
Ketorolac-Loaded PLGA-/PLA-Based Microparticles Stabilized by Hyaluronic Acid: Effects of Formulation Composition and Emulsification Technique on Particle Characteristics and Drug Release Behaviors
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Microparticle Preparation
2.2.1. Probe Sonication (PS) Technique
2.2.2. High-Speed Stirring (HSS) Technique
2.3. Characterization of Particles
2.3.1. Particle Size and Size Distribution
2.3.2. Zeta Potential
2.3.3. Drug Loading Content
2.3.4. Yield
2.3.5. Morphology Observation
2.4. Differential Scanning Calorimetry
2.5. X-ray Diffraction
2.6. In Vitro Release Study
2.7. Statistical Analysis
3. Results
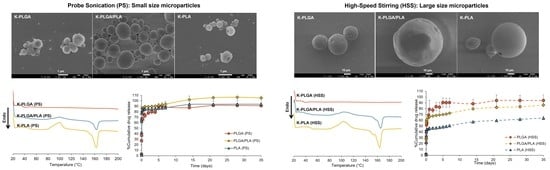
3.1. Physicochemical Properties of Microparticles Prepared by Different Emulsification Techniques
3.2. Differential Scanning Calorimetry (DSC)
3.3. X-ray Diffractometry (XRD)
3.4. In Vitro Drug Release Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.A.; Hinojosa, V.A.; Arriaga, M.A. 11—bioresorbable polymer microparticles in the medical and pharmaceutical fields. In Bioresorbable Polymers for Biomedical Applications; Perale, G., Hilborn, J., Eds.; Woodhead Publishing: Sawston, UK, 2017; pp. 229–264. [Google Scholar]
- Rosca, I.D.; Watari, F.; Uo, M. Microparticle formation and its mechanism in single and double emulsion solvent evaporation. J. Control. Release 2004, 99, 271–280. [Google Scholar] [CrossRef]
- Chen, W.; Palazzo, A.; Hennink, W.E.; Kok, R.J. Effect of particle size on drug loading and release kinetics of gefitinib-loaded PLGA microspheres. Mol. Pharm. 2017, 14, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Yin, B.; Ni, J.; Witherel, C.E.; Yang, M.; Burdick, J.A.; Wen, C.; Wong, S.H.D. Harnessing tissue-derived extracellular vesicles for osteoarthritis theranostics. Theranostics 2022, 12, 207–231. [Google Scholar] [CrossRef]
- Sen, R.; Hurley, J.A. Osteoarthritis. In Statpearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mahajan, A.; Verma, S.; Tandon, V. Osteoarthritis. J. Assoc. Physicians India 2005, 53, 634–641. [Google Scholar] [PubMed]
- Xu, J.; Qu, Y.; Li, H.; Zhu, A.; Jiang, T.; Chong, Z.; Wang, B.; Shen, P.; Xie, Z. Effect of intra-articular ketorolac versus corticosteroid injection for knee osteoarthritis: A retrospective comparative study. Orthop. J. Sports Med. 2020, 8, 2325967120911126. [Google Scholar] [CrossRef]
- Park, K.D.; Kim, T.K.; Bae, B.W.; Ahn, J.; Lee, W.Y.; Park, Y. Ultrasound guided intra-articular ketorolac versus corticosteroid injection in osteoarthritis of the hip: A retrospective comparative study. Skelet. Radiol. 2015, 44, 1333–1340. [Google Scholar] [CrossRef]
- Lee, S.C.; Rha, D.W.; Chang, W.H. Rapid analgesic onset of intra-articular hyaluronic acid with ketorolac in osteoarthritis of the knee. J. Back Musculoskelet. Rehabil. 2011, 24, 31–38. [Google Scholar] [CrossRef]
- Koh, S.H.; Lee, S.C.; Lee, W.Y.; Kim, J.; Park, Y. Ultrasound-guided intra-articular injection of hyaluronic acid and ketorolac for osteoarthritis of the carpometacarpal joint of the thumb: A retrospective comparative study. Medicine 2019, 98, e15506. [Google Scholar] [CrossRef]
- Badawi, A.A.; El-Laithy, H.M.; Nesseem, D.I.; El-Husseney, S.S. Pharmaceutical and medical aspects of hyaluronic acid–ketorolac combination therapy in osteoarthritis treatment: Radiographic imaging and bone mineral density. J. Drug Target. 2013, 21, 551–563. [Google Scholar] [CrossRef]
- Saraf, S.; Verma, A.; Tripathi, A.; Saraf, S. Fabrication and evaluation of sustained release microspheres of ketorolac tromethamine. Int. J. Pharm. Pharm. Sci. 2010, 2, 44–48. [Google Scholar]
- Mathew, S.T.; Devi, S.G.; Kv, S. Formulation and evaluation of ketorolac tromethamine-loaded albumin microspheres for potential intramuscular administration. AAPS PharmSciTech 2007, 8, 14. [Google Scholar] [CrossRef]
- Wagh, P.; Mujumdar, A.; Naik, J.B. Preparation and characterization of ketorolac tromethamine-loaded ethyl cellulose micro-/nanospheres using different techniques. Part. Sci. Technol. 2019, 37, 347–357. [Google Scholar] [CrossRef]
- Nagda, C.D.; Chotai, N.P.; Nagda, D.C.; Patel, S.B.; Patel, U.L. Preparation and characterization of spray-dried mucoadhesive microspheres of ketorolac for nasal administration. Curr. Drug Del. 2012, 9, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.R.; Trehan, A. Development, characterization, and evaluation of ketorolac tromethamine-loaded biodegradable microspheres as a depot system for parenteral delivery. Drug Deliv. 2008, 15, 365–372. [Google Scholar] [CrossRef]
- Sinha, V.R.; Trehan, A. Formulation, characterization, and evaluation of ketorolac tromethamine-loaded biodegradable microspheres. Drug Deliv. 2005, 12, 133–139. [Google Scholar] [CrossRef]
- Bhaskaran, S. Poly (lactic acid) microspheres of ketorolac tromethamine for patenteral controlled drug delivery system. Indian J. Pharm. Sci. 2001, 63, 538–540. [Google Scholar]
- Basu, S.K.; Kavitha, K.; Rupeshkumar, M. Evaluation of ketorolac tromethamine microspheres by chitosan/gelatin B complex coacervation. Sci. Pharm. 2010, 78, 79–92. [Google Scholar] [CrossRef]
- Giri, T.; Choudhary, C.; Ajaz, A.; Alexander, A.; Badwaik, H.; Tripathi, D. Prospects of pharmaceuticals and biopharmaceuticals loaded microparticles prepared by double emulsion technique for controlled delivery. Saudi Pharm. J. 2013, 21, 125–141. [Google Scholar] [CrossRef]
- Pubchem compound summary for cid 3826, ketorolac. In Pubchem [Internet]; National Library of Medicine (US), National Center for Biotechnology Information: Bethesda, MD, USA, 2004; Volume 2022.
- Zaghloul, N.; Mahmoud, A.A.; Elkasabgy, N.A.; El Hoffy, N.M. PLGA-modified syloid®-based microparticles for the ocular delivery of terconazole: In-vitro and in-vivo investigations. Drug Deliv. 2022, 29, 2117–2129. [Google Scholar] [CrossRef]
- Chantaburanan, T.; Teeranachaideekul, V.; Chantasart, D.; Jintapattanakit, A.; Junyaprasert, V.B. Effect of binary solid lipid matrix of wax and triglyceride on lipid crystallinity, drug-lipid interaction and drug release of ibuprofen-loaded solid lipid nanoparticles (SLN) for dermal delivery. J. Colloid Interface Sci. 2017, 504, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Selek, H.; Sahin, S.; Ercan, M.T.; Sargon, M.; Hincal, A.A.; Kas, H.S. Formulation and in vitro/in vivo evaluation of terbutaline sulphate incorporated in PLGA (25/75) and L-PLA microspheres. J. Microencaps. 2003, 20, 261–271. [Google Scholar]
- Bianco, A.W.; Constable, P.D.; Cooper, B.R.; Taylor, S.D. Pharmacokinetics of ketorolac tromethamine in horses after intravenous, intramuscular, and oral single-dose administration. J. Vet. Pharmacol. Ther. 2016, 39, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Ghauri, A.; Ghauri, I.; Elhissi, A.M.A.; Ahmed, W. Chapter 14—Characterization of cochleate nanoparticles for delivery of the anti-asthma drug beclomethasone dipropionate. In Advances in Medical and Surgical Engineering; Ahmed, W., Phoenix, D.A., Jackson, M.J., Charalambous, C.P., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 267–277. [Google Scholar]
- Kohli, R. Chapter 2—applications of strippable coatings for removal of surface contaminants. In Developments in Surface Contamination and Cleaning: Applications of Cleaning Techniques; Kohli, R., Mittal, K.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 49–96. [Google Scholar]
- Goedemoed, J.H.; Mense, E.H.G.; de Groot, K.; Claessen, A.M.E.; Scheper, R.J. Development of injectable antitumor microspheres based on polyphosphazene. J. Control. Release 1991, 17, 245–257. [Google Scholar] [CrossRef]
- Dinarvand, R.; Sepehri, N.; Manoochehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef]
- Carmagnola, I.; Nardo, T.; Gentile, P.; Tonda-Turo, C.; Mattu, C.; Cabodi, S.; Defilippi, P.; Chiono, V. Poly(lactic acid)-based blends with tailored physicochemical properties for tissue engineering applications: A case study. Int. J. Polymer. Mater. 2015, 64, 90–98. [Google Scholar] [CrossRef]
- Szuman, K.; Krucińska, I.; Boguń, M.; Draczyński, Z. PLA/PHA-biodegradable blends for pneumothermic fabrication of nonwovens. Autex Res. J. 2016, 16, 119–127. [Google Scholar] [CrossRef]
- Park, P.I.P.; Jonnalagadda, S. Predictors of glass transition in the biodegradable poly-lactide and poly-lactide-co-glycolide polymers. J. Appl. Polym. Sci. 2006, 100, 1983–1987. [Google Scholar] [CrossRef]
- Wang, Y.; Mano, J.F. Role of thermal history on the thermal behavior of poly(L-lactic acid) studied by DSC and optical microscopy. J. Therm. Anal. Calorim. 2005, 80, 171–175. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Liu, X.P.; Liu, Q.Q.; Liu, L.H. Influence of temperature and time on isothermal crystallization of poly-L-lactide. Int. J. Polym. Mater. Polym. Biomater. 2008, 57, 878–890. [Google Scholar] [CrossRef]
- Jurgensmeier, K.; Jurgensmeier, D.M.D.; Kunz, D.E.; Fuerst, P.G.; Warth, L.C.; Daines, S.B. Intra-articular injections of the hip and knee with triamcinolone vs ketorolac: A randomized controlled trial. J. Arthroplast. 2021, 36, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Won, Y.-Y. Phenomenology of the initial burst release of drugs from PLGA microparticles. ACS Biomater. Sci. Eng. 2020, 6, 6053–6062. [Google Scholar] [CrossRef]
- 5—Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Bruschi, M.L., Ed.; Woodhead Publishing: Sawston, UK, 2015; pp. 63–86. [Google Scholar]
- Busatto, C.; Pesoa, J.; Helbling, I.; Luna, J.; Estenoz, D. Effect of particle size, polydispersity and polymer degradation on progesterone release from PLGA microparticles: Experimental and mathematical modeling. Int. J. Pharm. 2018, 536, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Sansdrap, P.; Moës, A.J. In vitro evaluation of the hydrolytic degradation of dispersed and aggregated poly(DL-lactide-co-glycolide) microspheres. J. Control. Release 1997, 43, 47–58. [Google Scholar] [CrossRef]
- Gasmi, H.; Siepmann, F.; Hamoudi, M.C.; Danede, F.; Verin, J.; Willart, J.F.; Siepmann, J. Towards a better understanding of the different release phases from PLGA microparticles: Dexamethasone-loaded systems. Int. J. Pharm. 2016, 514, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Kim, T.H.; Park, T.G. Critical effect of freezing/freeze-drying on sustained release of FITC-dextran encapsulated within PLGA microspheres. Int. J. Pharm. 2004, 271, 207–214. [Google Scholar] [CrossRef]
- Kang, J.; Schwendeman, S.P. Pore closing and opening in biodegradable polymers and their effect on the controlled release of proteins. Mol. Pharm. 2007, 4, 104–118. [Google Scholar] [CrossRef]
- Rapier, C.E.; Shea, K.J.; Lee, A.P. Investigating PLGA microparticle swelling behavior reveals an interplay of expansive intermolecular forces. Sci. Rep. 2021, 11, 14512. [Google Scholar] [CrossRef]
- Park, T.G. Degradation of poly(lactic-co-glycolic acid) microspheres: Effect of copolymer composition. Biomaterials 1995, 16, 1123–1130. [Google Scholar] [CrossRef]
- Kenley, R.A.; Lee, M.O.; Mahoney, T.R., II; Sanders, L.M. Poly(lactide-co-glycolide) decomposition kinetics in vivo and in vitro. Macromolecules 1987, 20, 2398–2403. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Particle Size (µm) | Span | Zeta Potential (mV) | %Yield | %Drug Loading | %Entrapment Efficiency | |
|---|---|---|---|---|---|---|---|
| d (0.5) * | d (0.9) * | ||||||
| PLGA (PS) | 2.16 ± 0.00 | 5.89 ± 0.01 | 2.20 ± 0.01 | −3.9 ± 0.1 | 49.98 ± 8.59 | 8.83 ± 0.65 | 76.32 ± 5.58 |
| PLGA/PLA (PS) | 2.12 ± 0.11 | 19.20 ± 6.70 | 8.50 ± 2.83 | −3.5 ± 0.4 | 55.56 ± 4.13 | 9.24 ± 1.32 | 79.83 ± 11.38 |
| PLA (PS) | 1.36 ± 0.01 | 3.08 ± 0.19 | 1.47 ± 0.13 | −3.5 ± 0.1 | 55.83 ± 7.08 | 9.45 ± 0.34 | 81.63 ± 2.94 |
| PLGA (HSS) | 45.50 ± 0.06 | 64.76 ± 0.15 | 1.04 ± 0.00 | −4.4 ± 0.3 | 46.79 ± 6.81 | 8.93 ± 2.15 | 77.15 ± 18.61 |
| PLGA/PLA (HSS) | 67.43 ± 0.40 | 101.32 ± 1.32 | 0.97 ± 0.01 | −3.8 ± 0.2 | 58.18 ± 5.47 | 8.60 ± 0.37 | 74.28 ± 3.20 |
| PLA (HSS) | 60.88 ± 0.54 | 103.51 ± 1.67 | 1.44 ± 0.01 | −3.6 ± 0.2 | 50.29 ± 3.16 | 7.82 ± 0.56 | 67.52 ± 4.84 |
| Formulations | Tg (°C) | Tcc (°C) a | ΔHcc (J/g) | Tm (°C) b | ΔHm (J/g) |
|---|---|---|---|---|---|
| Ketorolac | 43.72 | ||||
| PLGA | 42.79 | ||||
| PLA | 53.29 | (110.67) 121.00 | 45.93 | (167.67) 173.00 | 45.48 |
| Blank microparticles | |||||
| PLGA (PS) | 41.80 | ||||
| PLGA/PLA (PS) | 40.82, 53.95 | 100.67 | 5.48 | 171.33 | 8.28 |
| PLA (PS) | 51.90 | (99.67) 106.67 | 11.49 | 172.67 | 15.00 |
| PLGA (HSS) | 45.70 | ||||
| PLGA/PLA (HSS) | 46.29, 58.72 | 117.33 | 20.82 | 174.00 | 20.06 |
| PLA (HSS) | 59.80 | 116.33 | 40.56 | 175.33 | 39.86 |
| Drug-loaded microparticles | |||||
| PLGA (PS) | 35.87 | ||||
| PLGA/PLA (PS) | 35.85, 48.01 | (84.93) 100.00 | 8.26 | (155.67) 163.00 | 12.11 |
| PLA (PS) | 48.73 | (88.33) 99.67 | 19.52 | (152.33) 162.67 | 21.82 |
| PLGA (HSS) | 34.94 | ||||
| PLGA/PLA (HSS) | 38.65, 48.95 | (90.67) 104.33 | 12.70 | (157.00) 165.67 | 12.58 |
| PLA (HSS) | 48.79 | (89.67) 102.67 | 20.56 | (153.67) 164.00 | 168.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wongrakpanich, A.; Khunkitchai, N.; Achayawat, Y.; Suksiriworapong, J. Ketorolac-Loaded PLGA-/PLA-Based Microparticles Stabilized by Hyaluronic Acid: Effects of Formulation Composition and Emulsification Technique on Particle Characteristics and Drug Release Behaviors. Polymers 2023, 15, 266. https://doi.org/10.3390/polym15020266
Wongrakpanich A, Khunkitchai N, Achayawat Y, Suksiriworapong J. Ketorolac-Loaded PLGA-/PLA-Based Microparticles Stabilized by Hyaluronic Acid: Effects of Formulation Composition and Emulsification Technique on Particle Characteristics and Drug Release Behaviors. Polymers. 2023; 15(2):266. https://doi.org/10.3390/polym15020266
Chicago/Turabian StyleWongrakpanich, Amaraporn, Nichakan Khunkitchai, Yanisa Achayawat, and Jiraphong Suksiriworapong. 2023. "Ketorolac-Loaded PLGA-/PLA-Based Microparticles Stabilized by Hyaluronic Acid: Effects of Formulation Composition and Emulsification Technique on Particle Characteristics and Drug Release Behaviors" Polymers 15, no. 2: 266. https://doi.org/10.3390/polym15020266
APA StyleWongrakpanich, A., Khunkitchai, N., Achayawat, Y., & Suksiriworapong, J. (2023). Ketorolac-Loaded PLGA-/PLA-Based Microparticles Stabilized by Hyaluronic Acid: Effects of Formulation Composition and Emulsification Technique on Particle Characteristics and Drug Release Behaviors. Polymers, 15(2), 266. https://doi.org/10.3390/polym15020266