
Influence of Thermal Treatment and Acetic Acid Concentration on the Electroactive Properties of Chitosan/PVA-Based Micro- and Nanofibers


Abstract
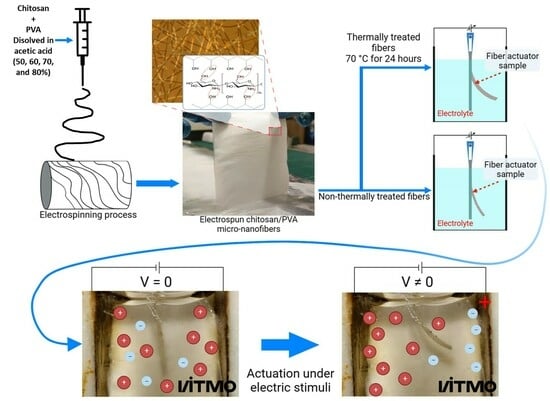
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chitosan and Chitosan/PVA Solutions Preparation
2.3. Rheological Properties and Conductivity of Polymeric Solutions
2.4. Chitosan/PVA Fiber Production by Electrospinning Method
2.5. Thermal Treatment
2.6. Morphology and Diameter Determination of Chitosan/PVA Electrospun Fibers
2.7. Fourier-Transform Infrared Spectroscopy FTIR Analysis
2.8. Thermogravimetric and Differential Scanning Calorimetry Analysis
2.9. Tensile Properties of Electrospun Fibers
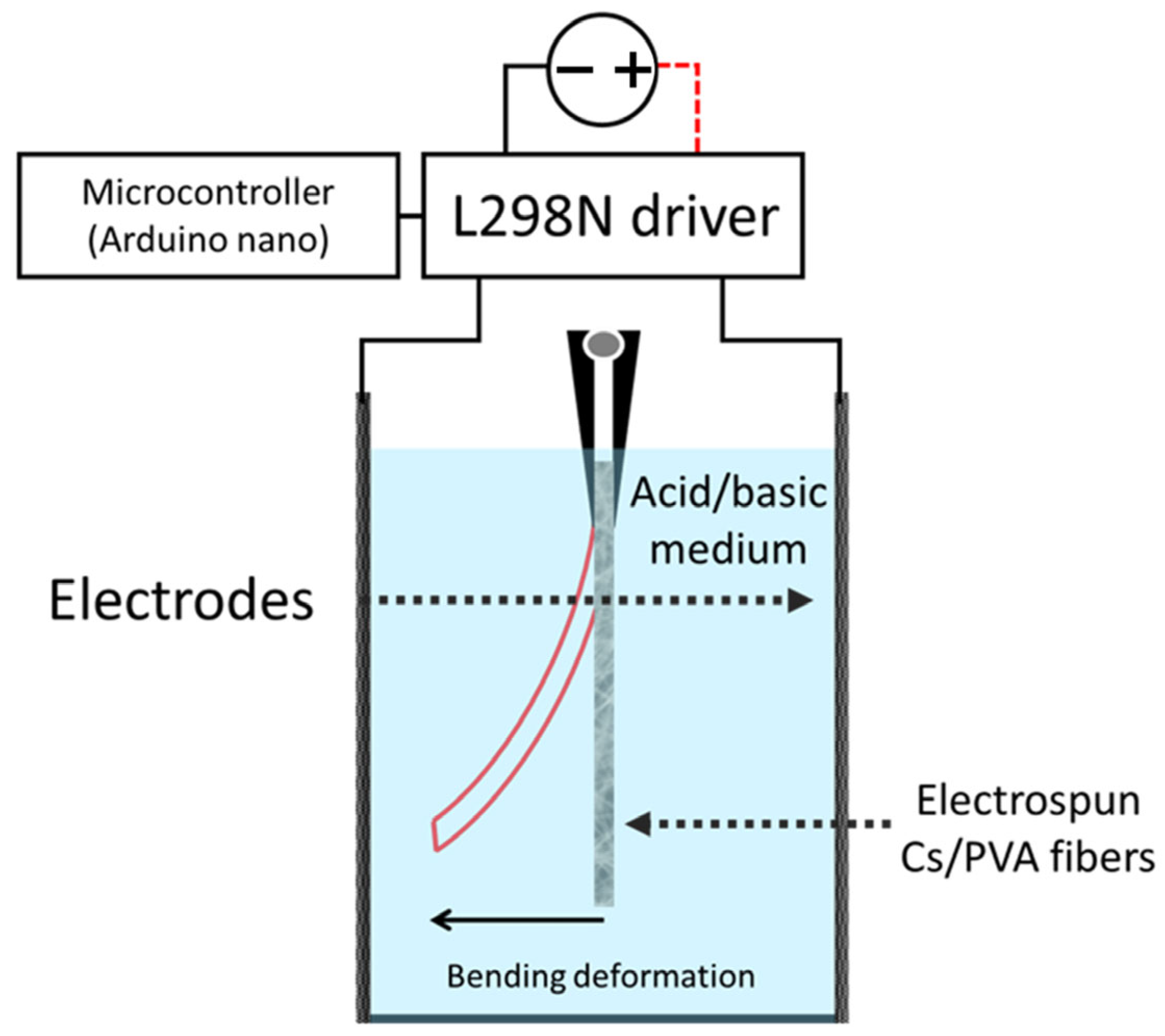
2.10. Electroactive Test Response
3. Results and Discussion
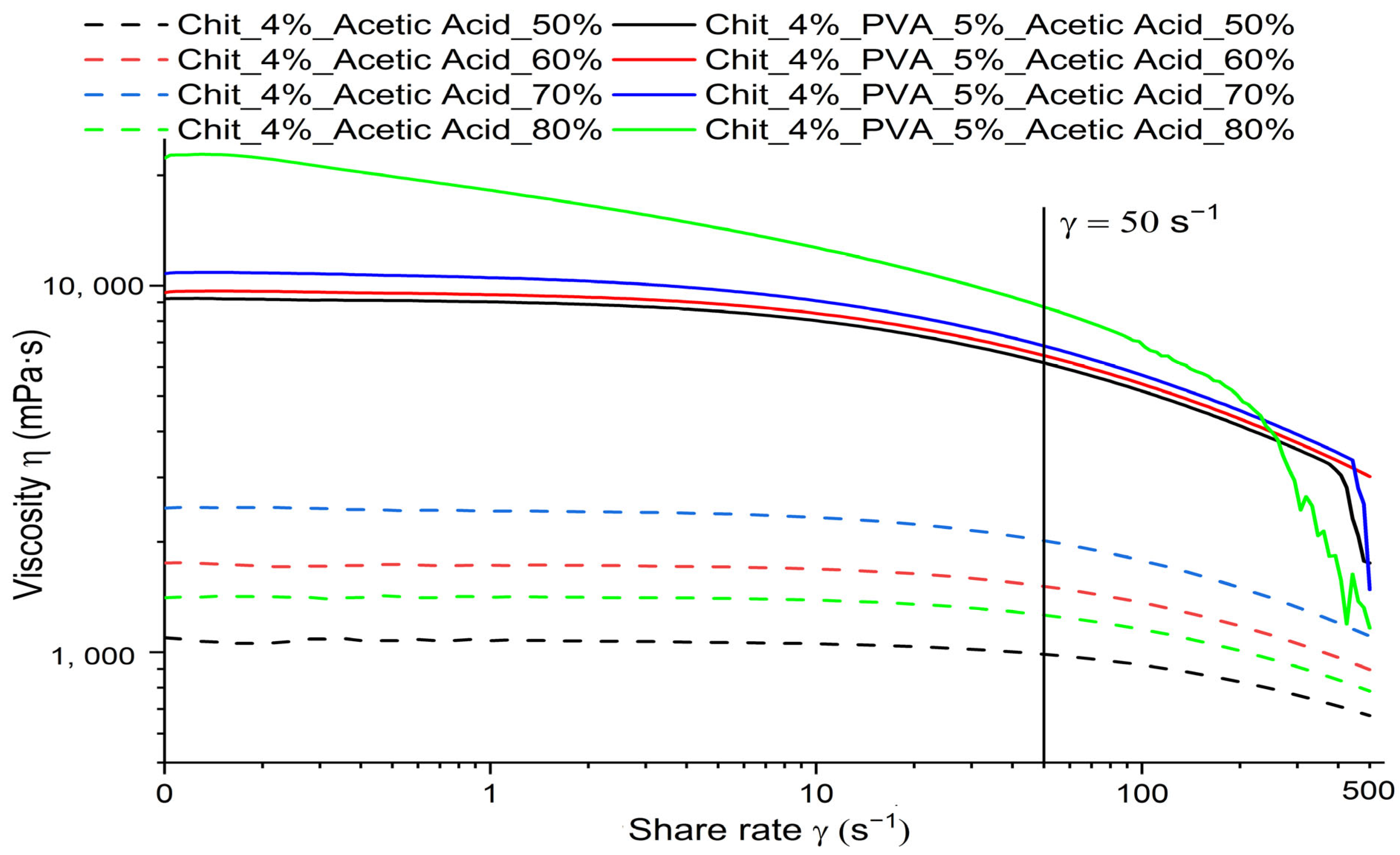
3.1. Rheological Properties and Conductivity of Polymeric Solutions
3.2. Morphology and Diameter Distribution of Chitosan/PVA Electrospun Fibers
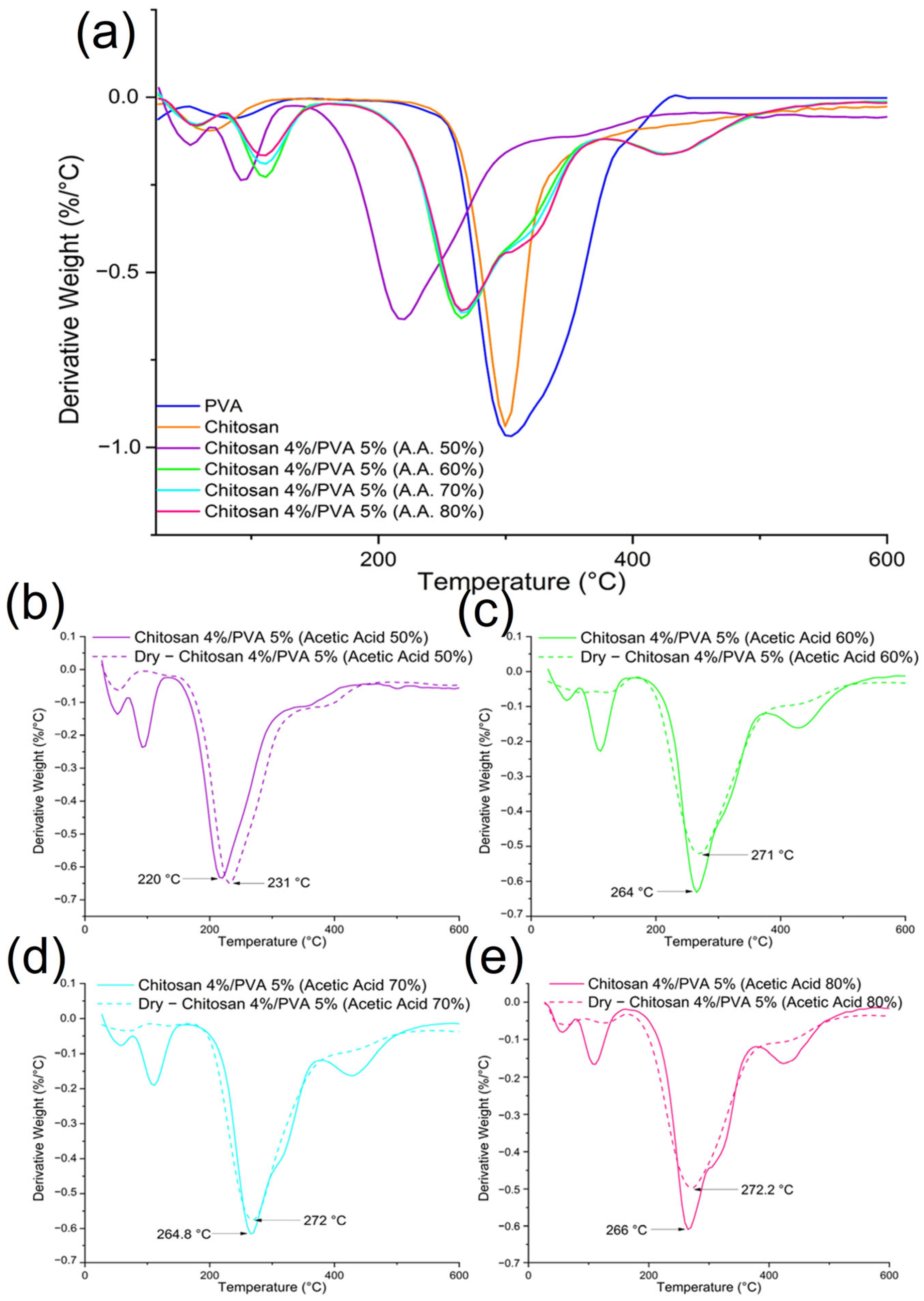
3.3. Thermogravimetric and Differential Scanning Calorimetry Analysis
3.4. Tensile Properties of Electrospun Fibers
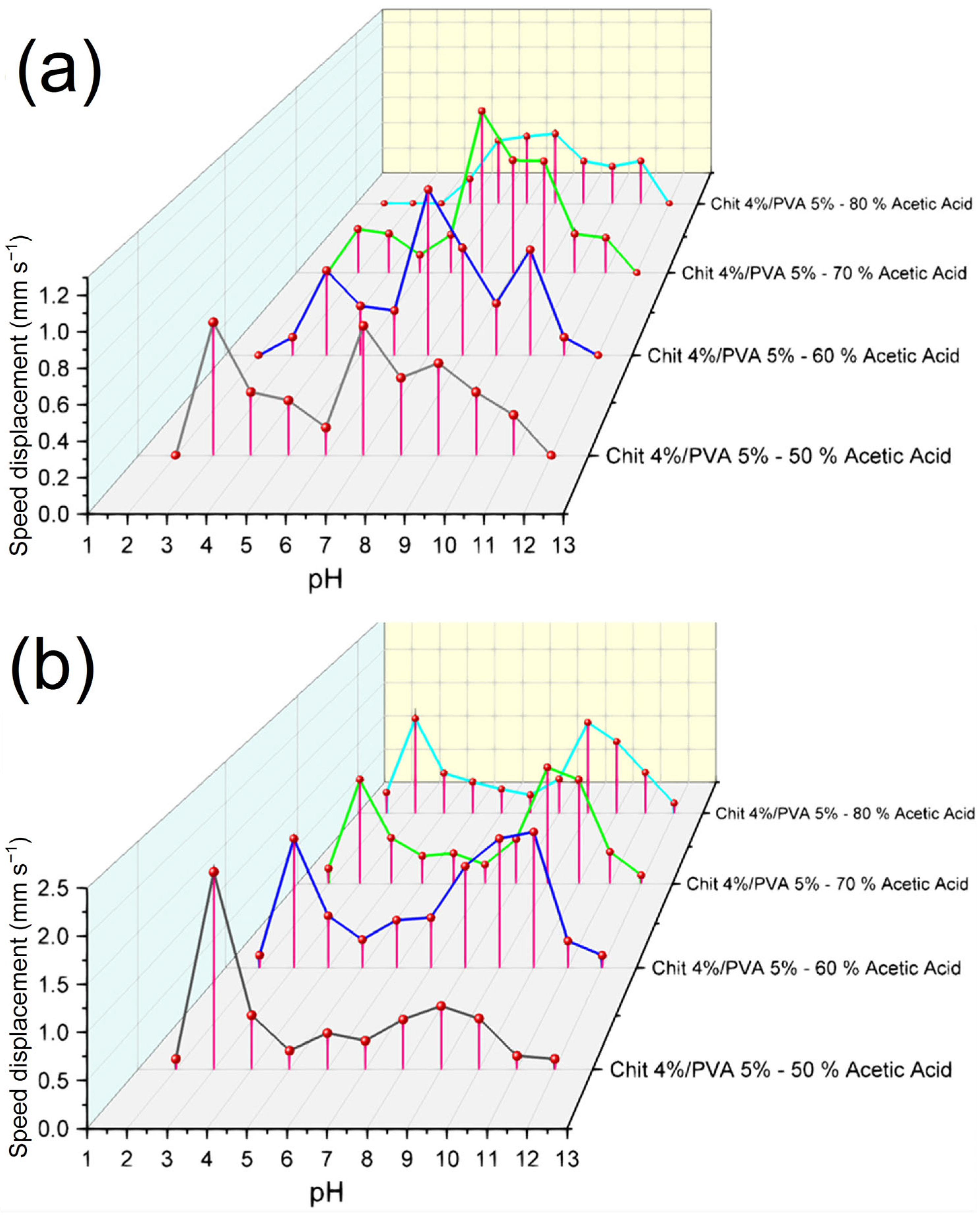
3.5. Electroactive Test Response
3.6. Fourier-Transform Infrared Spectroscopy FTIR Analysis
Spectra Deconvolution for the Determination of Intermolecular Hydrogen Bonding and Free Amine (—NH2) Variation Post-Thermal Treatment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bar-Cohen, Y. Biomimetics: Biologically Inspired Technologies; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Mirvakili, S.M.; Hunter, I.W. Artificial Muscles: Mechanisms, Applications, and Challenges. Adv. Mater. 2018, 30, 1704407. [Google Scholar] [CrossRef]
- Cianchetti, M.; Laschi, C.; Menciassi, A.; Dario, P. Biomedical applications of soft robotics. Nat. Rev. Mater. 2018, 3, 143–153. [Google Scholar] [CrossRef]
- Shintake, J.; Rosset, S.; Schubert, B.; Floreano, D.; Shea, H. Versatile Soft Grippers with Intrinsic Electroadhesion Based on Multifunctional Polymer Actuators. Adv. Mater. 2015, 28, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Thalman, C.; Artemiadis, P. A review of soft wearable robots that provide active assistance: Trends, common actuation methods, fabrication, and applications. Wearable Technol. 2020, 1, E3. [Google Scholar] [CrossRef]
- Tang, X.; Li, H.; Ma, T.; Yang, Y.; Luo, J.; Wang, H.; Jiang, P. A Review of Soft Actuator Motion: Actuation, Design, Manufacturing and Applications. Actuators 2022, 11, 331. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.; Li, Y. Pre-Charged Pneumatic Soft Gripper With Closed-Loop Control. IEEE Robot. Autom. Lett. 2019, 4, 1402–1408. [Google Scholar] [CrossRef]
- Yin, L.-J.; Zhao, Y.; Zhu, J.; Yang, M.; Zhao, H.; Pei, J.-Y.; Zhong, S.-L.; Dang, Z.-M. Soft, tough, and fast polyacrylate dielectric elastomer for non-magnetic motor. Nat. Commun. 2021, 12, 4517. [Google Scholar] [CrossRef]
- Cheng, N.G.; Gopinath, A.; Wang, L.; Iagnemma, K.; Hosoi, A.E. Thermally Tunable, Self-Healing Composites for Soft Robotic Applications. Macromol. Mater. Eng. 2014, 299, 1279–1284. [Google Scholar] [CrossRef]
- Houben, S.J.A.; Lugger, S.J.D.; van Raak, R.J.H.; Schenning, A.P.H.J. Schenning. A pH-Responsive Liquid Crystal Hydrogel Actuator with Calcium-Induced Reprogrammable Shape Fixing. ACS Appl. Polym. Mater. 2022, 4, 1298–1304. [Google Scholar] [CrossRef]
- Carpi, F.; Smela, E. (Eds.) Biomedical Applications of Electroactive Polymer Actuators; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Kim, O.; Shin, T.J.; Park, M.J. Fast low-voltage electroactive actuators using nanostructured polymer electrolytes. Nat. Commun. 2013, 4, 2208. [Google Scholar] [CrossRef]
- O’halloran, A.; O’malley, F.; McHugh, P. A review on dielectric elastomer actuators, technology, applications, and challenges. J. Appl. Phys. 2008, 104, 071101. [Google Scholar] [CrossRef]
- Bar-Cohen, Y. Electroactive Polymer (EAP) Actuators as Artificial Muscles: Reality, Potential, and Challenges; SPIE Press: Bellingham, WA, USA, 2004; Volume 136. [Google Scholar]
- Ghosh, T.; Das, T.; Purwar, R. Review of electrospun hydrogel nanofiber system: Synthesis, Properties and Applications. Polym. Eng. Sci. 2021, 61, 1887–1911. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, C.; Wiener, C.G.; Hao, J.; Shatas, S.; Weiss, R.; Vogt, B.D. Tough Stretchable Physically-Cross-linked Electrospun Hydrogel Fiber Mats. ACS Appl. Mater. Interfaces 2016, 8, 22774–22779. [Google Scholar] [CrossRef] [PubMed]
- Yarin, A.L.; Pourdehyhim, B.; Ramakrishna, S. Fundamentals and Applications of Micro- and Nanofibers; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Nayak, R.; Padhye, R.; Kyratzis, I.L.; Truong, Y.B.; Arnold, L. Recent advances in nanofibre fabrication techniques. Text. Res. J. 2011, 82, 129–147. [Google Scholar] [CrossRef]
- Beachley, V.; Wen, X. Polymer nanofibrous structures: Fabrication, biofunctionalization and cell interactions. Prog. Polym. Sci. J. 2010, 35, 868–892. [Google Scholar] [CrossRef] [PubMed]
- Alghoraibi, I.; Alomari, S. Different Methods for Nanofiber Design and Fabrication. In Handbook of Nanofibers; Barhoum, A., Bechelany, M., Makhlouf, A., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Mitchell, G.R. (Ed.) Electrospinning: Principles, Practice and Possibilities; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Gotti, C.; Sensini, A.; Zucchelli, A.; Carloni, R.; Focarete, M.L. Hierarchical fibrous structures for muscle-inspired soft-actuators: A review. Appl. Mater. Today 2020, 20, 100772. [Google Scholar] [CrossRef]
- Iacob, A.-T.; Drăgan, M.; Ionescu, O.-M.; Profire, L.; Ficai, A.; Andronescu, E.; Confederat, L.G.; Lupașcu, D. An Overview of Biopolymeric Electrospun Nanofibers Based on Polysaccharides for Wound Healing Management. Pharmaceutics 2020, 12, 983. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, M.; Dai, W.; Zhou, Y.; Xie, Z.; Liu, K.; Gao, L. Enhancement of Fe(III) to electro-response of starch hydrogel. Colloid. Polym. Sci. 2020, 298, 1533–1541. [Google Scholar] [CrossRef]
- Bernal, R.A.O.; Uspenskaya, M.V.; Olekhnovich, R.O. Biopolymers and its application as electroactive polymers. Proc. Voronezh State Univ. Eng. Technol. 2021, 83, 270–277. [Google Scholar] [CrossRef]
- Chauhan, D.; Dwivedi, J.; Sankararamakrishnan, N. Novel chitosan/PVA/zerovalent iron biopolymeric nanofibers with enhanced arsenic removal applications. Environ. Sci. Pollut. Res. 2014, 21, 9430–9442. [Google Scholar] [CrossRef]
- Romanazzi, G.; Gabler, F.M.; Margosan, D.; Mackey, B.E.; Smilanick, J.L.; Mancini, V.; Feliziani, E.; Servili, A.; Endeshaw, S.; Neri, D.; et al. Effect of chitosan dissolved in different acids on its ability to control postharvest gray mold of table grape. Phytopathology 2009, 99, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Hund, R.-D.; Aibibu, D.; Horakova, J.; Cherif, C. Pure Chitosan and Chitsoan/Chitosan Lactate Blended Nanofibres made by Single Step Electrospinning. Autex Res. J. 2013, 13, 128–133. [Google Scholar] [CrossRef]
- Pa, J.-H.; Yu, T.L. Light Scattering Study of Chitosan in Acetic Acid Aqueous Solutions. Macromol. Chem. Phys. 2001, 202, 985–991. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, H.; Yu, A.; Wen, D. Biodegradable Polylactide/Chitosan Blend Membranes. Biomacromolecules 2006, 7, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Zolfagharian, A.; Kaynak, A.; Khoo, S.Y.; Kouzani, A.Z. Polyelectrolyte Soft Actuators: 3D Printed Chitosan and Cast Gelatin. 3D Print. Addit. Manuf. 2018, 5, 138–150. [Google Scholar] [CrossRef]
- Shamsudeen, R.K.; Jayakumari, V.G.; Rajeswari, R.; Mukundan, T. Polyelectrolyte hydrogels of chitosan and polyacrylamide: A comparison of electroactive characteristics. Indian J. Eng. Mater. Sci. 2012, 19, 331–337. [Google Scholar]
- Pakravan, M.; Heuzey, M.-C.; Ajji, A. A fundamental study of chitosan/PEO electrospinning. Polymer 2011, 52, 4813–4824. [Google Scholar] [CrossRef]
- Liu, Y.; Park, M.; Shin, H.K.; Pant, B.; Park, S.-J.; Kim, H.-Y. Preparation and characterization of chitosan-based nanofibers by ecofriendly electrospinning. Mater. Lett. 2014, 132, 23–26. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Nie, J. Electrospinning of chitosan/poly(vinyl alcohol)/acrylic acid aqueous solutions. J. Appl. Polym. Sci. 2006, 102, 5692–5697. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Chen, H.-H.; Chang, S.-H.; Ni, T.-S. Pectin-chitosan-PVA nanofibrous scaffold made by electrospinning and its potential use as a skin tissue scaffold. J. Biomater. Sci. Polym. Ed. 2012, 24, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Kazeminava, F.; Javanbakht, S.; Nouri, M.; Adibkia, K.; Ganbarov, K.; Yousefi, M.; Ahmadi, M.; Gholizadeh, P.; Kafil, H.S. Electrospun nanofibers based on carboxymethyl cellulose/polyvinyl alcohol as a potential antimicrobial wound dressing. Int. J. Biol. Macromol. 2022, 214, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Bernal, R.A.O.; Olekhnovich, R.O.; Uspenskaya, M.V. Chitosan/PVA Nanofibers as Potential Material for the Development of Soft Actuators. Polymers 2023, 15, 2037. [Google Scholar] [CrossRef] [PubMed]
- D’Anniballe, R.; Zucchelli, A.; Carloni, R. The effect of morphology on poly(vinylidene fluoride-trifluoroethylene-chlorotrifluoroethylene)-based soft actuators: Films and electrospun aligned nanofiber mats. Sensors Actuators A Phys. 2021, 333, 113255. [Google Scholar] [CrossRef]
- Asai, H.; Okumura, T.; Sakamoto, H.; Nakane, K. Effect of polymer type on the performance of a nanofiber mat actuator. Polym. J. 2019, 51, 523–528. [Google Scholar] [CrossRef]
- Ebadi, S.V.; Semnani, D.; Fashandi, H.; Rezaei, B.; Fakhrali, A. Gaining insight into electrolyte solution effects on the electrochemomechanical behavior of electroactive PU/PPy nanofibers: Introducing a high-performance artificial muscle. Sens. Actuators B Chem. 2020, 305, 127519. [Google Scholar] [CrossRef]
- Miranda, D.O.; Dorneles, M.F.; Oréfice, R.L. One-step process for the preparation of fast-response soft actuators based on electrospun hybrid hydrogel nanofibers obtained by reactive electrospinning with in situ synthesis of conjugated polymers. Polymer 2020, 200, 122590. [Google Scholar] [CrossRef]
- Ismail, Y.A.; Shin, M.K.; Kim, S.J. A nanofibrous hydrogel templated electrochemical actuator: From single mat to a rolled-up structure. Sens. Actuators B Chem. 2009, 136, 438–443. [Google Scholar] [CrossRef]
- Arvanitoyannis, I.; Kolokuris, I.; Nakayama, A.; Yamamoto, N.; Aiba, S.-I. Physico-chemical studies of chitosan-poly(vinyl alcohol) blends plasticized with sorbitol and sucrose. Carbohydr. Polym. 1997, 34, 9–19. [Google Scholar] [CrossRef]
- Peppas, N.A.; Merrill, E.W. Differential scanning calorimetry of crystallized PVA hydrogels. J. Appl. Polym. Sci. 1976, 20, 1457–1465. [Google Scholar] [CrossRef]
- Delben, F.; Lapasin, R.; Pricl, S. Flow properties of N-(carboxymethyl) chitosan aqueous systems in the sol and gel domains. Int. J. Biol. Macromol. 1990, 12, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Maswal, M.; Dar, A.A. Rheological behavior of pH responsive composite hydrogels of chitosan and alginate: Characterization and its use in encapsulation of citral. Colloids Surf. B Biointerfaces 2018, 169, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, X.; Qiao, C.; Li, Y.; Li, T.; Xu, C. Effects of chitosan quaternary ammonium salt on the physicochemical properties of sodium carboxymethyl cellulose-based films. Carbohydr. Polym. 2018, 184, 37–46. [Google Scholar] [CrossRef]
- Tabernero, A.; Baldino, L.; Misol, A.; Cardea, S.; del Valle, E.M.M. Role of rheological properties on physical chitosan aerogels obtained by supercritical drying. Carbohydr. Polym. 2020, 233, 115850. [Google Scholar] [CrossRef] [PubMed]
- Abu-Jdayil, B.; Ghannam, M.; Alsayyed Ahmed, K.; Djama, M. The Effect of Biopolymer Chitosan on the Rheology and Stability of Na-Bentonite Drilling Mud. Polymers 2021, 13, 3361. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, D.P.; Inamdar, M.S. Aqueous Behaviour of Chitosan. Int. J. Polym. Sci. 2010, 2010, 288–294. [Google Scholar] [CrossRef]
- Kienzle-Sterzer, C.; Rodriguez-Sanchez, D.; Rha, C. Dilute solution behavior of a cationic polyelectrolyte. J. Appl. Polym. Sci. 1982, 27, 4467–4470. [Google Scholar] [CrossRef]
- Jia, Y.-T.; Gong, J.; Gu, X.-H.; Kim, H.-Y.; Dong, J.; Shen, X.-Y. Fabrication and characterization of poly (vinyl alcohol)/chitosan blend nanofibers produced by electrospinning method. Carbohydr. Polym. 2007, 67, 403–409. [Google Scholar] [CrossRef]
- Paipitak, K.; Pornpra, T.; Mongkontalang, P.; Techitdheer, W.; Pecharapa, W. Characterization of PVA-Chitosan Nanofibers Prepared by Electrospinning. Procedia Eng. 2011, 8, 101–105. [Google Scholar] [CrossRef]
- Nunthanid, J.; Puttipipatkhachorn, S.; Yamamoto, K.; Peck, G.E. Physical Properties and Molecular Behavior of Chitosan Films. Drug Dev. Ind. Pharm. 2001, 27, 143–157. [Google Scholar] [CrossRef]
- Çay, A.; Miraftab, M.; Kumbasar, E.P.A. Characterization and swelling performance of physically stabilized electrospun poly(vinyl alcohol)/chitosan nanofibers. Eur. Polym. J. 2014, 61, 253–262. [Google Scholar] [CrossRef]
- Wong, K.K.H.; Zinke-Allmang, M.; Wan, W. Effect of annealing on aqueous stability and elastic modulus of electrospun poly(vinyl alcohol) fibers. J. Mater. Sci. 2010, 45, 2456–2465. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Q.; Zhang, L.; Wang, S.; Harper, D.P.; Wu, Q.; Young, T.M. Preparation of electrospun nanofibrous poly(vinyl alcohol)/cellulose nanocrystals air filter for efficient particulate matter removal with repetitive usage capability via facile heat treatment. Chem. Eng. J. 2020, 399, 125768. [Google Scholar] [CrossRef]
- Kumar, S.; Koh, J. Physiochemical, Optical and Biological Activity of Chitosan-Chromone Derivative for Biomedical Applications. Int. J. Mol. Sci. 2012, 13, 6102–6116. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Su, W.Y.; Leu, T.L.; Yang, M.C. Evaluation of chitosan/PVA blended hydrogel membranes. J. Membr. Sci. 2004, 236, 39–51. [Google Scholar] [CrossRef]
- Andrade, J.; González-Martínez, C.; Chiralt, A. The Incorporation of Carvacrol into Poly (vinyl alcohol) Films Encapsulated in Lecithin Liposomes. Polymers 2020, 12, 497. [Google Scholar] [CrossRef] [PubMed]
- Koosha, M.; Mirzadeh, H. Electrospinning, mechanical properties, and cell behavior study of chitosan/PVA nanofibers. J. Biomed. Mater. Res. Part A 2015, 103, 3081–3093. [Google Scholar] [CrossRef]
- Kim, G.M.; Asran, A.S.; Michler, G.H.; Simon, P.; Kim, J.S. Electrospun PVA/HAp nanocomposite nanofibers: Biomimetics of mineralized hard tissues at a lower level of complexity. Bioinspiration Biomim. 2008, 3, 046003. [Google Scholar] [CrossRef]
- Bagherian Far, M.; Ziyadi, H. Fabrication of polyvinyl alcohol/kefiran nanofibers membrane using electrospinning. JPHCS 2016, 4, 211–2018. [Google Scholar]
- Gholipour, K.A.; Bahrami, S.H.; Nouri, M. Chitosan-poly(vinyl alcohol) blend nanofibers: Morphology, biological and antimicrobial properties. e-Polymers 2009, 9, 133. [Google Scholar] [CrossRef]
- Zhou, Y.S.; Yang, D.Z.; Nie, J. Effect of PVA content on morphology, swelling and mechanical property of crosslinked chitosan/PVA nanofiber. Plast. Rubber Compos. 2007, 36, 254–258. [Google Scholar] [CrossRef]
- Charernsriwilaiwat, N.; Rojanarata, T.; Ngawhirunpat, T.; Opanasopit, P. Electrospun chitosan/polyvinyl alcohol nanofibre mats for wound healing. Int. Wound J. 2014, 11, 215–222. [Google Scholar] [CrossRef]
- Albanna, M.Z.; Bou-Akl, T.H.; Blowytsky, O.; Walters, H.L.; Matthew, H.W. Chitosan fibers with improved biological and mechanical properties for tissue engineering applications. J. Mech. Behav. Biomed. Mater. 2013, 20, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Gartner, C.; López, B.L.; Sierra, L.; Graf, R.; Spiess, H.W.; Gaborieau, M. Interplay between Structure and Dynamics in Chitosan Films Investigated with Solid-State NMR, Dynamic Mechanical Analysis, and X-ray Diffraction. Biomacromolecules 2011, 12, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, B.-L.; Wang, L.-J.; Deng, Y.-H.; Zhou, S.-Y.; Feng, J.-W. Preparation, Structure and Properties of Acid Aqueous Solution Plasticized Thermoplastic Chitosan. Polymers 2019, 11, 818. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.N.; Morozkina, S.N.; Uspenskaya, M.V.; Olekhnovich, R.O. Fabrication of Polyvinyl Alcohol Nanofibers for the Delivery of Biologically Active Molecules. In 2022 IEEE-EMBS Conference on Biomedical Engineering and Sciences (IECBES); IEEE: Piscataway, NJ, USA, 2022; pp. 344–349. [Google Scholar]
- Wardhono, E.Y.; Pinem, M.P.; Susilo, S.; Siom, B.J.; Sudrajad, A.; Pramono, A.; Meliana, Y.; Guénin, E. Modification of Physio-Mechanical Properties of Chitosan-Based Films via Physical Treatment Approach. Polymers 2022, 14, 5216. [Google Scholar] [CrossRef] [PubMed]
- Es-Saheb, M.; Elzatahry, A. Post-Heat Treatment and Mechanical Assessment of Polyvinyl Alcohol Nanofiber Sheet Fabricated by Electrospinning Technique. Int. J. Polym. Sci. 2014, 2014, 357–362. [Google Scholar] [CrossRef]
- Sorlier, P.; Denuzière, A.; Viton, C.; Domard, A. Relation between the Degree of Acetylation and the Electrostatic Properties of Chitin and Chitosan. Biomacromolecules 2001, 2, 765–772. [Google Scholar] [CrossRef]
- Shiga, T.; Kurauchi, T. Deformation of polyelectrolyte gels under the influence of electric field. J. Appl. Polym. Sci. 1990, 39, 2305–2320. [Google Scholar] [CrossRef]
- Shiga, T.; Hirose, Y.; Okada, A.; Kurauchi, T. Bending of poly(vinyl alcohol)–poly(sodium acrylate) composite hydrogel in electric fields. J. Appl. Polym. Sci. 1992, 44, 249–253. [Google Scholar] [CrossRef]
- Shang, J.; Shao, Z.; Chen, X. Electrical Behavior of a Natural Polyelectrolyte Hydrogel: Chitosan/Carboxymethylcellulose Hydrogel. Biomacromolecules 2008, 9, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Mansur, H.S.; Sadahira, C.M.; Souza, A.N.; Mansur, A.A. FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater. Sci. Eng. C 2008, 28, 539–548. [Google Scholar] [CrossRef]
- Anicuta, S.G.; Dobre, L.; Stroescu, M.; Jipa, I. Fourier Transform Infrared (FTIR) spectroscopy for characterization of antimicrobial films containing chitosan. An. Univ. Oradea Fasc. Ecotoxicol. Zooteh. Tehnol. Ind. Aliment. 2010, 2010, 1234–1240. [Google Scholar]
- Rivero, S.; García, M.A.; Pinotti, A. Heat Treatment To Modify the Structural and Physical Properties of Chitosan-Based Films. J. Agric. Food Chem. 2012, 60, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.Y.; Wan, L.S.C. Heat Treatment of Chitosan Films. Drug Dev. Ind. Pharm. 1995, 21, 839–846. [Google Scholar] [CrossRef]
- Ardila, N.; Daigle, F.; Heuzey, M.-C.; Ajji, A. Antibacterial Activity of Neat Chitosan Powder and Flakes. Molecules 2017, 22, 100. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.; Ferreira, M.; Marvão, M.; Rocha, J. An optimised method to determine the degree of acetylation of chitin and chitosan by FTIR spectroscopy. Int. J. Biol. Macromol. 2002, 31, 1–8. [Google Scholar] [CrossRef]
- Gupta, K.; Jabrail, F.H. Effects of degree of deacetylation and cross-linking on physical characteristics, swelling and release behavior of chitosan microspheres. Carbohydr. Polym. 2006, 66, 43–54. [Google Scholar] [CrossRef]
- Kasaai, M.R. A review of several reported procedures to determine the degree of N-acetylation for chitin and chitosan using infrared spectroscopy. Carbohydr. Polym. 2008, 71, 497–508. [Google Scholar] [CrossRef]
- Branca, C.; D’Angelo, G.; Crupi, C.; Khouzami, K.; Rifici, S.; Ruello, G.; Wanderlingh, U. Role of the OH and NH vibrational groups in polysaccharide-nanocomposite interactions: A FTIR-ATR study on chitosan and chitosan/clay films. Polymer 2016, 99, 614–622. [Google Scholar] [CrossRef]
- Malek, A.; Abderraouf, G. Synthesis and characterization of the composite material PVA/Chitosan/5% sorbitol with different ratio of chitosan. Int. J. Mech. Mechatron. Eng. 2017, 17, 15–28. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acetic Acid Concentration | 50% | 60% | 70% | 80% | ||||
|---|---|---|---|---|---|---|---|---|
| Cs. | Cs/PVA | Cs. | Cs/PVA | Cs. | Cs/PVA | Cs. | Cs/PVA | |
| Viscosity (η) [mPa·s] | 986.93 | 6157 | 1512.5 | 6445 | 2014.2 | 6847.7 | 1262.8 | 8747.3 |
| Acetic Acid Concentration | 50% | 60% | 70% | 80% | ||||
|---|---|---|---|---|---|---|---|---|
| Cs. | Cs/PVA | Cs. | Cs/PVA | Cs. | Cs/PVA | Cs. | Cs/PVA | |
| Conductivity (µS cm−1) | 3558.4 | 2852.6 | 2695.2 | 2126.2 | 2395.4 | 1682.6 | 1245.6 | 999.6 |
| Sample | Chitosan (wt.%) | PVA (wt.%) | Acetic Acid (%) | Mean (µm) | Morphology |
|---|---|---|---|---|---|
| AA50 | 4 | 5 | 50 | 0.482 ± 0.006 | Uniform fiber formation |
| AA60 | 4 | 5 | 60 | 0.533 ± 0.01 | Uniform fiber formation |
| AA70 | 4 | 5 | 70 | 0.622 ± 0.0059 | Uniform fiber formation |
| AA80 | 4 | 5 | 80 | 0.793 ± 0.011 | Uniform fiber formation |
| Sample | Chitosan (wt.%) | PVA (wt.%) | First Mass Loss (%) 50–160 °C | Second Mass Loss (%) 175–370 °C | Third Mass Loss (%) 375–500 °C | First Stage 1st Peak (°C) | First Stage 2nd Peak (°C) | Second Stage (°C) | Third Stage (°C) |
|---|---|---|---|---|---|---|---|---|---|
| PVA | 4 | 90.53 | 81 | 304 | |||||
| Chit | 5 | 49.68 | 67 | 299 | |||||
| F-AA50 | 4 | 5 | 11.6 | 65.98 | 51 | 93 | 220 | ||
| F-AA60 | 4 | 5 | 11.2 | 58.18 | 15.86 | 56 | 100 | 264 | 428 |
| F-AA70 | 4 | 5 | 10.52 | 57.98 | 16.44 | 56 | 109 | 264.8 | 428 |
| F-AA80 | 4 | 5 | 9.58 | 59.27 | 15.81 | 56 | 108 | 266 | 427 |
| Sample | Chitosan (wt.%) | PVA (wt.%) | First Mass Loss (%) 50–160 °C | Second Mass Loss (%) 140–370 °C | Third Mass Loss (%) 375–500 °C | First Stage 1st Peak (°C) | First Stage 2nd Peak (°C) | Second Stage (°C) | Third Stage (°C) |
|---|---|---|---|---|---|---|---|---|---|
| F-AA50 | 4 | 5 | 2.42 | 71.07 | 52 | 231 | |||
| F-AA60 | 4 | 5 | 3.1 | 71.62 | 60 | 271 | |||
| F-AA70 | 4 | 5 | 3.26 | 71.35 | 60 | 125 | 272 | ||
| F-AA80 | 4 | 5 | 6.47 | 71.79 | 63 | 125 | 275 |
| Sample | Glass Transition t(g) | Melting Point t(m) | Degree of Crystallinity (%) | |||
|---|---|---|---|---|---|---|
| Not Dry | Dry | Not Dry | Dry | Not Dry | Dry | |
| F-AA50 | 37 | 164.2 | 165.15 | 1.43 | 1.87 | |
| F-AA60 | 37 | 166.0 | 166.4 | 1.77 | 1.96 | |
| F-AA70 | 37 | 165.5 | 166.8 | 1.86 | 2.25 | |
| F-AA80 | 39 | 164.45 | 164.97 | 1.30 | 1.90 | |
| Sample | Young’s Modulus (MPa) | Tensile Strength (MPa) | Elongation at Break (%) | |||
|---|---|---|---|---|---|---|
| Not Dry | Dry | Not Dry | Dry | Not Dry | Dry | |
| F-AA50 | 649.19 ± 52.3 | 592.3 ± 50.4 | 13.1 ± 1.22 | 10.34 ± 1.5 | 12.13 ± 1.2 | 7.5 ± 1.21 |
| F-AA60 | 474.8 ± 23.8 | 462.6 ± 28.25 | 5.8 ± 1.1 | 4.11 ± 1.01 | 13.68 ± 2.9 | 11.6 ± 3.19 |
| F-AA70 | 594.1 ± 53.6 | 472.1 ± 23.7 | 5.26 ± 0.69 | 4.6 ± 0.8 | 15.06 ± 0.36 | 14.3 ± 1.5 |
| F-AA80 | 545.92 ± 52.9 | 375.415 ± 80.4 | 11.43 ± 1 | 9.56 ± 2.6 | 5 ± 0.86 | 4.3 ± 0.74 |
| Sample | Types of Hydrogen Bond | Abbreviation | Wave Number (cm−1) | Relative Strength (%) | Relative Strength/% (Dried Samples) | |
|---|---|---|---|---|---|---|
| Acetic acid 50% | Primary ammonium | I | —NH+3 | ~3100 cm−1 | 3.93 | 3.2 |
| Intermolecular hydrogen bond | II | OH…ether O | ~3200 cm−1 | 8.7 | 5.2 | |
| Amide | III | —CONH— | ~3240 cm−1 | 30.1 | 33 | |
| Intermolecular association | IV | N2—H1…O5/N2—H2…O1 | ~3335 cm−1 | 27.5 | 28 | |
| Intramolecular association | V | O3H…O5/O3H…O6 | ~3366 cm−1 | 0.8 | 0.9 | |
| Free amine | VI | —NH2 | ~3408 cm−1 | 6.39 | 6.9 | |
| Multimer (Intermolecular association) | VII | O6H…N2 | ~3462 cm−1 | 22.13 | 22.6 | |
| Free hydroxyl | VIII | —OH | ~3580 cm−1 | 0.36 | 0.43 | |
| Acetic acid 60% | Primary ammonium | I | —NH+3 | ~3100 cm−1 | 4.4 | 4.2 |
| Intermolecular hydrogen bond | II | OH…ether O | ~3200 cm−1 | 10.27 | 8.2 | |
| Amide | III | —CONH— | ~3240 cm−1 | 029.3 | 30.2 | |
| Intermolecular association | IV | N2—H1…O5/N2—H2…O1 | ~3335 cm−1 | 27.46 | 29.15 | |
| Intramolecular association | V | O3H…O5/O3H…O6 | ~3366 cm−1 | 0.90 | 0.92 | |
| Free amine | VI | —NH2 | ~3408 cm−1 | 6.14 | 6.45 | |
| Multimer (Intermolecular association) | VII | O6H…N2 | ~3462 cm−1 | 20 | 21.1 | |
| Free hydroxyl | VIII | —OH | ~3580 cm−1 | 0.09 | 0.21 | |
| Acetic acid pse-70% | Primary ammonium | I | —NH+3 | ~3100 cm−1 | 4.21 | 3.66 |
| Intermolecular hydrogen bond | II | OH…ether O | ~3200 cm−1 | 9.6 | 6.69 | |
| Amide | III | —CONH— | ~3240 cm−1 | 29.7 | 32.1 | |
| Intermolecular association | IV | N2—H1…O5/N2—H2…O1 | ~3335 cm−1 | 26.6 | 27.9 | |
| Intramolecular association | V | O3H…O5/O3H…O6 | ~3366 cm−1 | 0.88 | 0.91 | |
| Free amine | VI | —NH2 | ~3408 cm−1 | 5.89 | 5.96 | |
| Multimer (Intermolecular association) | VII | O6H…N2 | ~3462 cm−1 | 21.32 | 23.35 | |
| Free hydroxyl | VIII | —OH | ~3580 cm−1 | 0.4 | 0.64 | |
| Acetic acid 80% | Primary ammonium | I | —NH+3 | ~3100 cm−1 | 4.5 | 4.0 |
| Intermolecular hydrogen bond | II | OH…ether O | ~3200 cm−1 | 10.53 | 6.23 | |
| Amide | III | —CONH— | ~3240 cm−1 | 27 | 32.44 | |
| Intermolecular association | IV | N2—H1…O5/N2—H2…O1 | ~3335 cm−1 | 27.6 | 28.9 | |
| Intramolecular association | V | O3H…O5/O3H…O6 | ~3366 cm−1 | 0.71 | 0.79 | |
| Free amine | VI | —NH2 | ~3408 cm−1 | 6.56 | 6.85 | |
| Multimer (Intermolecular association) | VII | O6H…N2 | ~3462 cm−1 | 21.08 | 21.77 | |
| Free hydroxyl | VIII | —OH | ~3580 cm−1 | 0.1 | 0.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernal, R.A.O.; Olekhnovich, R.O.; Uspenskaya, M.V. Influence of Thermal Treatment and Acetic Acid Concentration on the Electroactive Properties of Chitosan/PVA-Based Micro- and Nanofibers. Polymers 2023, 15, 3719. https://doi.org/10.3390/polym15183719
Bernal RAO, Olekhnovich RO, Uspenskaya MV. Influence of Thermal Treatment and Acetic Acid Concentration on the Electroactive Properties of Chitosan/PVA-Based Micro- and Nanofibers. Polymers. 2023; 15(18):3719. https://doi.org/10.3390/polym15183719
Chicago/Turabian StyleBernal, Rigel Antonio Olvera, Roman O. Olekhnovich, and Mayya V. Uspenskaya. 2023. "Influence of Thermal Treatment and Acetic Acid Concentration on the Electroactive Properties of Chitosan/PVA-Based Micro- and Nanofibers" Polymers 15, no. 18: 3719. https://doi.org/10.3390/polym15183719