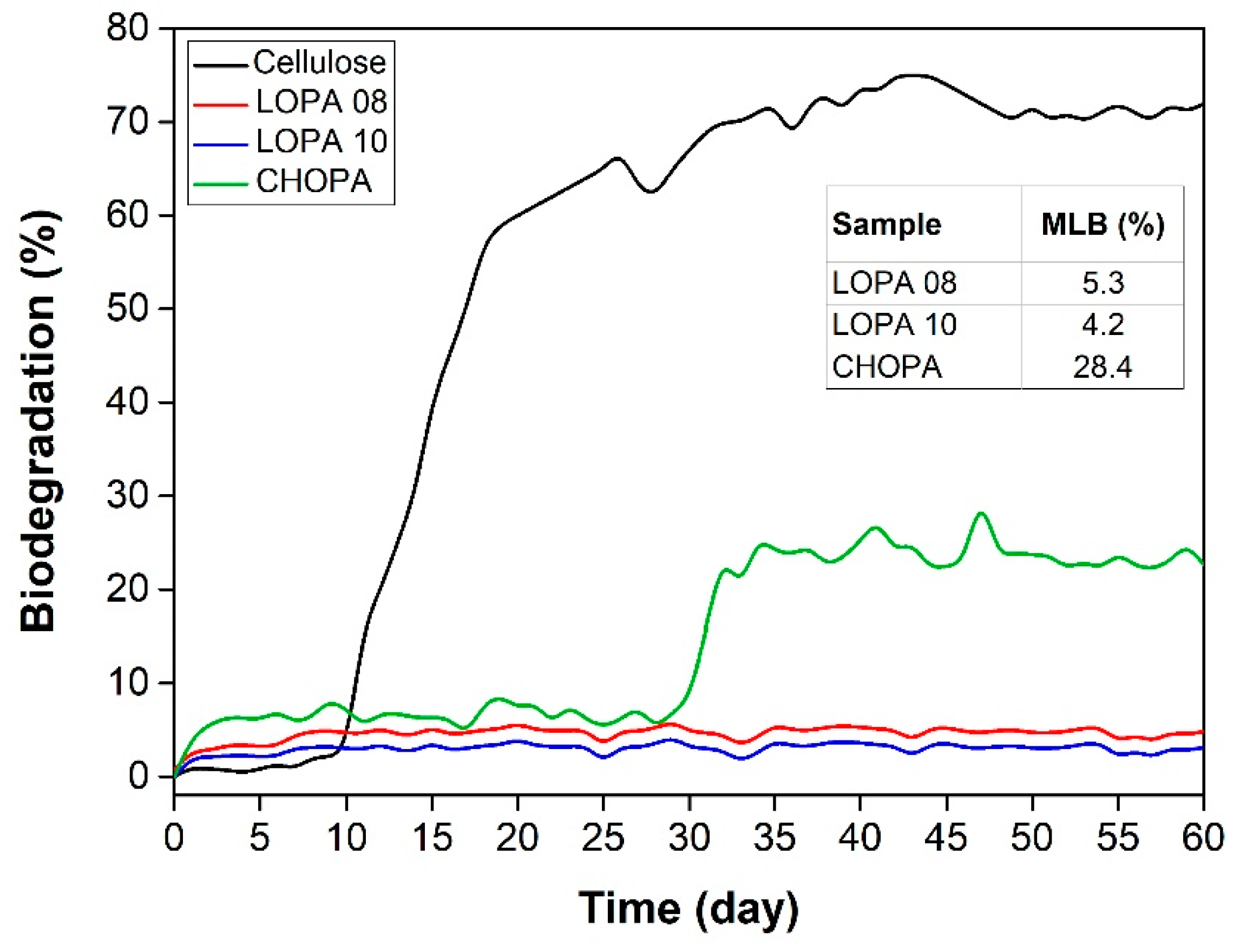
| LOPA 08 | 1/DMAP | 1 | 24 | 73 | 8.8 | 1.2 | 129 |
| LOPA 67 | 1/DMAP | 1 | 48 | 94 | 12.7 | 1.3 | 135 |
| LOPA 74 | 1/DMAP | 24 | 18 | 91 | 12.8 | 1.2 | 136 |
| LOPA 11 | 2/DMAP | 1 | 3 | / | / | / | / |
| LOPA 12 | 2/DMAP | 1 | 24 | 60 | 7.7 | 1.2 | 122 |
| LOPA 69 | 2/DMAP | 1 | 48 | 87 | 11.6 | 1.4 | 134 |
| LOPA 76 | 2/DMAP | 24 | 18 | 82 | 10.7 | 1.3 | 134 |
| LOPA 15 | 3/DMAP | 1 | 3 | 0.2 | / | / | / |
| LOPA 16 | 3/DMAP | 1 | 24 | 48 | 5.7 | 1.2 | 116 |
| LOPA 68 | 3/DMAP | 1 | 48 | 84 | 10.8 | 1.5 | 131 |
| LOPA 75 | 3/DMAP | 24 | 18 | 72 | 10.3 | 1.3 | 133 |
| LOPA 05 | 1/PPNCl | 1 | 3 | 20 | 5.7 | 1.2 | 118 |
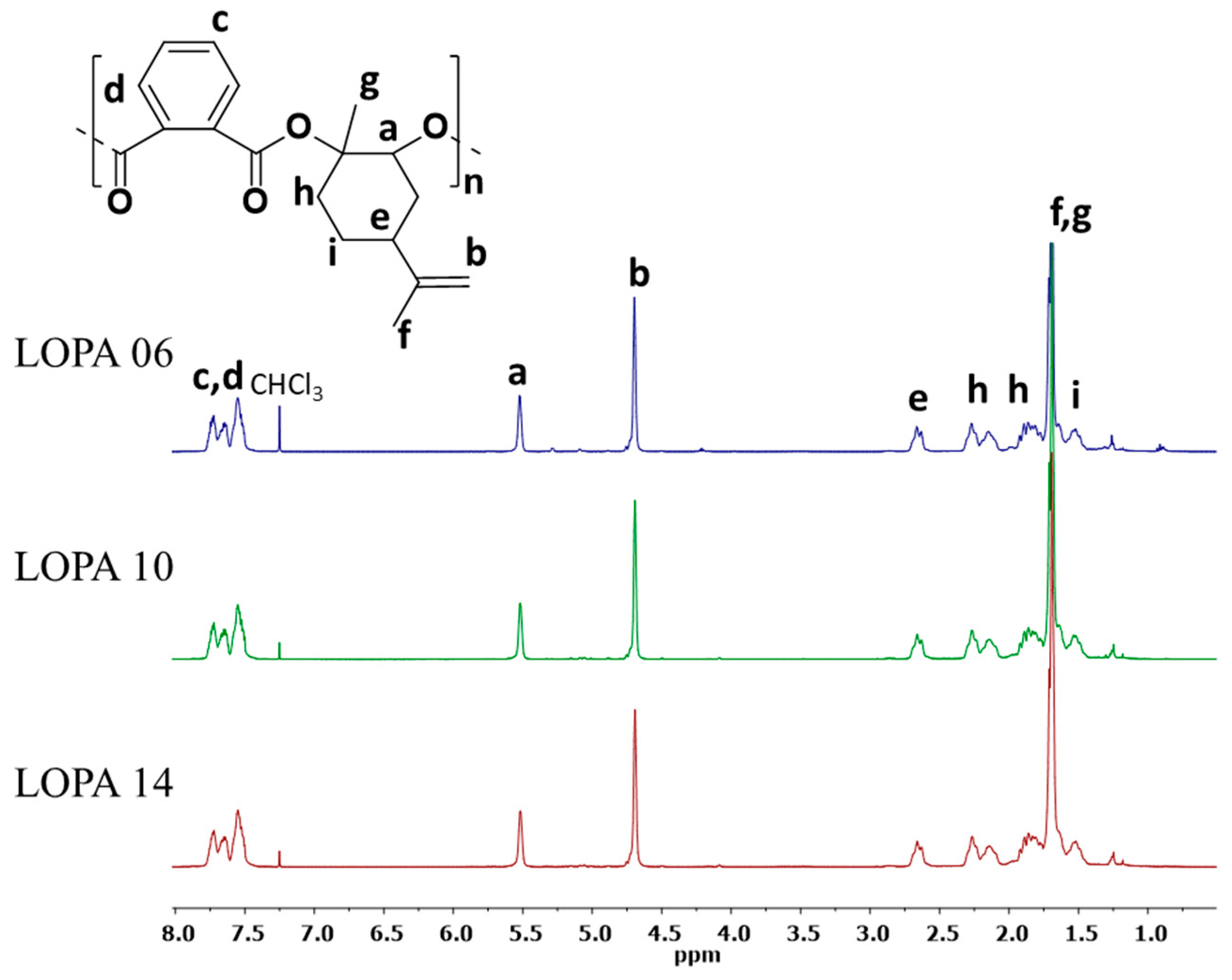
| LOPA 06 | 1/PPNCl | 1 | 24 | 71 | 10.4 | 1.2 | 131 |
| LOPA 70 | 1/PPNCl | 1 | 48 | 91 | 12.0 | 1.3 | 134 |
| LOPA 77 | 1/PPNCl | 24 | 18 | 92 | 12.5 | 1.2 | 135 |
| LOPA 09 | 2/PPNCl | 1 | 3 | 6 | 5.1 | 1.1 | 104 |
| LOPA 10 | 2/PPNCl | 1 | 24 | 83 | 9.7 | 1.2 | 129 |
| LOPA 72 | 2/PPNCl | 1 | 48 | 92 | 13.8 | 1.4 | 134 |
| LOPA 79 | 2/PPNCl | 24 | 18 | 86 | 14.1 | 1.3 | 136 |
| LOPA 13 | 3/PPNCl | 1 | 3 | 5 | 4.7 | 1.1 | 106 |
| LOPA 14 | 3/PPNCl | 1 | 24 | 80 | 8.2 | 1.2 | 126 |
| LOPA 71 | 3/PPNCl | 1 | 48 | 93 | 13.8 | 1.4 | 134 |
| LOPA 78 | 3/PPNCl | 24 | 18 | 91 | 12.8 | 1.3 | 136 |
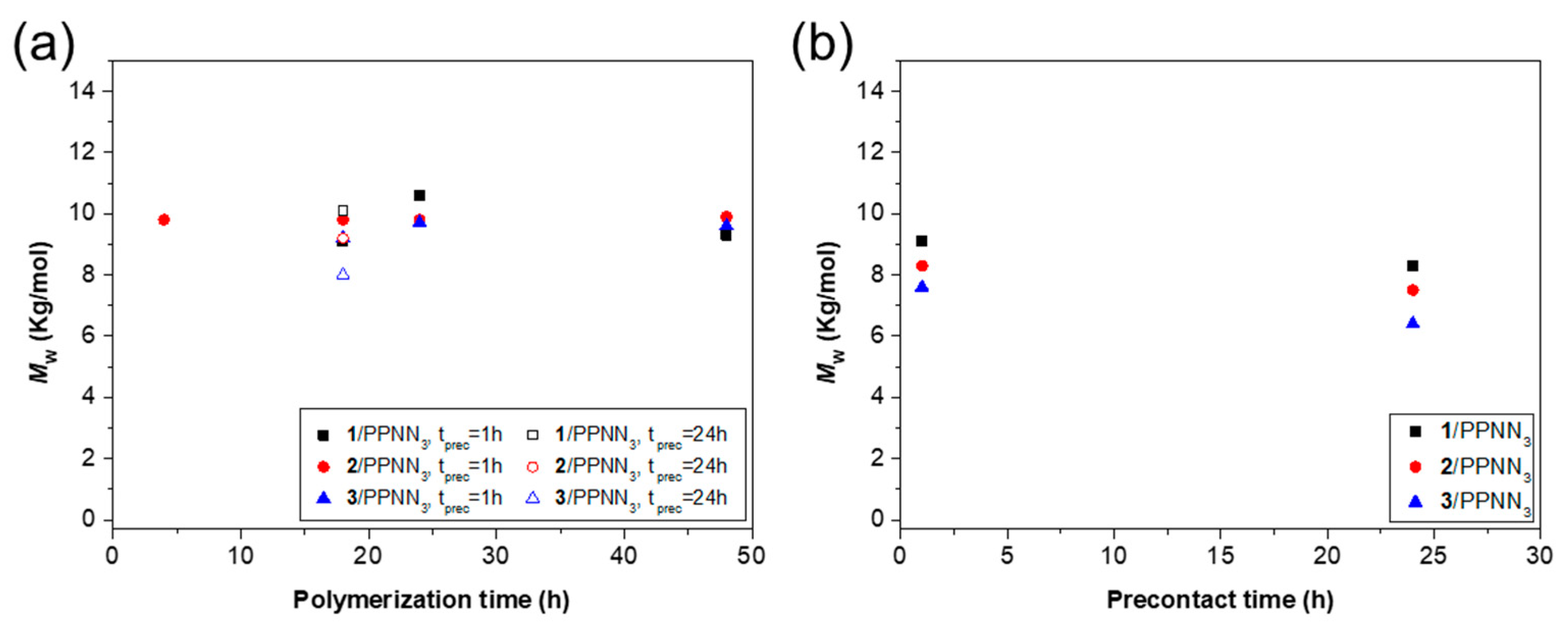
| LOPA 93 | 1/PPNN3 | 1 | 18 | 85 | 9.1 | 1.2 | 131 |
| LOPA 96 | 1/PPNN3 | 1 | 24 | 87 | 10.6 | 1.2 | 131 |
| LOPA 99 | 1/PPNN3 | 1 | 48 | 89 | 9.3 | 1.2 | 131 |
| LOPA 102 | 1/PPNN3 | 24 | 18 | 83 | 10.1 | 1.2 | 131 |
| LOPA 33 | 2/PPNN3 | 1 | 4 | 64 | 9.8 | 1.2 | 130 |
| LOPA 95 | 2/PPNN3 | 1 | 18 | 87 | 9.8 | 1.1 | 133 |
| LOPA 98 | 2/PPNN3 | 1 | 24 | 87 | 9.8 | 1.2 | 132 |
| LOPA 101 | 2/PPNN3 | 1 | 48 | 78 | 9.9 | 1.2 | 130 |
| LOPA 104 | 2/PPNN3 | 24 | 18 | 80 | 9.2 | 1.2 | 129 |
| LOPA 94 | 3/PPNN3 | 1 | 18 | 89 | 9.2 | 1.2 | 130 |
| LOPA 97 | 3/PPNN3 | 1 | 24 | 88 | 9.7 | 1.2 | 132 |
| LOPA 100 | 3/PPNN3 | 1 | 48 | 86 | 9.6 | 1.2 | 132 |
| LOPA 103 | 3/PPNN3 | 24 | 18 | 67 | 8.0 | 1.3 | 126 |
| Entry | Thiol | %Functionalization % b | Yield % | Mn (kg/mol) | Tg (°C) | T5% (°C) | Tmax (°C) |
|---|---|---|---|---|---|---|---|
| LOPA 98 c | - | - | - | 8.2 | 132 | 253 | 271 |
| LOPASH 04 | M3MP | 63 | 75 | 7.8 | 95 | 247 | 265 |
| LOPASH 02 | B3MP | 41 | 68 | 8.3 | 71 | 248 | 268 |
| LOPASH 03 | I83MP | 17 | 58 | 8.1 | 86 | 249 | 270 |
| Entry | Catalyst/ C\Cocatalyst | Yield (%) | Mw (kg/mol) | Đ | Tg (° C) | T5% (° C) | Tmax (° C) |
|---|---|---|---|---|---|---|---|
| VCHOPA 01 | 1/DMAP | 92 | 22.5 | 1.2 | 130 | 323 | 368 |
| VCHOPA 04 | 1/PPNCl | 92 | 23.1 | 1.2 | 129 | 319 | 369 |
| VCHOPA 07 | 1/PPNN3 | 86 | 22.1 | 1.2 | 130 | 319 | 369 |
| VCHOPA 03 | 2/DMAP | 64 | 7.4 | 1.7 | 122 | 304 | 364 |
| VCHOPA 06 | 2/PPNCl | 82 | 13.8 | 1.6 | 128 | 326 | 368 |
| VCHOPA 09 | 2/PPNN3 | 83 | 13.2 | 1.8 | 126 | 324 | 368 |
| VCHOPA 02 | 3/DMAP | 38 | 6.8 | 1.9 | 121 | 307 | 370 |
| VCHOPA 05 | 3/PPNCl | 69 | 10.8 | 1.6 | 126 | 326 | 368 |
| VCHOPA 08 | 3/PPNN3 | 65 | 10.7 | 1.8 | 127 | 322 | 368 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silvano, S.; Proverbio, M.; Vignali, A.; Bertini, F.; Boggioni, L. High-Glass-Transition Polyesters Produced with Phthalic Anhydride and Epoxides by Ring-Opening Copolymerization (ROCOP). Polymers 2023, 15, 2801. https://doi.org/10.3390/polym15132801
Silvano S, Proverbio M, Vignali A, Bertini F, Boggioni L. High-Glass-Transition Polyesters Produced with Phthalic Anhydride and Epoxides by Ring-Opening Copolymerization (ROCOP). Polymers. 2023; 15(13):2801. https://doi.org/10.3390/polym15132801
Chicago/Turabian StyleSilvano, Selena, Matteo Proverbio, Adriano Vignali, Fabio Bertini, and Laura Boggioni. 2023. "High-Glass-Transition Polyesters Produced with Phthalic Anhydride and Epoxides by Ring-Opening Copolymerization (ROCOP)" Polymers 15, no. 13: 2801. https://doi.org/10.3390/polym15132801
APA StyleSilvano, S., Proverbio, M., Vignali, A., Bertini, F., & Boggioni, L. (2023). High-Glass-Transition Polyesters Produced with Phthalic Anhydride and Epoxides by Ring-Opening Copolymerization (ROCOP). Polymers, 15(13), 2801. https://doi.org/10.3390/polym15132801