

Improving Sustainability through Covalent Adaptable Networks in the Recycling of Polyurethane Plastics

 , , ,
, , ,  and
and
Abstract
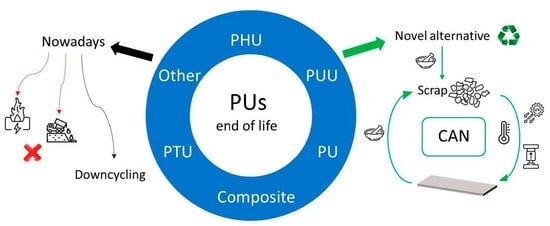
:
1. Introduction
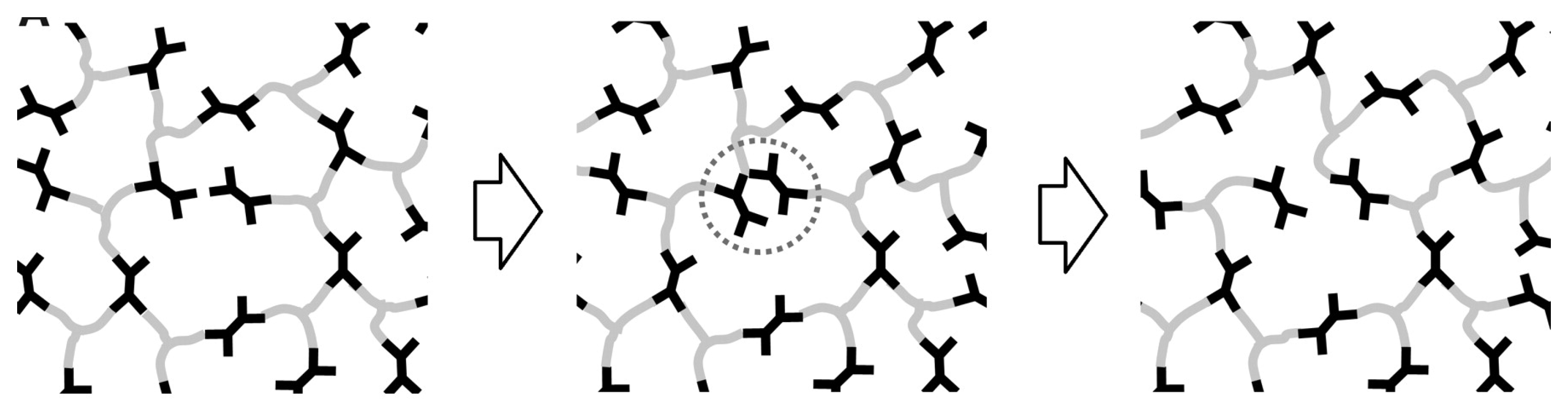
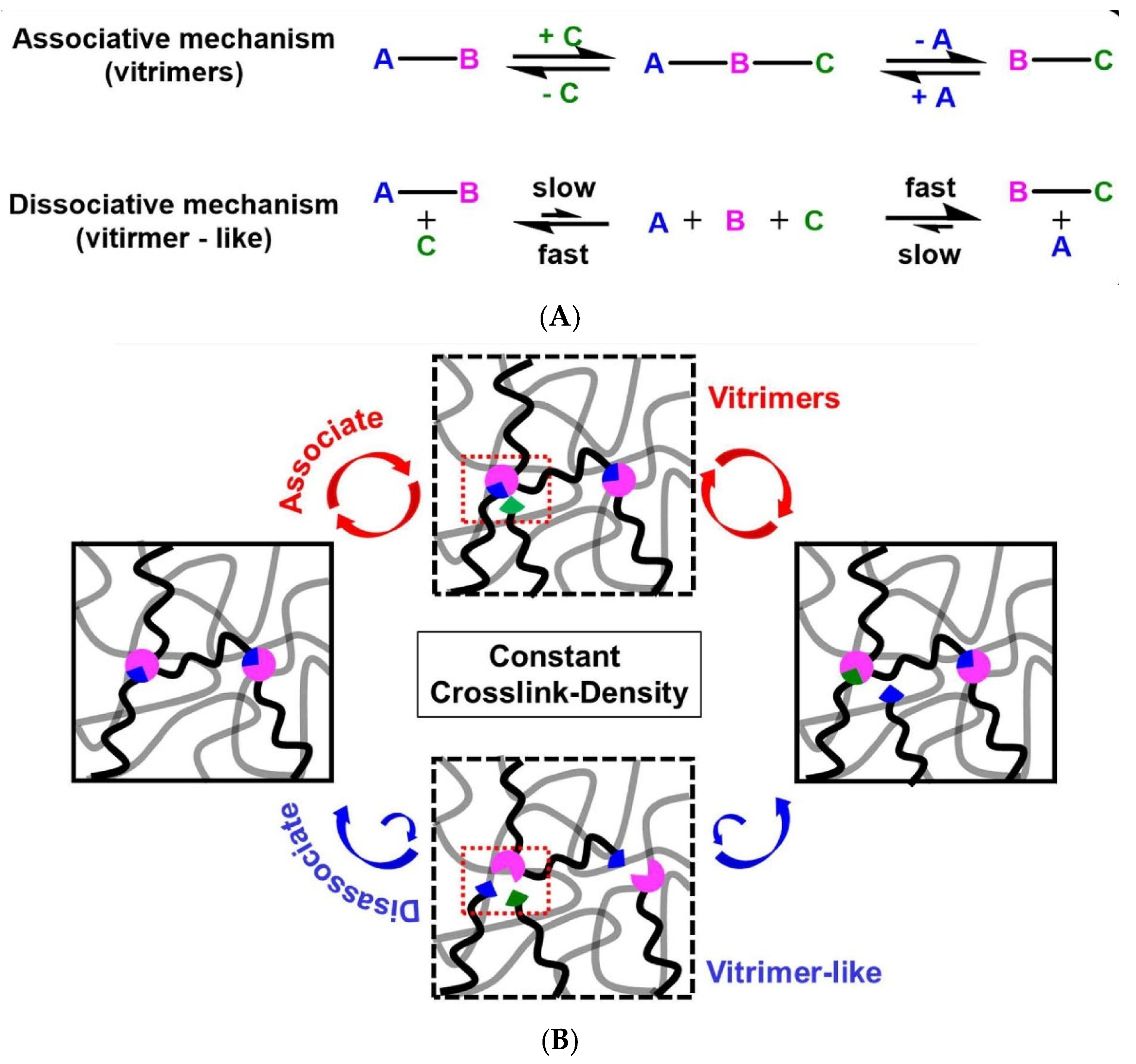
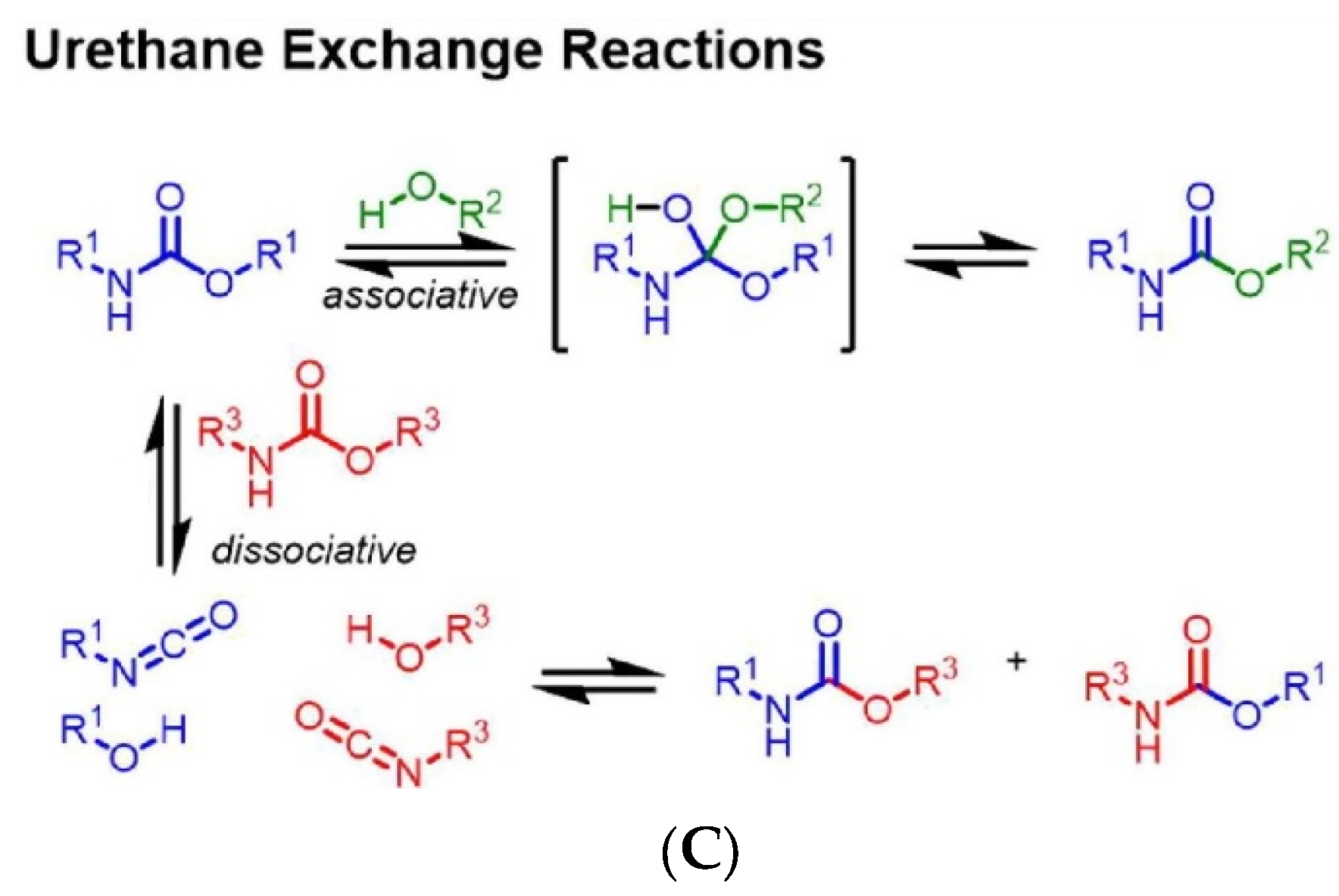
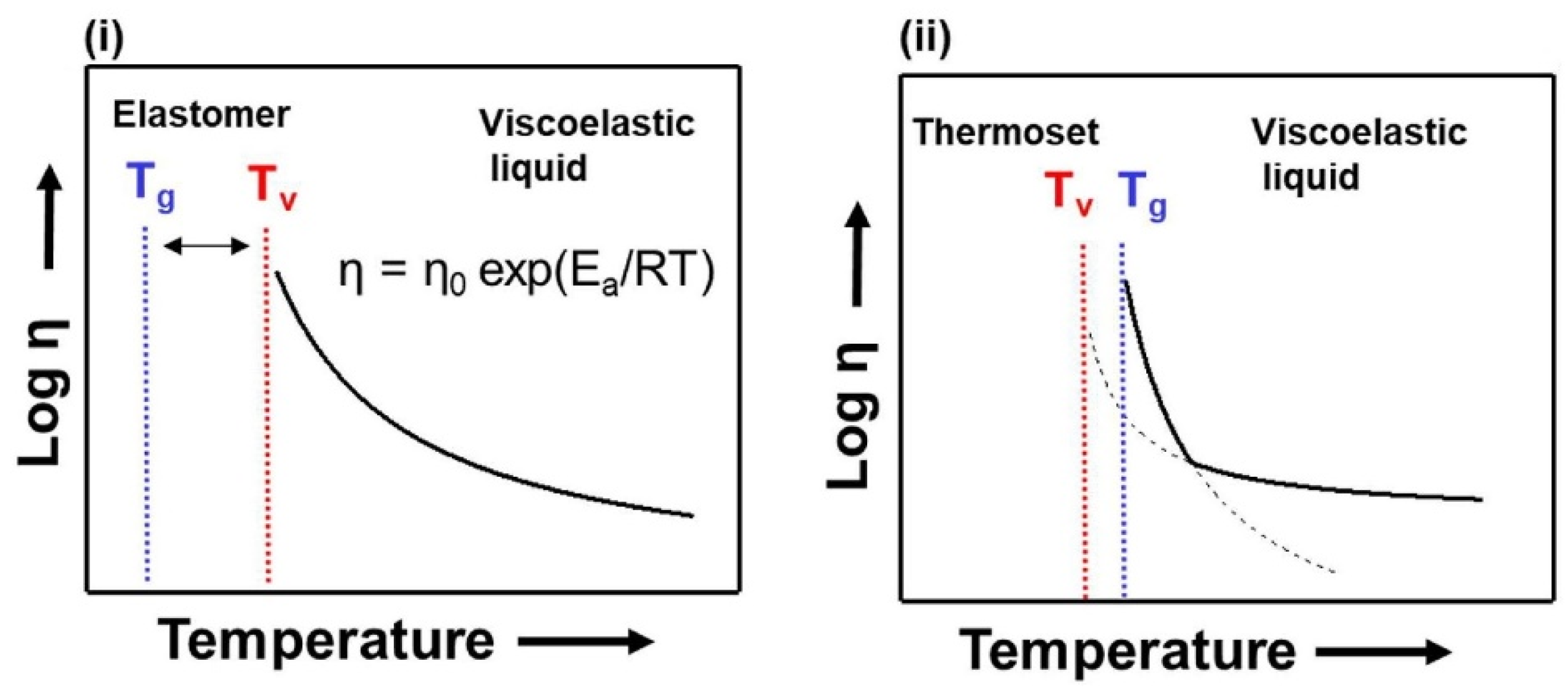
2. Considerations on CANs
3. Polyurethanes
3.1. Thermosets
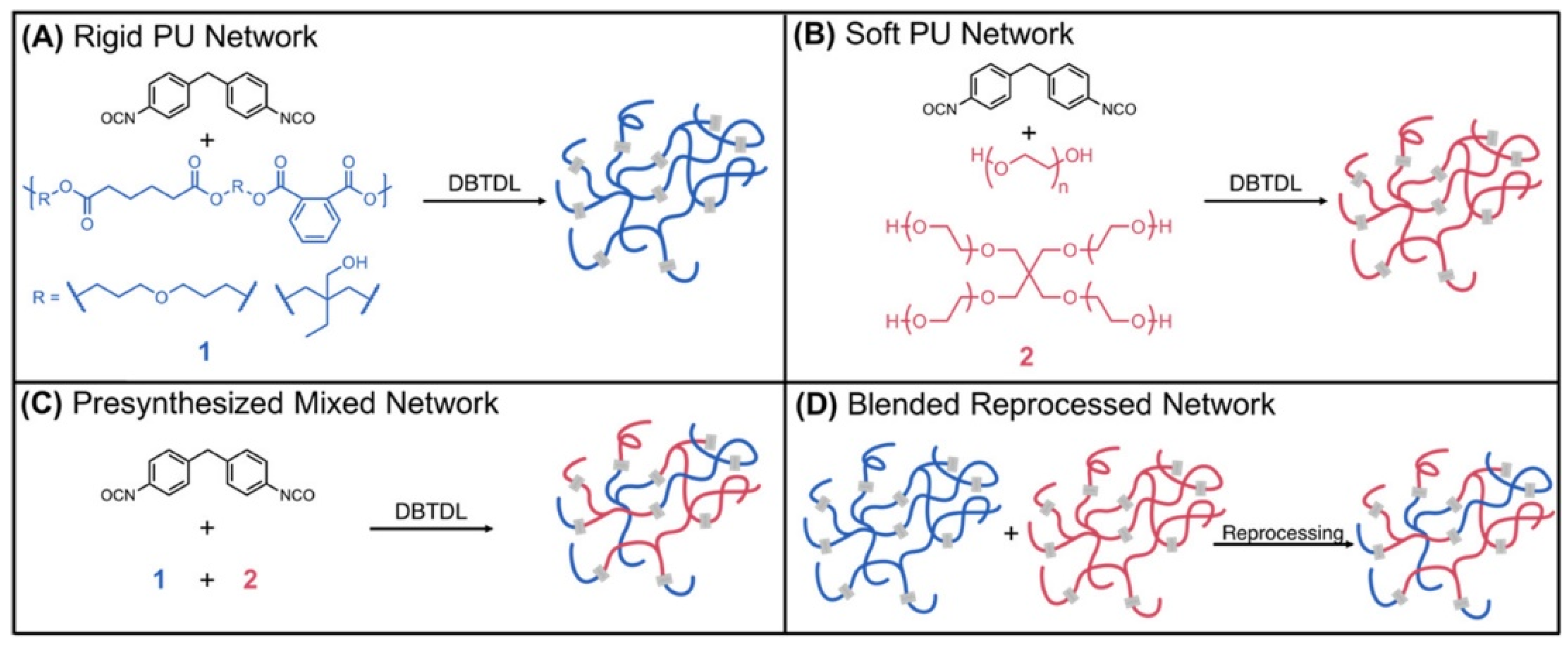

3.2. Elastomers
3.3. Foams
4. Polyhydroxyurethanes
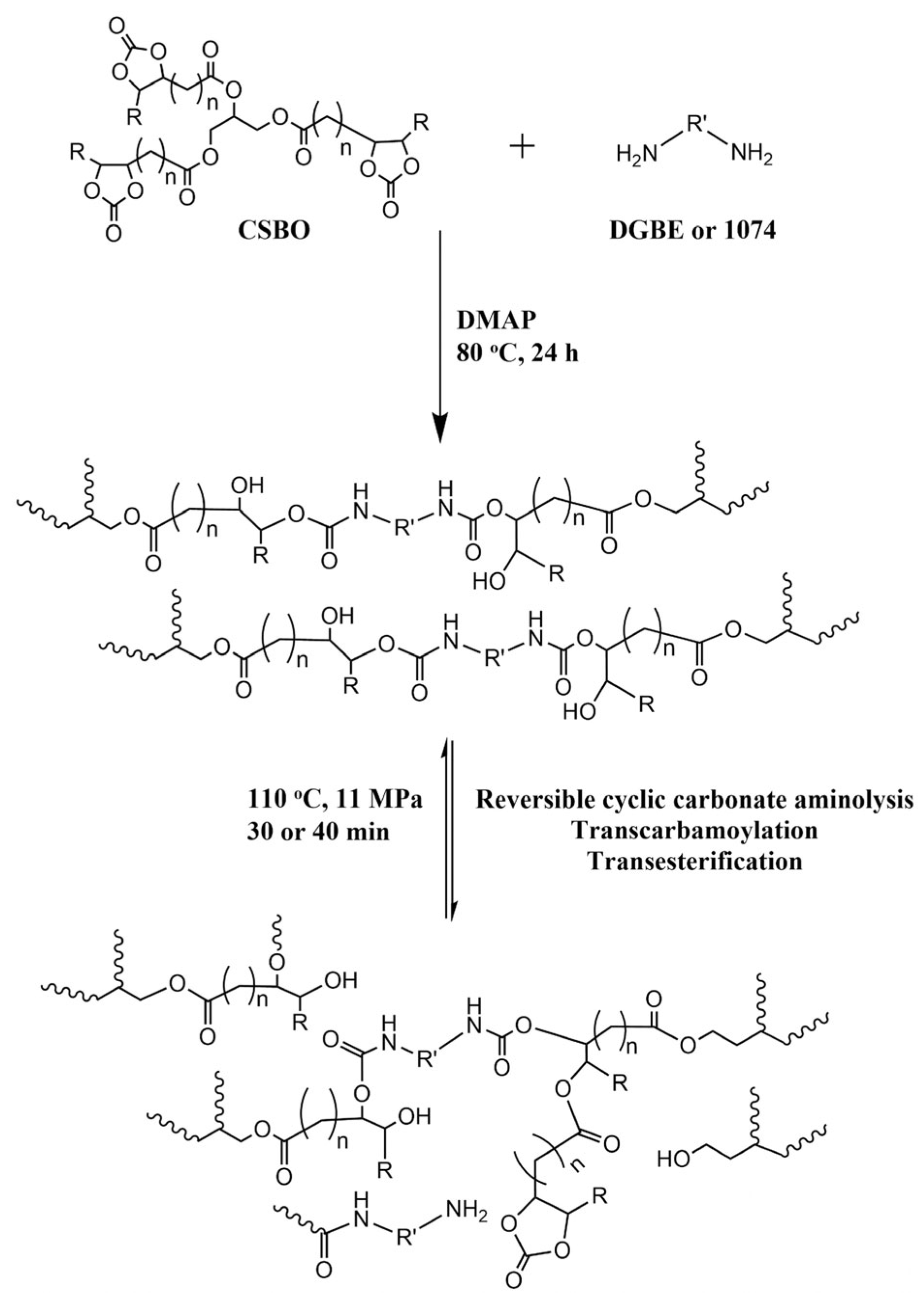
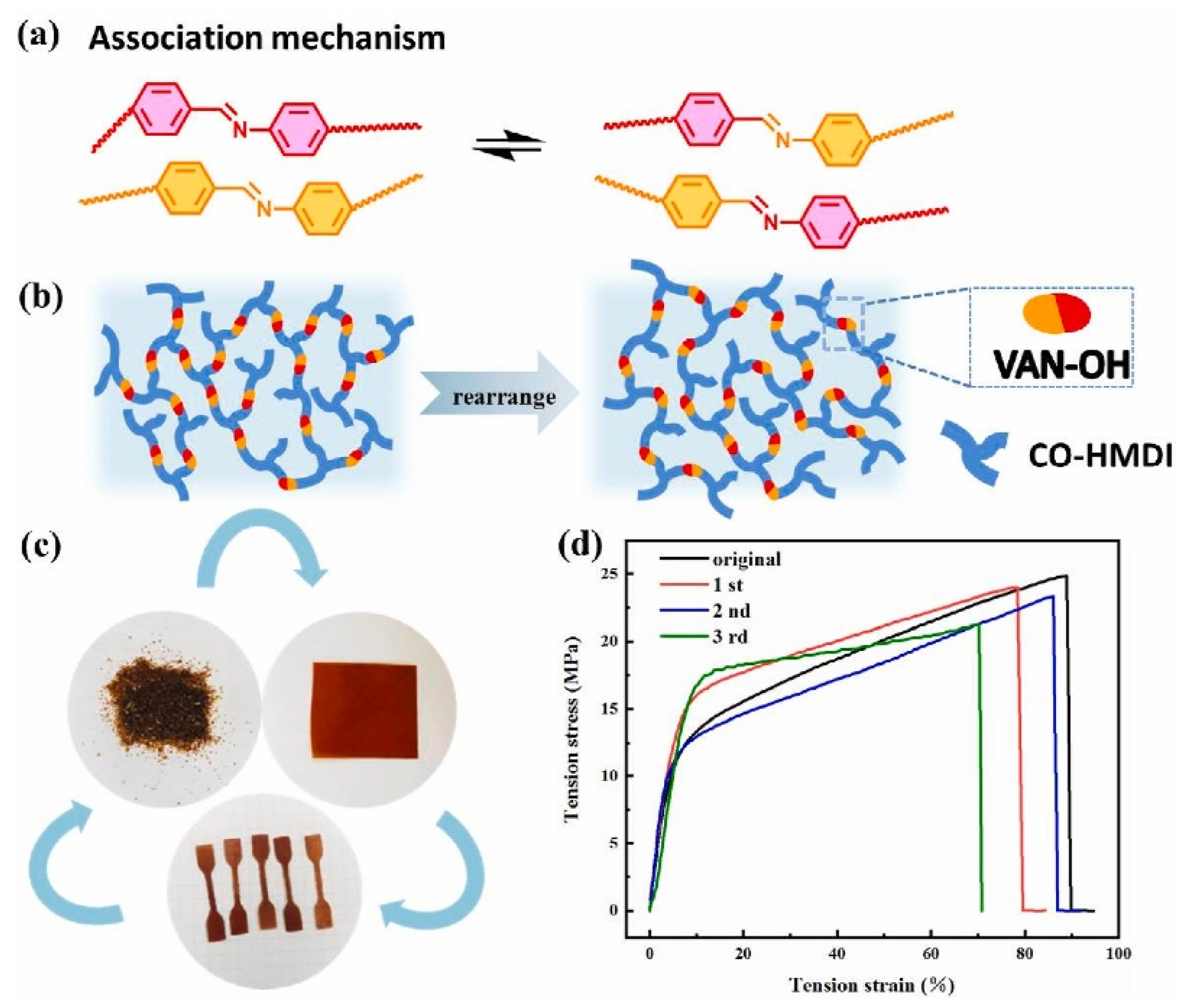
5. Poly(urethane-urea)
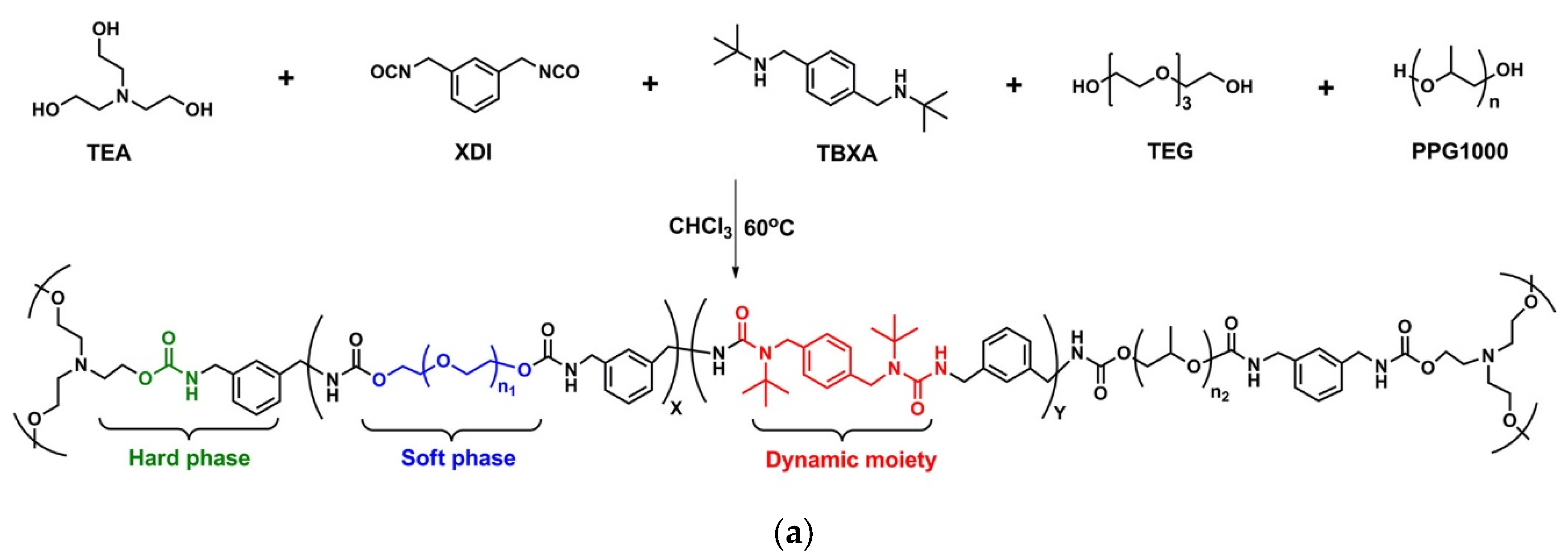
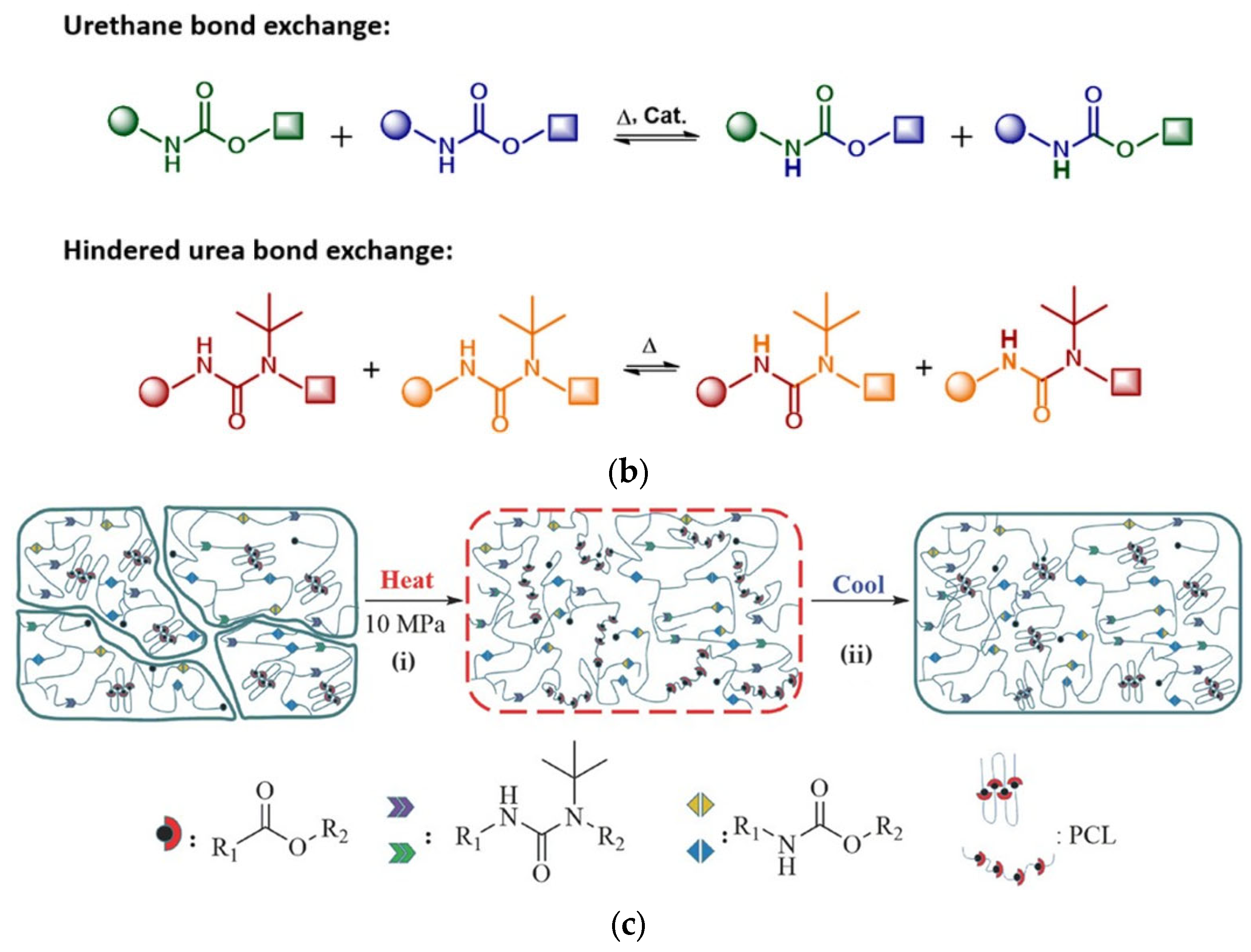
6. Polythiourethanes

7. Composites
8. Others
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3HDI | Hexamethylene diisocyanate trimer |
| BDB | 2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] |
| BDO | 1,4-Butanediol |
| BHMF | 2,5-bis-(hydroxymethyl)furan |
| CAN | Covalent Adaptable Network |
| CASE | Coatings, adhesives, sealants, and elastomers |
| CSBO | Carbonated soybean oil |
| CTO | Castor oil |
| D-A | Dies-Alder |
| DABCO | 1,4-diazabicyclo [2.2.2] octane |
| DBDA | N,N’-di-tert-butylethylenediamine |
| DBTDL | Dibutyltin dilaurate |
| DCM | Dichloromethane |
| DDSQ | Double-decker silsesquioxane |
| DMA | Dynamic Mechanical Analysis |
| FTIR | Fourier Transformed Infrared Spectroscopy |
| GLY | Glycerine |
| HDI | Hexamethylene diisocyanate |
| HEDS | 2-hydroxyethyl disulphide |
| HPS | Bis(4-hydroxyphenyl) disulphide |
| HTBD | Hydroxy-terminated polybutadiene |
| IPDI | Isophorone diisocyanate |
| IR | Infrared |
| MDI | Methylene diphenyl diisocyanate |
| NCC | nanocrystalline cellulose |
| NIR | Near Infrared |
| NPs | Nano particles |
| PCL | Poly(ε-caprolactone) |
| PEG | Polyethylene glycol |
| PHU | Polyhydroxyurethanes |
| POU | Poly(oxime-urethane) |
| PPG | Poly(propylene glycol) |
| PTMEG | Polytetramethylene ether glycol |
| PTU | Polythiourethanes |
| PU | Polyurethanes |
| PUF | Polyurethanes foam |
| PUU | Poly(urethane-urea) |
| PVPy | N-vinylpyrrolidone |
| ROMP | Ring-opening metathesis polymerisation |
| SEC | Sorbitol ether carbonate |
| TDI | Toluene diisocyanate |
| TEG | Tetraethylene glycol |
| TMMP | Trimethylolpropane tris(3-mercaptopropionate) |
| TMP | Trimethylolpropane |
| UPy | Ureidopyrimidinone |
| UV | Ultraviolet |
References
- Plastics-the Facts 2022 OCTOBER 2022. Available online: https://plasticseurope.org/ (accessed on 15 June 2023).
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Otto, B. Das Di-Isocyanat-Polyadditionsverfahren (Polyurethane). Angew. Chem. 1947, 59, 257–288. [Google Scholar]
- Zia, K.M.; Anjum, S.; Zuber, M.; Mujahid, M.; Jamil, T. Synthesis and molecular characterization of chitosan based polyurethane elastomers using aromatic diisocyanate. Int. J. Biol. Macromol. 2014, 66, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane types, synthesis and applications-a review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef]
- Simón, D.; Borreguero, A.M.; de Lucas, A.; Rodríguez, J.F. Recycling of polyurethanes from laboratory to industry, a journey towards the sustainability. Waste Manag. 2018, 76, 147–171. [Google Scholar] [CrossRef]
- Kemona, A.; Piotrowska, M. Polyurethane recycling and disposal: Methods and prospects. Polymers 2020, 12, 1752. [Google Scholar] [CrossRef]
- Charlon, M.; Heinrich, B.; Matter, Y.; Couzigné, E.; Donnio, B.; Avérous, L. Synthesis, structure and properties of fully biobased thermoplastic polyurethanes, obtained from a diisocyanate based on modified dimer fatty acids, and different renewable diols. Eur. Polym. J. 2014, 61, 197–205. [Google Scholar] [CrossRef]
- Serrano, A.; Borreguero, A.M.; Garrido, I.; Rodríguez, J.F.; Carmona, M. Reducing heat loss through the building envelope by using polyurethane foams containing thermoregulating microcapsules. Appl. Therm. Eng. 2016, 103, 226–232. [Google Scholar] [CrossRef]
- Dutta, S.; Karak, N.; Jana, T. Evaluation of Mesua ferrea L. seed oil modified polyurethane paints. Prog. Org. Coat. 2009, 65, 131–135. [Google Scholar] [CrossRef]
- Dutta, S.; Karak, N. Synthesis, characterization of poly(urethane amide) resins from Nahar seed oil for surface coating applications. Prog. Org. Coat. 2005, 53, 147–152. [Google Scholar] [CrossRef]
- Deng, R.; Davies, P.; Bajaj, A.K. Flexible polyurethane foam modelling and identification of viscoelastic parameters for automotive seating applications. J. Sound. Vib. 2003, 262, 391–417. [Google Scholar] [CrossRef]
- Mukherjee, M.; Gurusamy-Thangavelu, S.A.; Chelike, D.K.; Alagumalai, A.; Das, B.N.; Jaisankar, S.N.; Mandal, A.B. Biodegradable polyurethane foam as shoe insole to reduce footwear waste: Optimization by morphological physicochemical and mechanical properties. Appl. Surf. Sci. 2020, 499, 143966. [Google Scholar] [CrossRef]
- Mekewi, M.A.; Ramadan, A.M.; ElDarse, F.M.; Rehim, M.H.A.; Mosa, N.A.; Ibrahim, M.A. Preparation and characterization of polyurethane plasticizer for flexible packaging applications: Natural oils affirmed access. Egypt. J. Pet. 2017, 26, 9–15. [Google Scholar] [CrossRef]
- Garrido, M.; Correia, J.R.; Keller, T. Effect of service temperature on the shear creep response of rigid polyurethane foam used in composite sandwich floor panels. Constr. Build. Mater. 2016, 118, 235–244. [Google Scholar] [CrossRef]
- Daemi, H.; Rajabi-Zeleti, S.; Sardon, H.; Barikani, M.; Khademhosseini, A.; Baharvand, H. A robust super-tough biodegradable elastomer engineered by supramolecular ionic interactions. Biomaterials 2016, 84, 54–63. [Google Scholar] [CrossRef]
- Uscátegui, Y.L.; Díaz, L.E.; Valero, M.F. In vitro and in vivo biocompatibility of polyurethanes synthesized with castor oil polyols for biomedical devices. J. Mater. Res. 2019, 34, 519–531. [Google Scholar] [CrossRef]
- Datta, J.; Kopczyńska, P.; Simón, D.; Rodríguez, J.F. Thermo-Chemical Decomposition Study of Polyurethane Elastomer through Glycerolysis Route with Using Crude and Refined Glycerine as a Transesterification Agent. J. Polym. Environ. 2018, 26, 166–174. [Google Scholar] [CrossRef]
- Deng, Y.; Dewil, R.; Appels, L.; Ansart, R.; Baeyens, J.; Kang, Q. Reviewing the thermo-chemical recycling of waste polyurethane foam. J. Environ. Manag. 2021, 278, 111527. [Google Scholar] [CrossRef]
- Zia, K.M.; Bhatti, H.N.; Bhatti, I.A. Methods for polyurethane and polyurethane composites recycling and recovery: A review. React. Funct. Polym. 2007, 67, 675–692. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, Y.; Wang, D.; Yan, M.; Zhang, J.; Zhang, P.; Ding, T.; Chen, L.; Chen, C. Current technologies for plastic waste treatment: A review. J. Clean. Prod. 2021, 282, 124523. [Google Scholar] [CrossRef]
- Simón, D.; Borreguero, A.M.; De Lucas, A.; Rodríguez, J.F. Glycolysis of viscoelastic flexible polyurethane foam wastes. Polym. Degrad. Stab. 2015, 116, 23–35. [Google Scholar] [CrossRef]
- Heiran, R.; Ghaderian, A.; Reghunadhan, A.; Sedaghati, F.; Thomas, S.; Haghighi, A.H. Glycolysis: An efficient route for recycling of end of life polyurethane foams. J. Polym. Res. 2021, 28, 22. [Google Scholar] [CrossRef]
- Gama, N.; Godinho, B.; Marques, G.; Silva, R.; Barros-Timmons, A.; Ferreira, A. Recycling of polyurethane scraps via acidolysis. Chem. Eng. J. 2020, 395, 125102. [Google Scholar] [CrossRef]
- Zhao, L.; Semetey, V. Recycling Polyurethanes through Transcarbamoylation. ACS Omega 2021, 6, 4175–4183. [Google Scholar] [CrossRef] [PubMed]
- Beran, R.; Zarybnicka, L.; Machova, D. Recycling of rigid polyurethane foam: Micro-milled powder used as active filler in polyurethane adhesives. J. Appl. Polym. Sci. 2020, 137, 49095. [Google Scholar] [CrossRef]
- Howard, G.T. Biodegradation of polyurethane: A review. Int. Biodeterior. Biodegrad. 2002, 49, 245–252. [Google Scholar] [CrossRef]
- Cregut, M.; Bedas, M.; Durand, M.; Thouand, G. New insights into polyurethane biodegradation and realistic prospects for the development of a sustainable waste recycling process. Biotechnol. Adv. 2013, 31, 1634–1647. [Google Scholar] [CrossRef]
- Offenbach, J.A.; Tobolsky, A.V. Chemical relaxation of stress in polyurethane elastomers. J. Colloid Sci. 1956, 11, 39–47. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef]
- Zheng, J.; Png, Z.M.; Ng, S.H.; Tham, G.X.; Ye, E.; Goh, S.S.; Loh, X.J.; Li, Z. Vitrimers: Current research trends and their emerging applications. Mater. Today 2021, 51, 586–625. [Google Scholar] [CrossRef]
- Nellepalli, P.; Patel, T.; Oh, J.K. Dynamic Covalent Polyurethane Network Materials: Synthesis and Self-Healability. Macromol. Rapid Commun. 2021, 42, 2100391. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Xu, Y.; Zhao, Q.; Xie, T. Dynamic Covalent Polymer Networks: A Molecular Platform for Designing Functions beyond Chemical Recycling and Self-Healing. Chem. Rev. 2021, 121, 1716–1745. [Google Scholar] [CrossRef]
- Kloxin, C.J.; Scott, T.F.; Adzima, B.J.; Bowman, C.N. Covalent adaptable networks (CANs): A unique paradigm in cross-linked polymers. Macromolecules 2010, 43, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Elling, B.R.; Dichtel, W.R. Reprocessable Cross-Linked Polymer Networks: Are Associative Exchange Mechanisms Desirable? ACS Cent. Sci. 2020, 6, 1488–1496. [Google Scholar] [CrossRef]
- Yu, K.; Shi, Q.; Li, H.; Jabour, J.; Yang, H.; Dunn, M.L.; Wang, T.; Qi, H.J. Interfacial welding of dynamic covalent network polymers. J. Mech. Phys. Solids 2016, 94, 1–17. [Google Scholar] [CrossRef]
- Snyder, R.L.; Fortman, D.J.; De Hoe, G.X.; Hillmyer, M.A.; Dichtel, W.R. Reprocessable Acid-Degradable Polycarbonate Vitrimers. Macromolecules 2018, 51, 389–397. [Google Scholar] [CrossRef]
- Yue, L.; Bonab, V.S.; Yuan, D.; Patel, A.; Karimkhani, V.; Manas-Zloczower, I. Vitrimerization: A Novel Concept to Reprocess and Recycle Thermoset Waste via Dynamic Chemistry. Glob. Chall. 2019, 3, 1800076. [Google Scholar] [CrossRef]
- Shi, J.; Zheng, T.; Guo, B.; Xu, J. Solvent-free thermo-reversible and self-healable crosslinked polyurethane with dynamic covalent networks based on phenol-carbamate bonds. Polymer 2019, 181, 121788. [Google Scholar] [CrossRef]
- Chen, X.; Hu, S.; Li, L.; Torkelson, J.M. Dynamic Covalent Polyurethane Networks with Excellent Property and Cross-Link Density Recovery after Recycling and Potential for Monomer Recovery. Appl. Polym. Mater. 2020, 2, 2093–2101. [Google Scholar] [CrossRef]
- Swartz, J.L.; Sheppard, D.T.; Haugstad, G.; Dichtel, W.R. Blending Polyurethane Thermosets Using Dynamic Urethane Exchange. Macromolecules 2021, 54, 11126–11133. [Google Scholar] [CrossRef]
- Shi, J.; Zheng, T.; Zhang, Y.; Guo, B.; Xu, J. Polymer Chemistry phenol-carbamate bonds: Properties affected by. Polym. Chem. 2021, 12, 2421–2432. [Google Scholar] [CrossRef]
- Li, J.; Ning, Z.; Yang, W.; Yang, B.; Zeng, Y. Hydroxyl-Terminated Polybutadiene-Based Polyurethane with Self-Healing and Reprocessing Capabilities. ACS Omega 2022, 7, 10156–10166. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Li, J.; Hu, C.; Yang, B.; Ning, Z. Sustainable Polyurethane Networks with High Self-Healing and Mechanical Properties Based on Dual Dynamic Covalent Bonds. Macromol. Chem. Phys. 2023, 224, 2200322. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Mechanically Robust.Self-Healable.and Highly Stretchable “Living” Crosslinked Polyurethane Based on a Reversible C-C Bond. Adv. Funct. Mater. 2018, 28, 1706050. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, Y.; Li, J.; Zhang, K. Catalyst-free room-temperature self-healing polymer networks based on dynamic covalent quinone methide-secondary amine chemistry. Polym. Chem. 2021, 12, 6161–6166. [Google Scholar] [CrossRef]
- Yan, P.; Zhao, W.; Fu, X.; Liu, Z.; Kong, W.; Zhou, C.; Lei, J. Multifunctional polyurethane-vitrimers completely based on transcarbamoylation of carbamates: Thermally-induced dual-shape memory effect and self-welding†. RSC Adv. 2017, 7, 26858–26866. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, C.; Song, F.; Shang, Q.; Hu, Y.; Jia, P. Castor-oil-based, robust, self-healing, shape memory, and reprocessable polymers enabled by dynamic hindered urea bonds and hydrogen bonds. Chem. Eng. J. 2022, 429, 131848. [Google Scholar] [CrossRef]
- Xie, D.; Lu, D.; Zhao, X.; Li, Y.; Zeng, J. Sustainable and malleable polyurethane networks from castor oil and vanillin with tunable mechanical properties. Ind. Crops Prod. 2021, 174, 114198. [Google Scholar] [CrossRef]
- Shi, J.; Zheng, T.; Zhang, Y.; Guo, B.; Xu, J. Reprocessable Cross-Linked Polyurethane with Dynamic and Tunable Phenol–Carbamate Network. Sustain. Chem. Eng. 2020, 8, 1207–1218. [Google Scholar] [CrossRef]
- Zhang, C.; Liang, H.; Liang, D.; Lin, Z.; Chen, Q.; Feng, P.; Wang, Q. Renewable Castor-Oil-based Waterborne Polyurethane Networks: Simultaneously Showing High Strength, Self-Healing, Processability and Tunable Multishape Memory. Angew. Chem. Int. Ed. 2021, 60, 4289–4299. [Google Scholar] [CrossRef]
- Liang, R.; Zhang, H.; Wang, Y.; Ye, J.; Guo, L.; He, L.; Li, X.; Qiu, T.; Tuo, X. Dual dynamic network system constructed by waterborne polyurethane for improved and recoverable performances. Chem. Eng. J. 2022, 442, 136204. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Yao, W.; Dong, Y.; Zhang, B.; Dong, X.; Fang, H.; Zhang, G.; Ding, Y. Dynamically Cross-Linked Waterborne Polyurethanes: Transalkylation Exchange of C-N Bonds toward High Performance and Reprocessable Thermosets. ACS Appl. Polym. Mater. 2022, 4, 5920–5926. [Google Scholar] [CrossRef]
- Li, J.; Yang, W.; Ning, Z.; Yang, B.; Zeng, Y. Sustainable Polyurethane Networks Based on Rosin with Reprocessing Performance. Polymers 2021, 13, 3538. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cha, I.; Yoon, Y.; Cha, B.J.; Yang, J.; Kim, Y.D.; Song, C. Facile Mechanochemical Synthesis of Malleable Biomass-Derived Network Polyurethanes and Their Shape-Memory Applications. Sustain. Chem. Eng. 2021, 9, 2952–6961. [Google Scholar] [CrossRef]
- Wen, Z.; Raquez, J. Catalyst-free reprocessable crosslinked biobased polybenzoxazine-polyurethane based on dynamic carbamate chemistry. J. Appl. Polym. Sci. 2022, 139, 52120. [Google Scholar] [CrossRef]
- Li, Q.; Ma, S.; Li, P.; Wang, B.; Feng, H.; Lu, N.; Wang, S.; Liu, Y.; Xu, X.; Zhu, J. Biosourced Acetal and Diels-Alder Adduct Concurrent Polyurethane Covalent Adaptable Network. Macromolecules 2021, 54, 1742–1753. [Google Scholar] [CrossRef]
- Sun, Y.; Sheng, D.; Wu, H.; Tian, X.; Xie, H.; Shi, B. Bio-based vitrimer-like polyurethane based on dynamic imine bond with antibacterial ability. Polymer 2021, 233, 124208. [Google Scholar] [CrossRef]
- Debnath, S.; Tiwary, S.K.; Ojha, U. Dynamic Carboxylate Linkage Based Reprocessable and Self-Healable Segmented Polyurethane Vitrimers Displaying Creep Resistance Behavior and Triple Shape Memory Ability. Appl. Polym. Mater. 2021, 3, 2166–2177. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, X.Y.; Zheng, M.; Moorlag, C.; Yang, J.; Wang, Z.L. 3D printing of thermoreversible polyurethanes with targeted shape memory and precise in situ self-healing properties. J. Mater. Chem. A Mater. 2019, 7, 6972–6984. [Google Scholar] [CrossRef]
- Lu-Ping, W.; Ming-Guang, Z.; Jing-Cheng, H.; Xu, W. Insertion of Supramolecular Segments into Covalently Crosslinked Polyurethane Networks towards the Fabrication of Recyclable. Chin. J. Polym. Sci. 2022, 40, 321–330. [Google Scholar] [CrossRef]
- Hu, S.; He, S.; Wang, Y.; Wu, Y.; Shou, T.; Yin, D. Self-repairable.recyclable and heat-resistant polyurethane for high-performance automobile tires. Nano Energy 2022, 95, 107012. [Google Scholar] [CrossRef]
- Jia, Y.; Guan, Q.; Zhang, L.; Neisiany, R.E.; Yan, N.; Li, Y.; You, Z. A fluorine-rich phenolic polyurethane elastomer with excellent self-healability and reprocessability and its applications for wearable electronics. Sci. China Mater. 2022, 65, 2553–2564. [Google Scholar] [CrossRef]
- Dong, J.; Yan, H.; Lv, X.; Wang, Z.; Rao, Z.; Zhu, B.; Wu, J.; Zhou, Y.; Chen, H. Reprocessable polyurethane elastomers based on reversible ketal exchange: Dielectric properties and water resistance. J. Mater. Chem. C Mater. 2022, 11, 1369–1380. [Google Scholar] [CrossRef]
- Xu, B.; Yin, Q.; Han, F.; Cheng, J.; Zhao, J.; Zhang, J. A Bio-based healable/renewable polyurethane elastomer derived from L-Tyrosine/Vanillin/Dimer acid. Chem. Eng. Sci. 2022, 258, 117736. [Google Scholar] [CrossRef]
- Sheppard, D.T.; Jin, K.; Hamachi, L.S.; Dean, W.; Fortman, D.J.; Fortman, D.J.; Ellison, C.J.; Dichtel, W.R. Reprocessing Postconsumer Polyurethane Foam Using Carbamate Exchange Catalysis and Twin-Screw Extrusion. ACS Cent. Sci. 2020, 6, 921–927. [Google Scholar] [CrossRef]
- Wang, X.; Lu, M.; Zeng, J.; Weng, Y.; Li, Y. Malleable and thermally recyclable polyurethanefoam. Green. Chem. 2021, 23, 307–313. [Google Scholar] [CrossRef]
- Liu, W.; Chang, Y.C.; Hao, C.; Liu, H.; Zhang, J.; Mielewski, D.; Kiziltas, A. Improving Thermal Reprocessability of Commercial Flexible Polyurethane Foam by Vitrimer Modification of the Hard Segments. ACS Appl. Polym. Mater. 2022, 4, 5056–5067. [Google Scholar] [CrossRef]
- Colodny, P.C.; Tobolsky, A.V. Chemorheological Study of Polyurethan Elastomers. J. Am. Chem. Soc. 1957, 79, 4320–4323. [Google Scholar] [CrossRef]
- Hu, S.; Chen, X.; Torkelson, J.M. Biobased Reprocessable Polyhydroxyurethane Networks: Full Recovery of Crosslink Density with Three Concurrent Dynamic Chemistries. ACS Sustain. Chem. Eng. 2019, 7, 10025–10034. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Cramer, C.J.; Hillmyer, M.A.; Dichtel, W.R. Mechanically Activated, Catalyst-Free Polyhydroxyurethane Vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022. [Google Scholar] [CrossRef]
- Fortman, D.J.; Brutman, J.P.; Hillmyer, M.A.; Dichtel, W.R. Structural effects on the reprocessability and stress relaxation of crosslinked polyhydroxyurethanes. J. Appl. Polym. Sci. 2017, 134, 44984. [Google Scholar] [CrossRef]
- Fortman, D.J.; Snyder, R.L.; Sheppard, D.T.; Dichtel, W.R. Rapidly Reprocessable Cross-Linked Polyhydroxyurethanes Based on Disulfide Exchange. ACS Macro Lett. 2018, 7, 1226–1231. [Google Scholar] [CrossRef]
- Zhou, Z.; Zeng, Y.; Yu, C.; Li, Q.; Zhang, F. Intrinsically self-healing and stretchy poly(urethane-urea) elastomer based on dynamic urea bonds and thiol-ene click reaction. Mater. Chem. Phys. 2021, 267, 124642. [Google Scholar] [CrossRef]
- Erice, A.; de Luzuriaga, A.R.; Matxain, J.M.; Ruipérez, F.; Asua, J.M.; Grande, H.J.; Rekondo, A. Reprocessable and recyclable crosslinked poly(urea-urethane)s based on dynamic amine/urea exchange. Polymer 2018, 145, 127–136. [Google Scholar] [CrossRef]
- Fang, Z.; Zheng, N.; Zhao, Q.; Xie, T. Healable, Reconfigurable, Reprocessable Thermoset Shape Memory Polymer with Highly Tunable Topological Rearrangement Kinetics. ACS Appl. Mater. Interfaces 2017, 9, 22077–22082. [Google Scholar] [CrossRef]
- Wang, X.-Z.; Xie, D.-M.; Zhao, X.-L.; Li, Y.-D.; Zeng, J.-B. Sustainable, Malleable, and Recyclable Castor Oil-Derived Poly(urethane urea) Networks with Tunable Mechanical Properties and Shape Memory Performance Based on Dynamic Piperazine–Urea Bonds. Macromolecules 2022, 55, 2243–2251. [Google Scholar] [CrossRef]
- Zhang, J.; Shang, Q.; Hu, Y.; Zhu, G.; Huang, J.; Yu, X.; Cheng, J.; Liu, C.; Chen, J.; Feng, G.; et al. Castor-oil-based UV-curable hybrid coatings with self-healing, recyclability, removability, and hydrophobicity. Prog. Org. Coat. 2022, 165, 106742. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, Y.; Zheng, Z.; Ding, X. Reconfigurable and Reprocessable Thermoset Shape Memory Polymer with Synergetic Triple Dynamic Covalent Bonds. Macromol. Rapid Commun. 2018, 39, 1800128. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Y.; Ying, H.; Jing, X.; Wang, B.; Zhang, Y.; Cheng, J. Recyclable, self-healable, and highly malleable poly(urethane-urea)s with improved thermal and mechanical performances. ACS Appl. Mater. Interfaces 2020, 12, 35403–35414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, C.; Tan, J.; Wang, M.; Liu, Z.; Ren, Y.; Xue, Y.; Zhang, Q. Double Modification of Poly(urethane-urea): Toward Healable, Tear-Resistant, and Mechanically Robust Elastomers for Strain Sensors. ACS Appl. Mater. Interfaces 2022, 15, 2134–2146. [Google Scholar] [CrossRef]
- Jaffrennou, B.; Droger, N.; Méchin, F.; Halary, J.L.; Pascault, J.P. Characterization, structural transitions and properties of a tightly crosslinked polythiourethane network for optical applications. E-Polymers 2005, 5, 081. [Google Scholar] [CrossRef]
- Gamardella, F.; Ramis, X.; De la Flor, S.; Serra, À. Preparation of poly(thiourethane) thermosets by controlled thiol-isocyanate click reaction using a latent organocatalyst. React. Funct. Polym. 2019, 134, 174–182. [Google Scholar] [CrossRef]
- Yue, H.; Zhou, J.; Huang, M.; Hao, C.; Hao, R.; Dong, C.; He, S.; Liu, H.; Liu, W.; Zhu, C. Recyclable, reconfigurable, thermadapt shape memory polythiourethane networks with multiple dynamic bonds for recycling of carbon fiber-reinforced composites. Polymer 2021, 237, 124358. [Google Scholar] [CrossRef]
- Gamardella, F.; De la Flor, S.; Ramis, X.; Serra, A. Recyclable poly(thiourethane) vitrimers with high Tg. Influence of the isocyanate structure. React. Funct. Polym. 2020, 151, 104574. [Google Scholar] [CrossRef]
- Li, C.; Li, H.; Tan, J.; Xue, Y.; Liu, Q.; Yang, Y.; Wang, C.; Zhang, Q. Constructing segregated thermoset composite via Pickering emulsion and dynamic polythiourethanes. Compos. Sci. Technol. 2022, 218, 109215. [Google Scholar] [CrossRef]
- Wen, Z.; Han, X.; Fairbanks, B.D.; Yang, K.; Bowman, C.N. Development of thiourethanes as robust, reprocessable networks. Polymer 2020, 202, 122715. [Google Scholar] [CrossRef]
- Li, L.; Chen, X.; Torkelson, J.M. Reprocessable Polymer Networks via Thiourethane Dynamic Chemistry: Recovery of Cross-link Density after Recycling and Proof-of-Principle Solvolysis Leading to Monomer Recovery. Macromolecules 2019, 52, 8207–8216. [Google Scholar] [CrossRef]
- Fan, C.J.; Wen, Z.B.; Xu, Z.Y.; Xiao, Y.; Wu, D.; Yang, K.K.; Wang, Y.Z. Adaptable Strategy to Fabricate Self-Healable and Reprocessable Poly(thiourethane-urethane) Elastomers via Reversible Thiol-Isocyanate Click Chemistry. Macromolecules 2020, 53, 4284–4293. [Google Scholar] [CrossRef]
- Cui, C.; Chen, X.; Ma, L.; Zhong, Q.; Li, Z.; Mariappan, A.; Zhang, Q.; Cheng, Y.; He, G.; Chen, X.; et al. Polythiourethane covalent adaptable networks for strong and reworkable adhesives and fully recyclable carbon fiber-reinforced composites. ACS Appl. Mater. Interfaces 2020, 12, 47975–47983. [Google Scholar] [CrossRef]
- Erice, A.; de Luzuriaga, A.R.; Azcune, I.; Fernandez, M.; Calafel, I.; Grande, H.J.; Rekondo, A. New injectable and self-healable thermoset polythiourethane based on S-aromatic thiourethane dissociative exchange mechanism. Polymer 2020, 196, 122461. [Google Scholar] [CrossRef]
- Zhao, B.; Mei, H.; Hang, G.; Li, L.; Zheng, S. Polyurethanes Reinforced with Polyethylene Nanocrystals: Synthesis, Triple Shape Memory, and Reprocessing Properties. Macromolecules 2022, 55, 4076–4090. [Google Scholar] [CrossRef]
- Yang, S.; Wang, S.; Du, X.; Cheng, X.; Wang, H.; Du, Z. Polymer Chemistry inorganic—Organic hybrid material as crosslinker. Polym. Chem. 2020, 11, 1161–1170. [Google Scholar] [CrossRef]
- Zhao, B.; Mei, H.; Hang, G.; Li, L.; Zheng, S. Shape recovery and reprocessable polyurethanes crosslinked with double decker silsesquioxane via Diels-Alder reaction. Polymer 2021, 230, 124042. [Google Scholar] [CrossRef]
- Li, L.; Zhao, B.; Wang, H.; Gao, Y.; Hu, J.; Zheng, S. Nanocomposites of polyhydroxyurethane with Fe3O4 nanoparticles: Synthesis, shape memory and reprocessing properties. Compos. Sci. Technol. 2021, 215, 109009. [Google Scholar] [CrossRef]
- Du, W.; Jin, Y.; Shi, L.; Shen, Y.; Lai, S. NIR-light-induced thermoset shape memory polyurethane composites with self-healing and recyclable functionalities. Compos. Part B Eng. 2020, 195, 108092. [Google Scholar] [CrossRef]
- Qi, G.; Yang, W.; Puglia, D.; Wang, H.; Xu, P.; Dong, W.; Zheng, T.; Ma, P. Hydrophobic.UV resistant and dielectric polyurethane-nanolignin composites with good reprocessability. Mater. Des. 2020, 196, 109150. [Google Scholar] [CrossRef]
- Huang, J.; Wang, H.; Liu, W.; Huang, J.; Yang, D.; Qiu, X.; Zhao, L.; Hu, F.; Feng, Y. Solvent-free synthesis of high-performance polyurethane elastomer based on low-molecular-weight alkali lignin. Int. J. Biol. Macromol. 2023, 225, 1505–1516. [Google Scholar] [CrossRef]
- Li, X.; Chen, X.; Zhang, S.; Yin, Y.; Wang, C. UV-resistant transparent lignin-based polyurethane elastomer with repeatable processing performance. Eur. Polym. J. 2021, 159, 110763. [Google Scholar] [CrossRef]
- Wang, H.; Huang, J.; Liu, W.; Huang, J.; Yang, D.; Qiu, X.; Zhang, J. Tough and Fast Light-Controlled Healable Lignin-Containing Polyurethane Elastomers. Macromolecules 2022, 55, 8629–8641. [Google Scholar] [CrossRef]
- Zhao, W.; Liang, Z.; Feng, Z.; Xue, B.; Xiong, C.; Duan, C.; Ni, Y. New Kind of Lignin/Polyhydroxyurethane Composite: Green Synthesis, Smart Properties, Promising Applications, and Good Reprocessability and Recyclability. ACS Appl. Mater. Interfaces 2021, 13, 28938–28948. [Google Scholar] [CrossRef]
- Ge, W.; Zhao, B.; Li, L.; Nie, K.; Zheng, S. Nanocomposites of polyhydroxyurethane with nanocrystalline cellulose: Synthesis, thermomechanical and reprocessing properties. Eur. Polym. J. 2021, 149, 110287. [Google Scholar] [CrossRef]
- Gao, W.; Bie, M.; Quan, Y.; Zhu, J.; Zhang, W. Self-healing, reprocessing and sealing abilities of polysulfide-based polyurethane. Polymer 2018, 151, 27–33. [Google Scholar] [CrossRef]
- Feng, H.; Zheng, N.; Peng, W.; Ni, C.; Song, H.; Zhao, Q.; Xie, T. Upcycling of dynamic thiourea thermoset polymers by intrinsic chemical strengthening. Nat. Commun. 2022, 13, 397. [Google Scholar] [CrossRef]
- Lei, Y.; Fu, X.; Jiang, L.; Liu, Z.; Lei, J. Oxime–Urethane Structure-Based Dynamically Crosslinked Polyurethane with Robust Reprocessing Properties. Macromol. Rapid Commun. 2022, 43, 2100781. [Google Scholar] [CrossRef]
- Liu, W.X.; Zhang, C.; Zhang, H.; Zhao, N.; Yu, Z.X.; Xu, J. Oxime-Based and Catalyst-Free Dynamic Covalent Polyurethanes. J. Am. Chem. Soc. 2017, 139, 8678–8684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Long, J.; Jia, L.; Gao, Q.; Fan, H.; Xiang, J. Self-healing and reprocess of crosslinked polyurethane based on dynamic oxime-carbamate bond. J. Appl. Polym. Sci. 2023, 140, e53478. [Google Scholar] [CrossRef]
- Liu, W.; Ge, W.; Mei, H.; Hang, G.; Li, L.; Zheng, S. Poly(hydroxyurethane-co-thiourethane)s cross-linked with disulfide bonds: Synthesis via isocyanate-free approach, thermomechanical and reprocessing properties. J. Polym. Sci. 2022, 60, 2756–2768. [Google Scholar] [CrossRef]
- Lagron, A.B.; El-Zaatari, B.M.; Hamachi, L.S. Characterization Techniques to Assess Recyclability in Dynamic Polymer Networks. Front. Mater. 2022, 9, 915296. [Google Scholar] [CrossRef]
- Namathoti, S.; Ravindra, R.K.; Rama, R.S. A review on progress in magnetic, microwave, ultrasonic responsive Shape-memory polymer composites. Mater. Today Proc. 2022, 56, 1182–1191. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miravalle, E.; Bracco, P.; Brunella, V.; Barolo, C.; Zanetti, M. Improving Sustainability through Covalent Adaptable Networks in the Recycling of Polyurethane Plastics. Polymers 2023, 15, 3780. https://doi.org/10.3390/polym15183780
Miravalle E, Bracco P, Brunella V, Barolo C, Zanetti M. Improving Sustainability through Covalent Adaptable Networks in the Recycling of Polyurethane Plastics. Polymers. 2023; 15(18):3780. https://doi.org/10.3390/polym15183780
Chicago/Turabian StyleMiravalle, Edoardo, Pierangiola Bracco, Valentina Brunella, Claudia Barolo, and Marco Zanetti. 2023. "Improving Sustainability through Covalent Adaptable Networks in the Recycling of Polyurethane Plastics" Polymers 15, no. 18: 3780. https://doi.org/10.3390/polym15183780
APA StyleMiravalle, E., Bracco, P., Brunella, V., Barolo, C., & Zanetti, M. (2023). Improving Sustainability through Covalent Adaptable Networks in the Recycling of Polyurethane Plastics. Polymers, 15(18), 3780. https://doi.org/10.3390/polym15183780