


Viscoelastic Properties of Polypropylene during Crystallization and Melting: Experimental and Phenomenological Modeling


Abstract
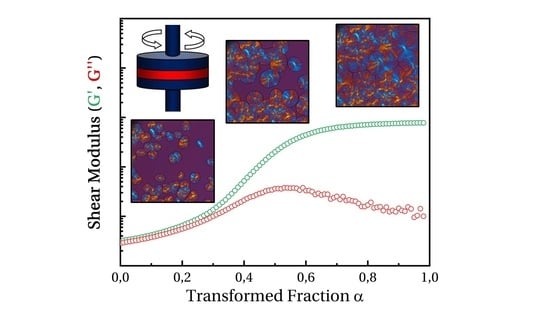
:
1. Introduction
- The first is the transformed volume fraction, which is related to the amount of crystalline phase.
- The second is related to the number of spherulites.
- The third is related to the average radius of spherulites over time.
- The fourth is chosen to assess the level of impingement of spherulites.
- Finally, these parameters were correlated to the rheological measurements and a phenomenological model was proposed and validated. Therefore, storage and loss moduli could be reproduced during melting–crystallization cycles.
2. Materials and Methods
2.1. Materials
2.2. Differential Scanning Calorimetry
2.3. Polarized Light Microscopy
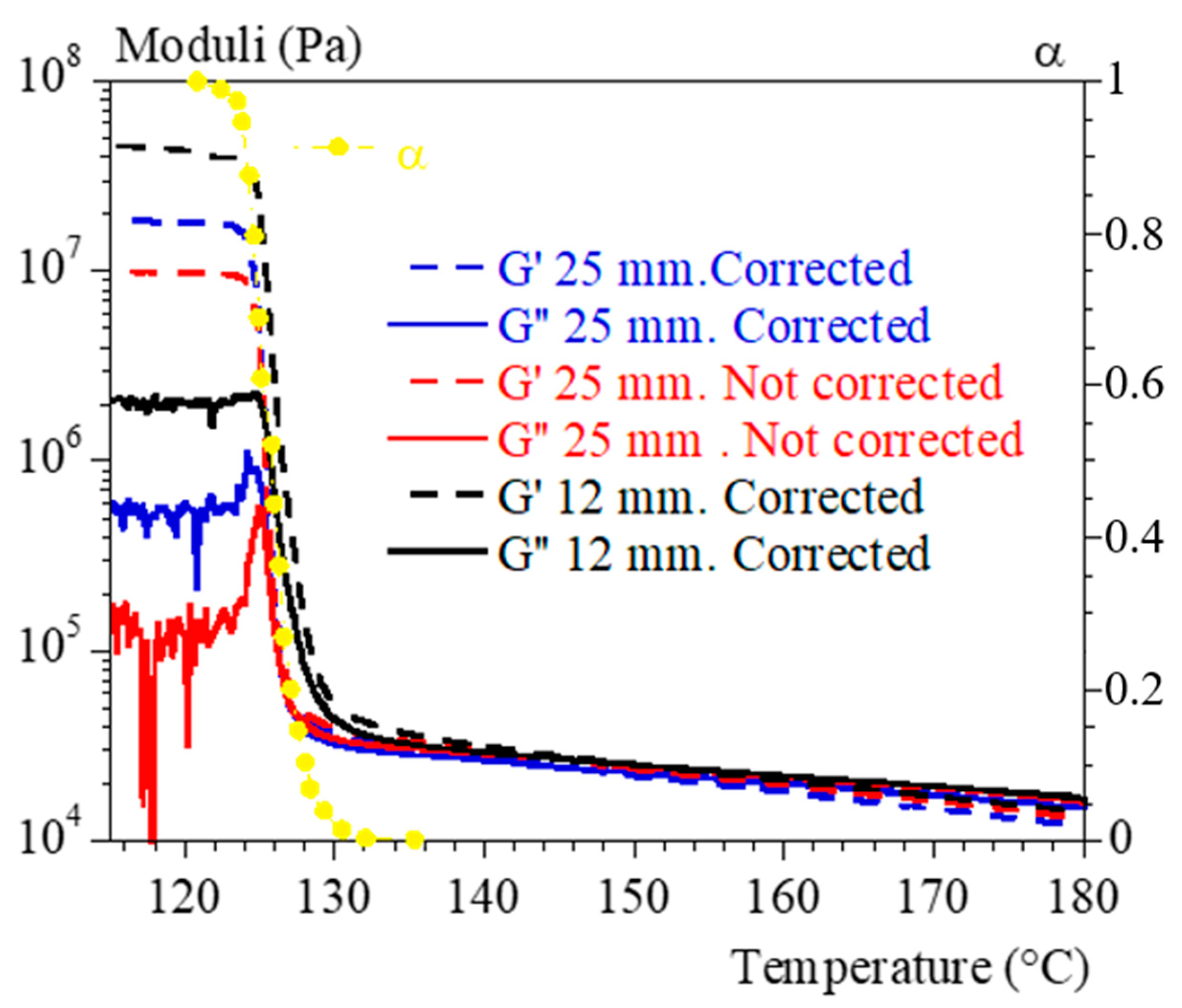
2.4. Rheological Analyses
3. Theoretical
3.1. State of the Art
3.2. Overall Crystallization Kinetics
- In the molten phase, some nuclei are activated according to nucleation kinetics, i.e., a probability per unit of time that one of them will become a growing entity.
- Growth starts at once, without delay.
- Only nuclei that are in a liquid region can be activated.
- Entities stop growing when they touch, i.e., no overlapping is allowed.
- All the entities grow at the same rate in all accessible directions (spheres in the space, disk on a plane, etc.).
- Total volume stays constant (isovolumic assumption).
3.3. Microstructure Description
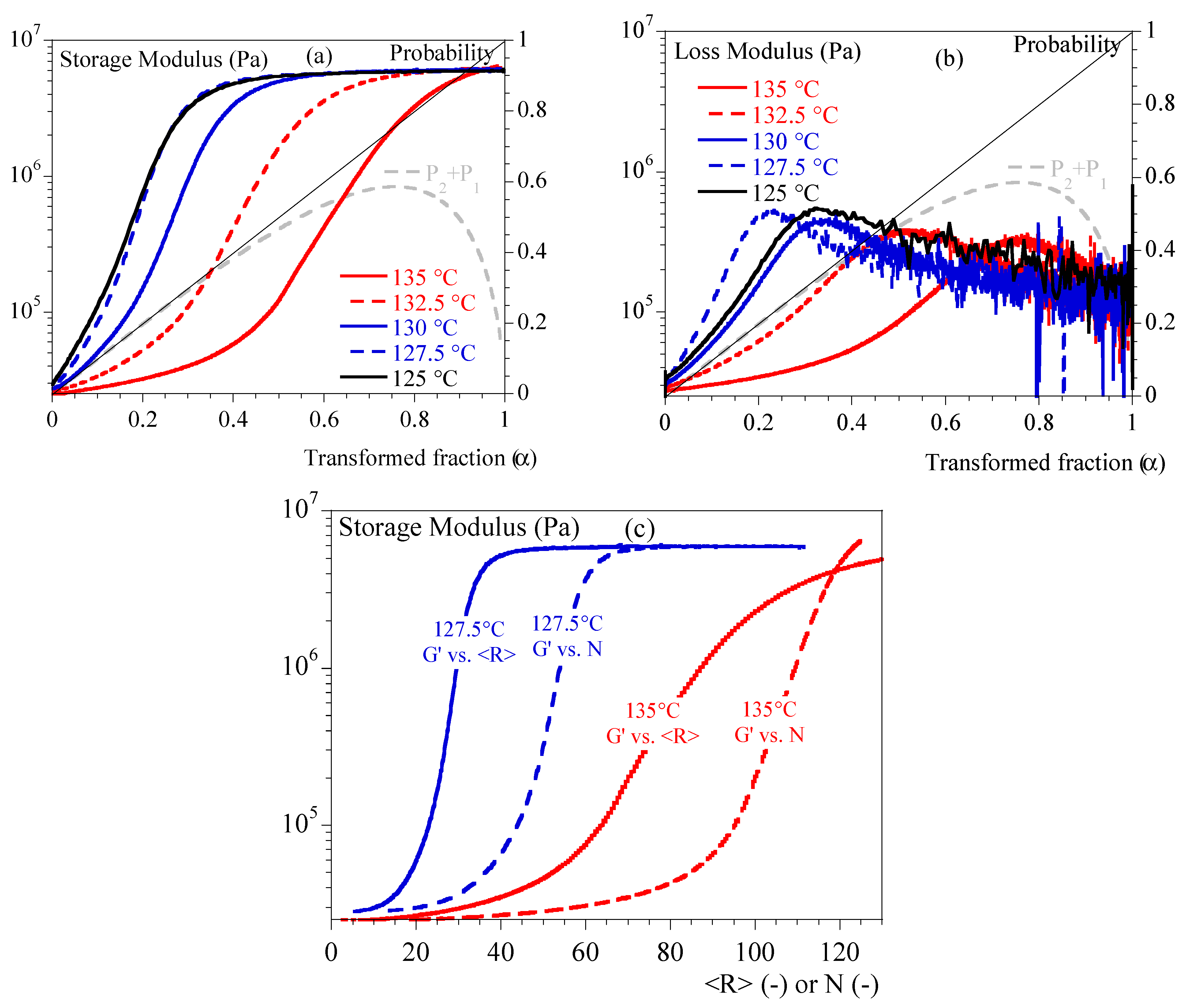
- Below 10 to 15% transformed volume fraction (), crystallization results mainly from the growth of statistically individual spherulites. During that period, the microstructure can be seen as individual semi-crystalline spheres per unit volume in a soft matrix. The largest radius is R. The size distribution and the average radius depend on nucleation kinetics.
- From 15 to 40%, impingement of spherulites cannot be neglected. Obviously, Poisson’s analysis does not allow evaluation of the number of impingements. It only allows one to say that it is probable that impingements had occurred. Then, microstructure results from single spherulites and isolated aggregates.
- From 40%, presumably, coalescence takes place. Progressively, the microstructure turns into a solid “skeleton” with embedded liquid pockets.
4. Results and Discussion
4.1. Methodology
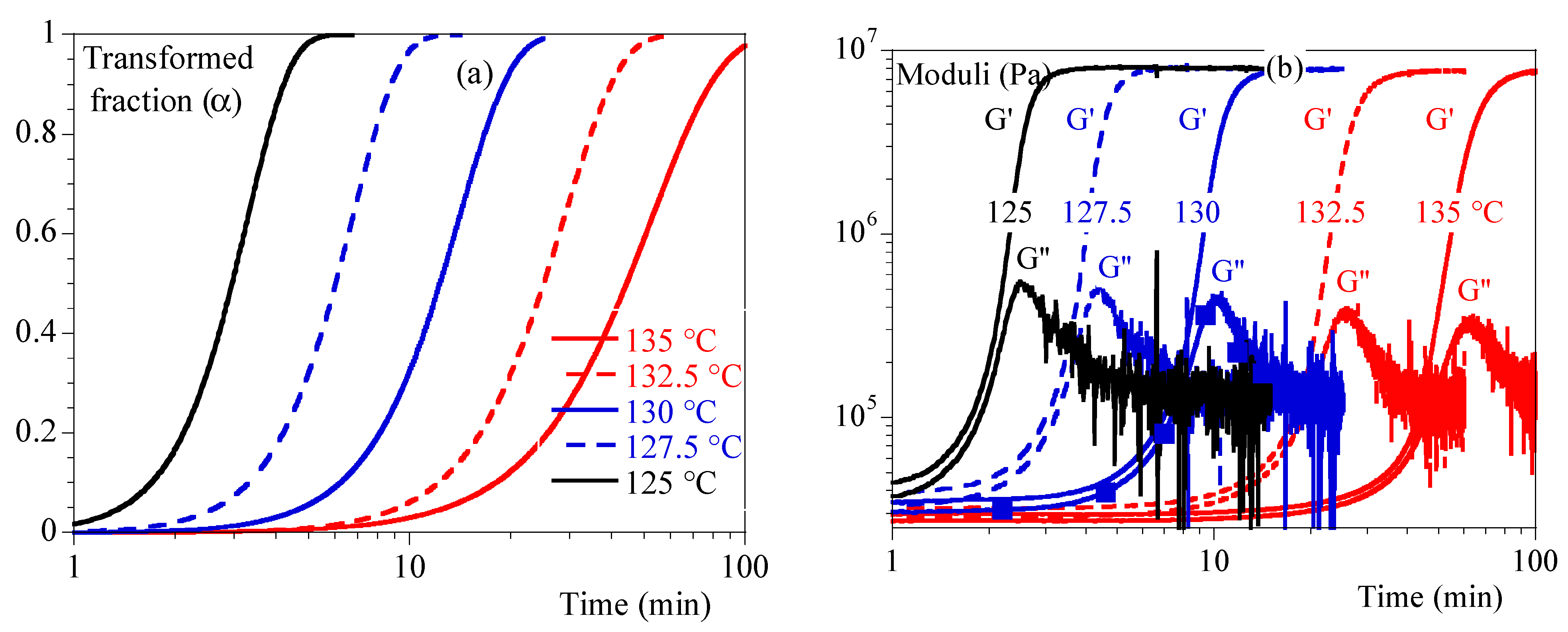
- Second, the isothermal crystallizations were performed using DSC and the transformed volume fraction, , was recorded as a function of time and temperature.
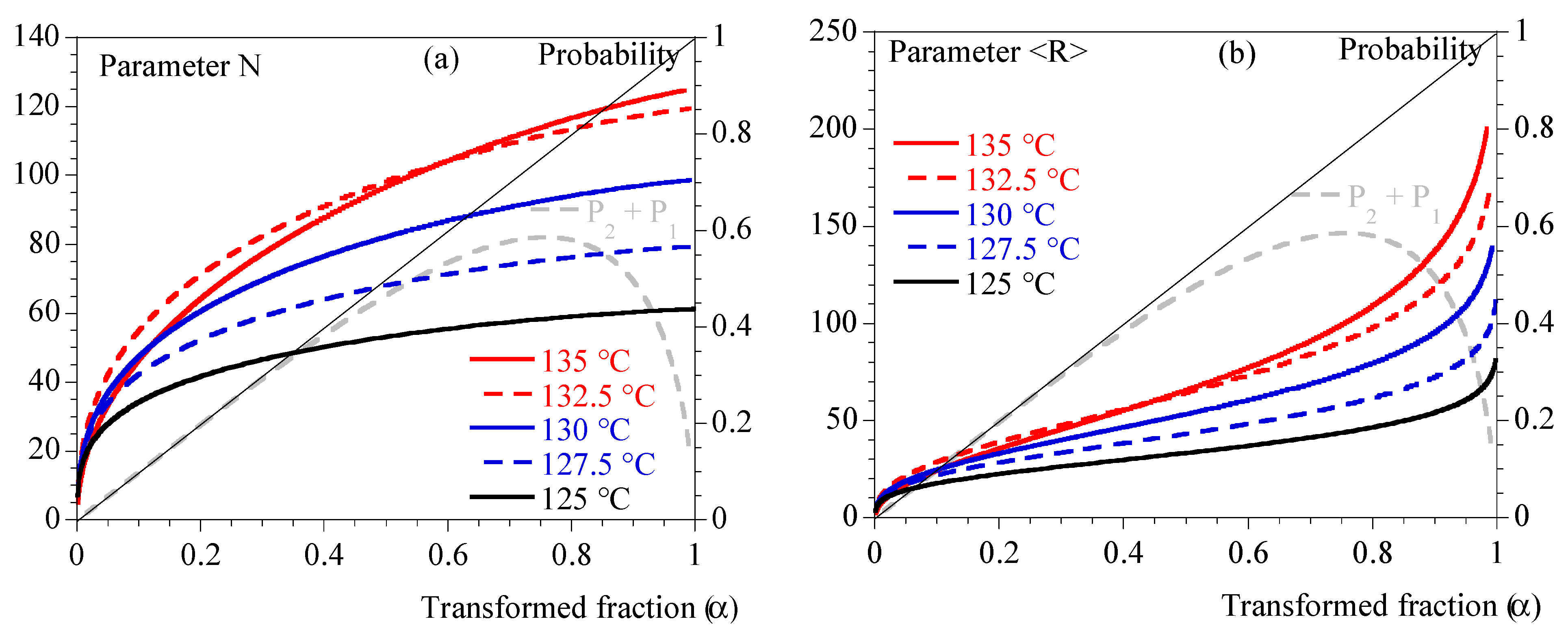
- Third, the parameters, N (Equation (12)) and <R> (Equation (15)) were calculated thanks to numerical integration. For their parts, and were deduced from .
- Fourth, isothermal rheological measurements were correlated to the parameters (see section below). A model was deduced. At this stage, storage and loss moduli were considered independently.
- Fifth, Avrami’s kinetics parameter and exponent (i.e., and n) were deduced from . n was averaged at 3 and was estimated from and (Equations (10) and (11)). Results are presented in Table 1.
- Finally, was averaged at 1.96 × 10−6 µm−3 min3 to model crystallization under a constant cooling rate. Both the rheology and crystallization models were merged to assess extrapolation of results to predict rheological measurements carried out under constant cooling and heating rates.
4.2. Rheology in Isothermal Conditions
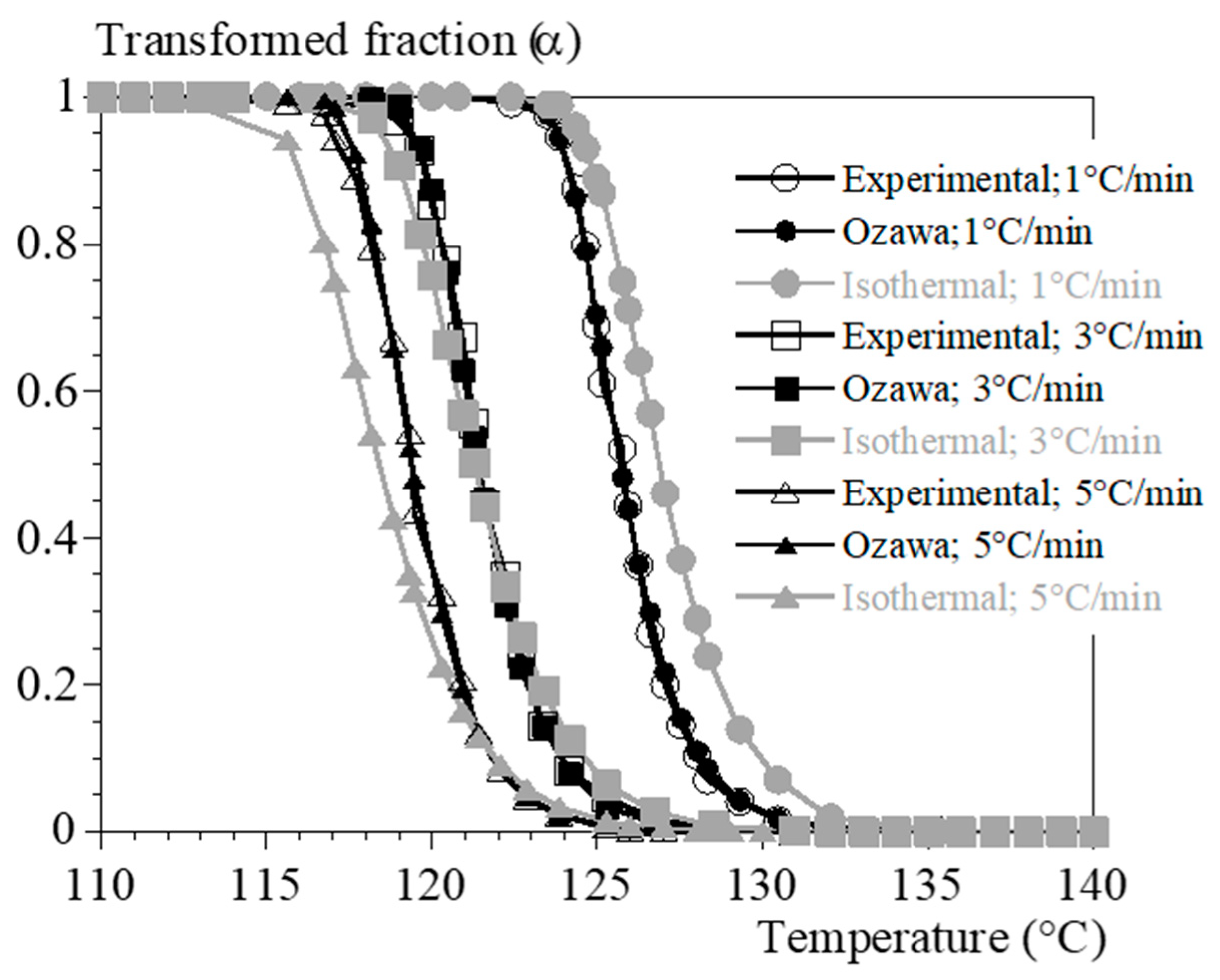
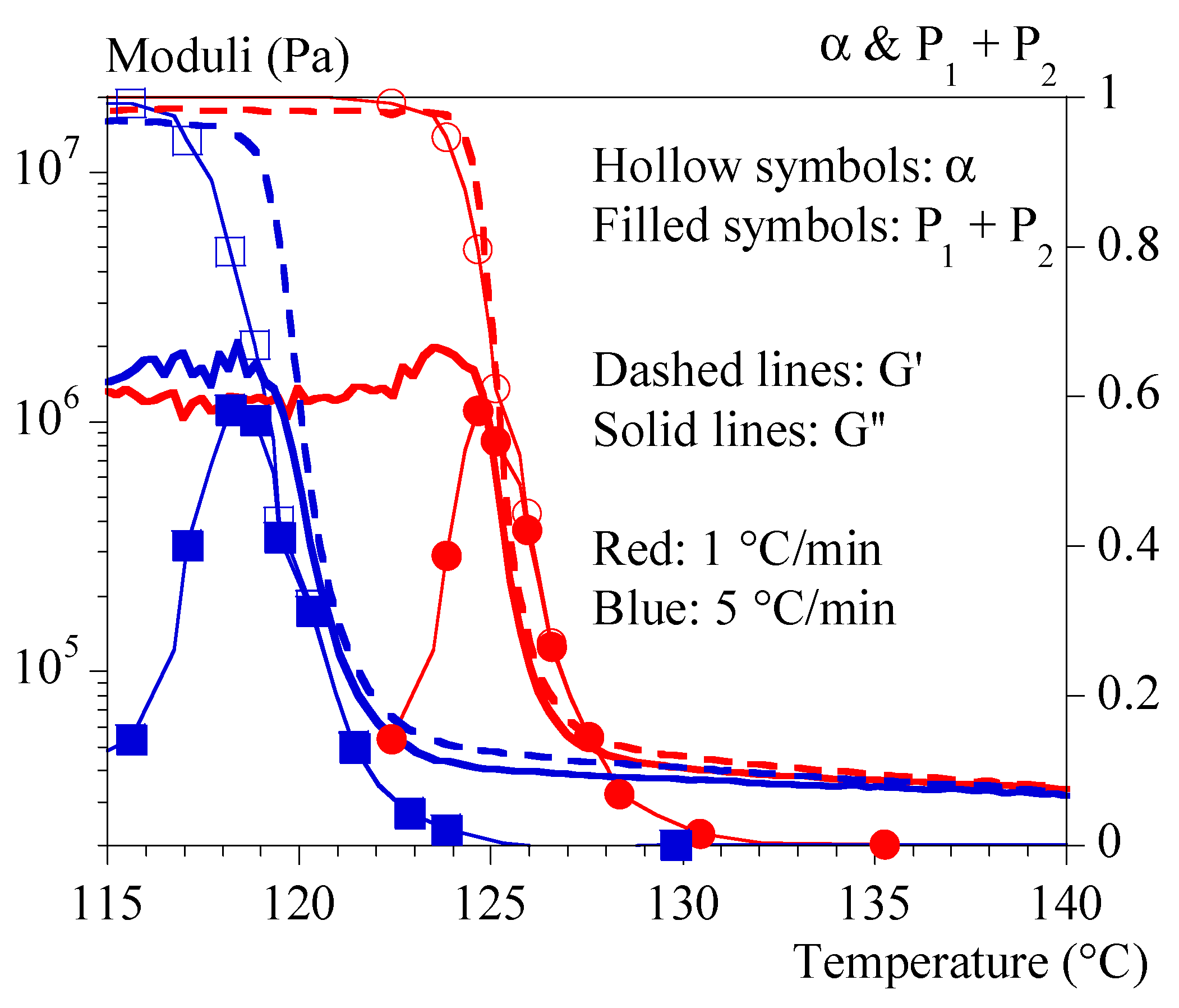
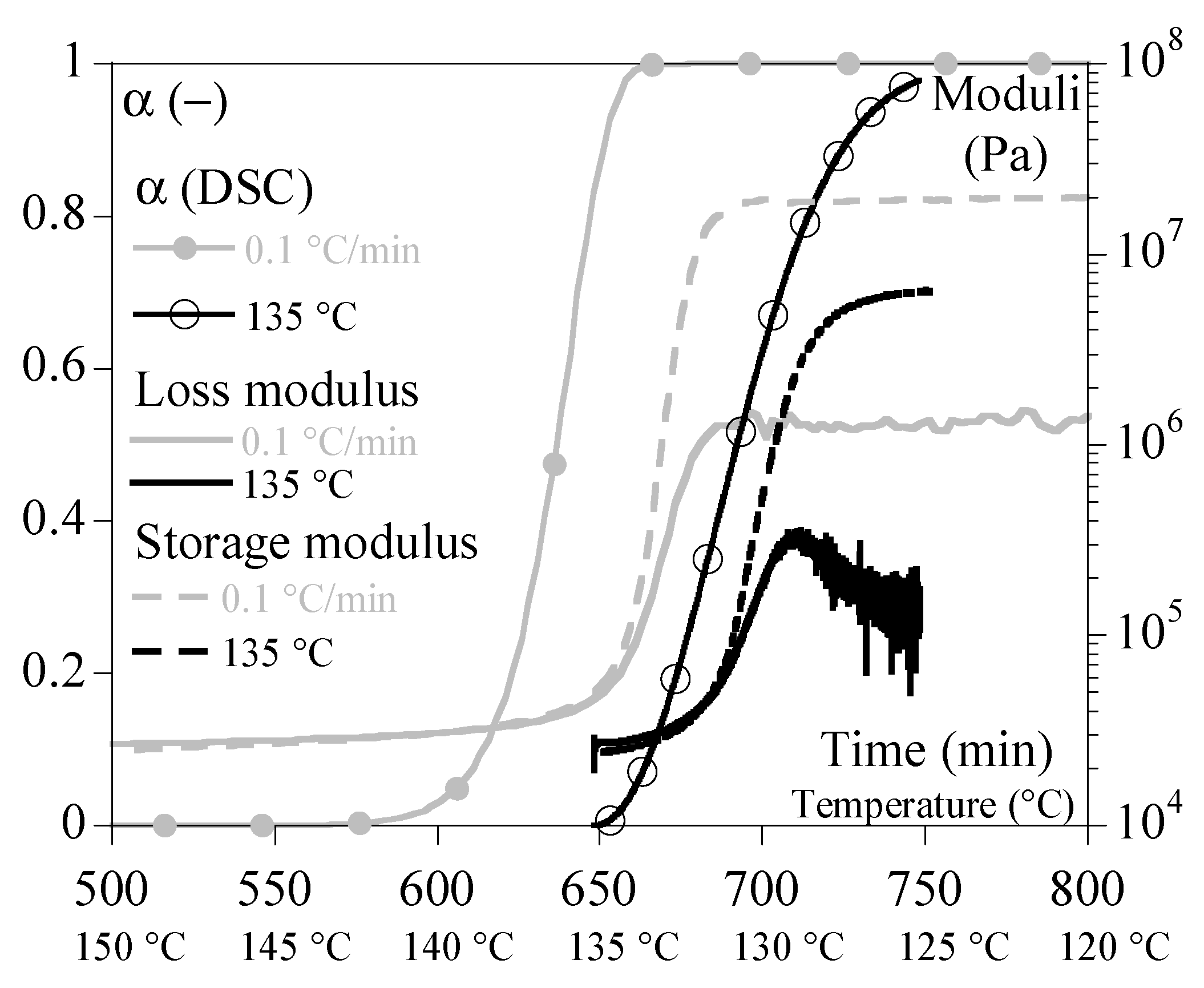
4.3. Rheology under Non-Isothermal Conditions
5. Modeling
5.1. Methodology
5.2. Rheology vs. Crystallization
5.3. Rheology vs. Melting
6. Results
- 1-
- by using only the test at 0.1 °C/min and interpolating the crystallization kinetics with Ozawa’s formalism (Equation (10));
- 2-
- by combining the test at 0.1 °C/min and the isothermal tests and interpolating the kinetics of not isothermal crystallization with the formalism of Ozawa (Equation (10)).
- by using the coefficient 1 under the conditions of identification 1 (Ozawa crystallization, hollow squares);
- by using the coefficient 1 and interpolating the crystallizations during the validation tests from the isothermal tests (hollow circles);
- by using the coefficient 2 under the conditions of identification 2 (Ozawa crystallization, filled squares);
- by using the coefficient 2 and interpolating the crystallizations during the validation tests from the isothermal tests (filled circles).
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A



References
- Biskas, H.; Stavropoulos, P.; Chryssolouros, G. Additive manufacturing methods and modelling; a critical review. Int. J. Adv. Manuf. Technol. 2016, 83, 389–405. [Google Scholar] [CrossRef]
- Haudin, J.-M.; Boyer, S.A.E. Crystallization of Polymers in Processing Conditions: An Overview. Int. Polym. Process. 2017, 32, 545–554. [Google Scholar] [CrossRef]
- Sreejith, P.; Kannan, K.; Rajagopal, K.R. A thermodynamic framework for the additive manufacturing of crystallizing polymers. Part I: A theory that accounts for phase change, shrinkage, warpage and residual stress. Int. J. Eng. Sci. 2023, 183, 103789. [Google Scholar] [CrossRef]
- Boutahar, K.; Carrot, C.; Guillet, J. Crystallization of polyolefins from rheological measurements–relation between the transformed fraction and the dynamic moduli. Macromolecules 1998, 31, 1921–1929. [Google Scholar] [CrossRef]
- Pogodina, N.V.; Winter, H.H. Crystallization as a Physical Gelation Process. Macromolecules 1998, 31, 8164. [Google Scholar] [CrossRef]
- Lamberti, G.; Peters, G.; Titomanlio, G. Crystallinity and Linear Rheological Properties of Polymers. Int. Polym. Proc. 2007, 22, 303. [Google Scholar] [CrossRef]
- Han, S.; Wang, K.K. Shrinkage prediction for slowly-crystallizing thermoplastic polymers in injection molding. Int. Polym. Proc. 1997, 3, 228. [Google Scholar] [CrossRef]
- Pantani, R.; Speranza, V.; Titomanlio, G. Simultaneous morphological and rheological measurements on polypropylene: Effect of crystallinity on viscoelastic parameters. J. Rheol. 2015, 59, 377–390. [Google Scholar] [CrossRef]
- Aris-Brosou, M.; Vincent, M.; Agassant, J.-F.; Billon, N. Viscoelastic rheology in the melting and crystallization domain: Application to polypropylene copolymers. J. Appl. Polym. Sci. 2017, 134, 44690. [Google Scholar] [CrossRef]
- Keith, H.D.; Vadimsky, R.G.; Padden, F.J., Jr. Crystallization of isotactic polystyrene from solution. J. Polym. Sci. Part. A-2 Polym. Phys. 1970, 8, 1687–1696. [Google Scholar] [CrossRef]
- Keith, H.D.; Padden, F.J., Jr.; Vadimsky, R.G. Intercrystalline links: Critical evaluation. J. Appl. Phys. 1971, 42, 4585–4592. [Google Scholar] [CrossRef]
- Padden, F.J., Jr.; Keith, H.D. Mechanism for lamellar branching in isotactic polypropylene. J. Appl. Phys. 1973, 44, 1217–1223. [Google Scholar] [CrossRef]
- Keith, H.D.; Padden, F.J., Jr. Twisting orientation and the role of transient states in polymer crystallization. Polymer 1984, 25, 28–42. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Weeks, J.J. Melting process and the equilibrium melting temperature of polychlorotriuoroethylene. J. Res. Nat. Bur. Stand. Sect. A Phys. Chem. 1962, 66A, 13–28. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Miller, R.L. Kinetics of crystallization from the melt and chain folding in polyethylene fractions revisited: Theory and experiment. Polymer 1997, 38, 3151–3212. [Google Scholar] [CrossRef]
- Lauritzen, J.I., Jr.; Hoffman, J.D. Extension of theory of growth of chain-folded polymer crystals to large undercoolings. J. Appl. Phys. 1973, 44, 4340–4352. [Google Scholar] [CrossRef]
- Piorkowskaa, E.; Galeski, A.; Haudin, J.-M. Critical assessment of overall crystallization kinetics theories and predictions. Prog. Polym. Sci. 2006, 31, 549–575. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. I. General theory. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. II. Transformation–time relations for random distribution of nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of phase change. III. Granulation, phase change and microstructure. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Johnson, W.A.; Mehl, R.F. Reaction kinetics in process of nucleation and growth. Trans. AIME 1939, 135, 416–458. [Google Scholar]
- Kolmogoroff, A.N. K statisticheskoi teorii kristallizacii metallov. Izvestiya Akad. Nauk. SSSR Ser. Math. 1937, 1, 1355–1359. [Google Scholar]
- Evans, U.R. The laws of expanding circles and spheres in relation to the lateral growth of surface films and the grainsizeof metals. Trans. Faraday Soc. 1945, 41, 365–375. [Google Scholar] [CrossRef]
- Nakamura, K.; Watanabe, T.; Katayama, K.; Amano, T. Some aspects of nonisothermal crystallization of polymers. I. Relationship between crystallization temperature, crystallinity and cooling conditions. J. Appl. Polym. Sci. 1972, 16, 1077–1091. [Google Scholar] [CrossRef]
- Nakamura, K.; Katayama, K.; Amano, T. Some aspects of nonisothermal crystallization of polymers. II. Consideration of the Isokinetic condition. J. Appl. Polym. Sci. 1973, 17, 1031–1941. [Google Scholar] [CrossRef]
- Ozawa, T. Kinetics of non-isothermal crystallization. Polymer 1971, 12, 150–158. [Google Scholar] [CrossRef]
- Billon, N.; Barq, P.; Haudin, J. Modelling of the cooling of semi-crystalline polymers during their processing. Int. Polym. Proc. 1991, 6, 348–355. [Google Scholar] [CrossRef]
- Haudin, J.-M.; Chenot, J.-L. Numerical and physical modeling of polymer crystallization—Part I: Theoretical and numerical analysis. Int. Polym. Process 2004, 19, 267–274. [Google Scholar] [CrossRef]
- Schneider, W.; Koppl, A.; Berger, J. Non-Isothermal crystallization of polymers. Int. Polym. Process. 1998, 2, 151–154. [Google Scholar] [CrossRef]
- Kerner, E. The Elastic and Thermo-elastic Properties of Composite Media. Proc. Phys. Soc. B 1956, 69B, 808–813. [Google Scholar] [CrossRef]
- Van Dommelen, J.A.W.; Parks, D.M.; Boyce, M.C.; Brekelmans, W.A.M.; Baaijens, F.P.T. Micromechanical modeling of the elasto-viscoplastic behavior of semi-crystalline polymers. J. Mech. Phys. Solids 2003, 51, 519–541. [Google Scholar] [CrossRef]
- Bedoui, F.; Diani, J.; Regnier, G.; Seiler, W. Micromechanical modeling of isotropic elastic behavior of semicrystalline polymers. Acta Mater. 2006, 54, 1513–1523. [Google Scholar] [CrossRef]
- Luo, Y.-M.; Detrez, F.; Chevalier, L.; Lu, X.; Roland, S. Multiscale framework for estimation of elastic properties of poly ethylene terephthalate from the crystallization temperature. Mech. Mater. 2023, 181, 104617. [Google Scholar] [CrossRef]
- Monasse, B.; Haudin, J.-M. Thermal dependence of nucleation and growth rate in polypropylene by non-isothermal calorimetry. Colloid. Polym. Sci. 1986, 264, 117–122. [Google Scholar] [CrossRef]
- Billon, N.; Haudin, J.-M. Determination of nucleation rate in polymers using isothermal crystallization experiments and computer simulation. Colloid. Polym. Sci. 1993, 271, 343–356. [Google Scholar] [CrossRef]
- Billon, N.; Henaff, V.; Haudin, J.-M. Transcrystallinity effects in high-density polyethylene. II. determination of kinetics parameters. J. Appl. Polym. Sci. 2002, 86, 734–742. [Google Scholar] [CrossRef]
- Hoffman, J.D. Regime III crystallization in melt-crystallized polymers: The variable cluster model of chain folding. Polymer 1983, 24, 3–26. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Frolen, L.J.; Ross, G.S.; Lauritzen, J.I., Jr. On the growth rate of spherulites and axiaites from the melt in polyethylene fractions: Regime I and regime II crystallization. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1975, 79A, 671. [Google Scholar] [CrossRef]
- Lachlan, D.M. An equation for the conductivity of binary mixtures with anisotropic grain structures. J. Phys. C Solid. State Phys. 1987, 20, 865–877. [Google Scholar] [CrossRef]
- Roy, D.; Audus, D.; Migler, K. Rheology of crystallizing polymers: The role of spherulitic superstructures, gap height, and nucleation densities. J. Rheol. 2019, 63, 851–862. [Google Scholar] [CrossRef]
- Schröter, K.; Hutcheson, S.A.; Shi, X.; Mandanici, A.; McKenna, G.B. Dynamic shear modulus of glycerol: Corrections due to instrument compliance. J. Chem. Phys. 2006, 125, 214507. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, O.V. Small-diameter parallel plate rheometry: A simple technique for measuring rheological properties of glass-forming liquids in shear. Rheol. Acta 2017, 56, 661–671. [Google Scholar] [CrossRef]
- Liu, C.Y.; Yao, M.; Garritano, R.G.; Franck, A.J.; Bailly, C. Instrument compliance effects revisited: Linear viscoelastic measurements. Rheol. Acta 2011, 50, 537–546. [Google Scholar] [CrossRef]


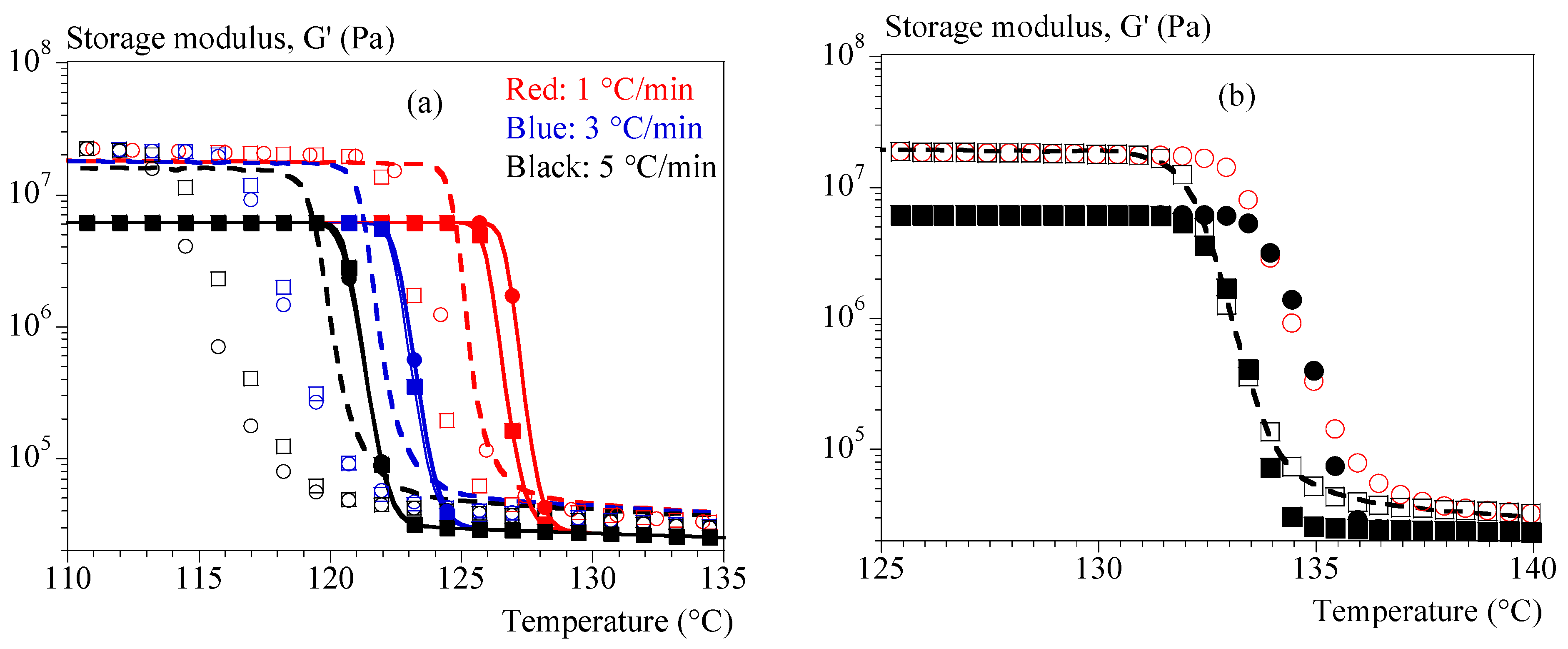
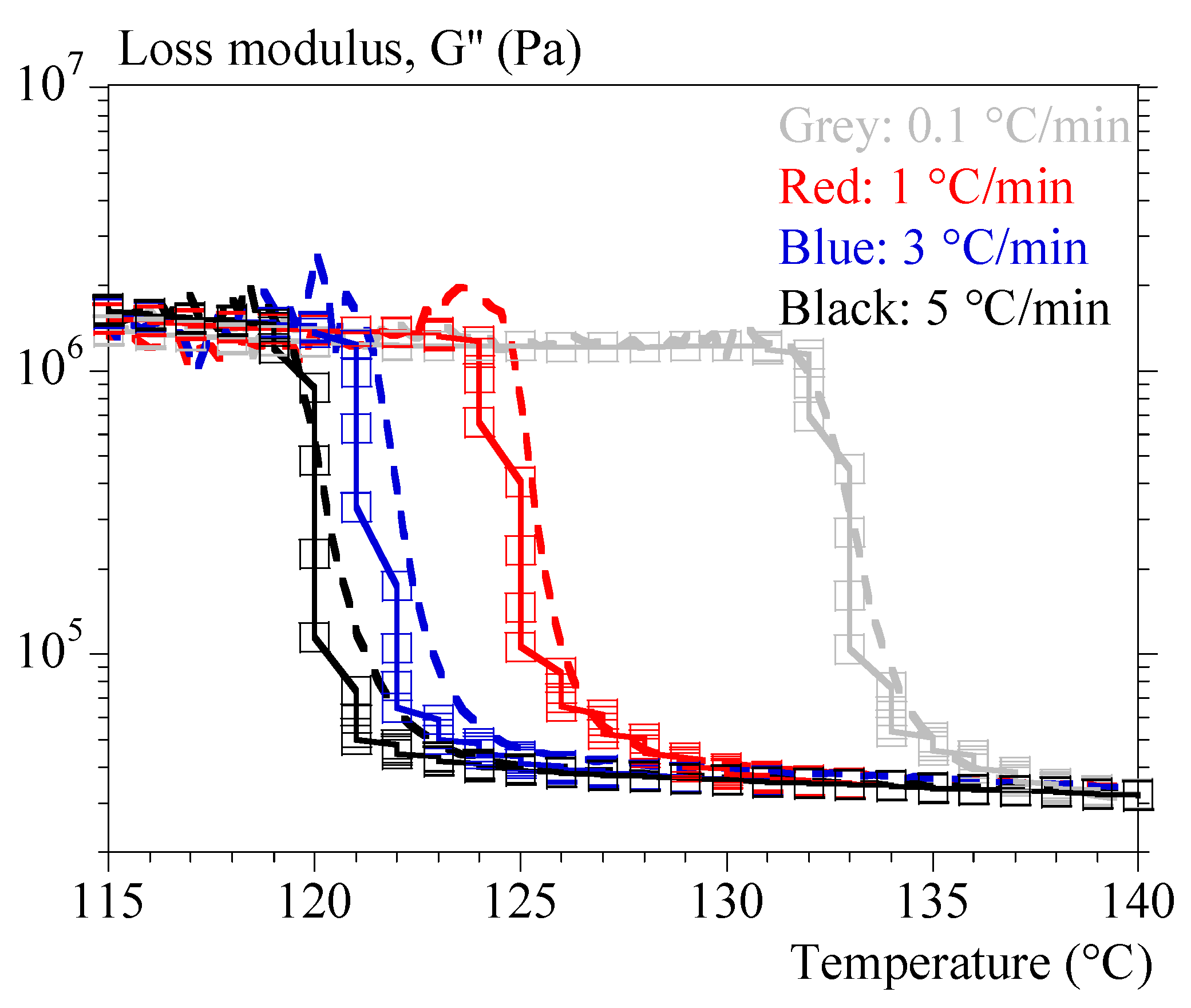
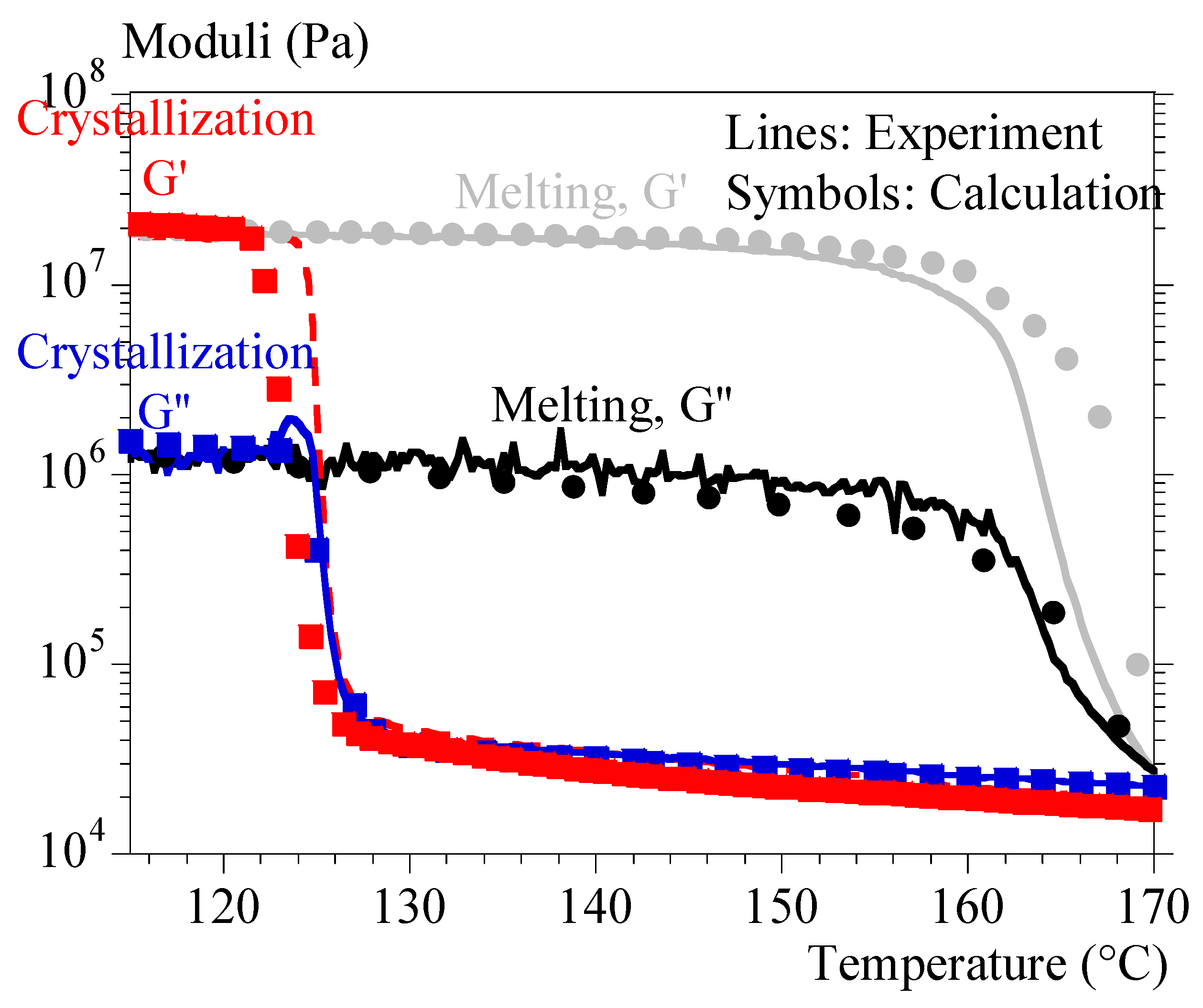

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature (°C) | 125 | 127.5 | 130 | 132.5 | 135 |
(µm/min) | 20.8 | 13.2 | 7.93 | 4.68 | 2.69 |
(min−n) | 1.77 × 10−2 | 2.46 × 10−3 | 7.08 × 10−4 | 1.59 × 10−4 | 2.52×10−4 |
| N | 3.39 | 3.15 | 2.75 | 2.60 | 2.09 |
| for n = 3 (min−3) | 4.00 × 10−5 | 8.33 × 10−5 | 9.12 × 10−4 | 4.51 × 10−3 | 2.81 × 10−2 |
(µm−3 min3) | 3.13 × 10−6 | 2.00 × 10−6 | 1.78 × 10−6 | 7.96 × 10−7 | 2.10 × 10−6 |
| 135 (°C) | 132.5 (°C) | 130 (°C) | 127.5 (°C) | 125 (°C) | 0.1 °C/min | Combined | ||
| (MPa) | ||||||||
| (MPa) | 7.04 | 6.16 | 5.98 | 5.86 | 5.98 | 0.22 | 6.64 | |
| 3.99 | 3.14 | 2.01 | 1.85 | 1.93 | 0.23 | 0.767 | ||
| (-) | 0.323 | 0.355 | 0.408 | 0.447 | 0.455 | 0.71 | 0.023 | |
| (-) | 0.0879 | 0.0663 | 0.0593 | 0.059 | 0.0544 | −0.035 | −276 | |
| (-) | 11.4 | 10.6 | 8.22 | 8.05 | 8.04 | 4.28 | 1.35 | |
| (-) | 4.18 | 4.67 | 6.96 | 8.16 | 6.03 | 0.24 | 0.0015 | |
| (MPa) | ||||||||
| (MPa) | 0.0936 | 0.1082 | 0.1254 | 0.1800 | 0.0809 | 5.32 × 10−12 | NA | |
| (MPa) | 0.047 | 0.048 | 0.050 | 0.051 | 0.053 | 2.28 × 10−4 | NA | |
| (-) | 199,577 | 3,943,412 | 18,398,585 | 20,373,492 | 16,690,372 | 6.57 | NA | |
| (-) | 5.03 | 3.77 | 3.21 | 2.44 | 2.87 | 3.97 | NA | |
| (-) | 0.848 | 1.752 | 2.897 | 3.692 | 2.977 | 0.0485 | NA | |
| (-) | 6.95 | 5.22 | 4.73 | 4.54 | 3.91 | 3.99 | NA | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Billon, N.; Castellani, R.; Bouvard, J.-L.; Rival, G. Viscoelastic Properties of Polypropylene during Crystallization and Melting: Experimental and Phenomenological Modeling. Polymers 2023, 15, 3846. https://doi.org/10.3390/polym15183846
Billon N, Castellani R, Bouvard J-L, Rival G. Viscoelastic Properties of Polypropylene during Crystallization and Melting: Experimental and Phenomenological Modeling. Polymers. 2023; 15(18):3846. https://doi.org/10.3390/polym15183846
Chicago/Turabian StyleBillon, Noëlle, Romain Castellani, Jean-Luc Bouvard, and Guilhem Rival. 2023. "Viscoelastic Properties of Polypropylene during Crystallization and Melting: Experimental and Phenomenological Modeling" Polymers 15, no. 18: 3846. https://doi.org/10.3390/polym15183846
APA StyleBillon, N., Castellani, R., Bouvard, J.-L., & Rival, G. (2023). Viscoelastic Properties of Polypropylene during Crystallization and Melting: Experimental and Phenomenological Modeling. Polymers, 15(18), 3846. https://doi.org/10.3390/polym15183846