
Synthesis and Characterization of Statistical and Block Copolymers of n-Hexyl Isocyanate and 3-(Triethoxysilyl) Propyl Isocyanate via Coordination Polymerization
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Poly[3-(Triethoxysilyl)propyl Isocyanate], PTESPI
2.3. General Synthesis of Statistical Copolymers of HIC and TESPI, PHIC-Stat-PTESPI
2.4. General Synthesis of Block Copolymers of HIC and TESPI, PTESPI-b-PHIC
2.5. Characterization Techniques
3. Results and Discussion
3.1. Polymerization of TESPI via Coordination Polymerization
3.2. Statistical Copolymers
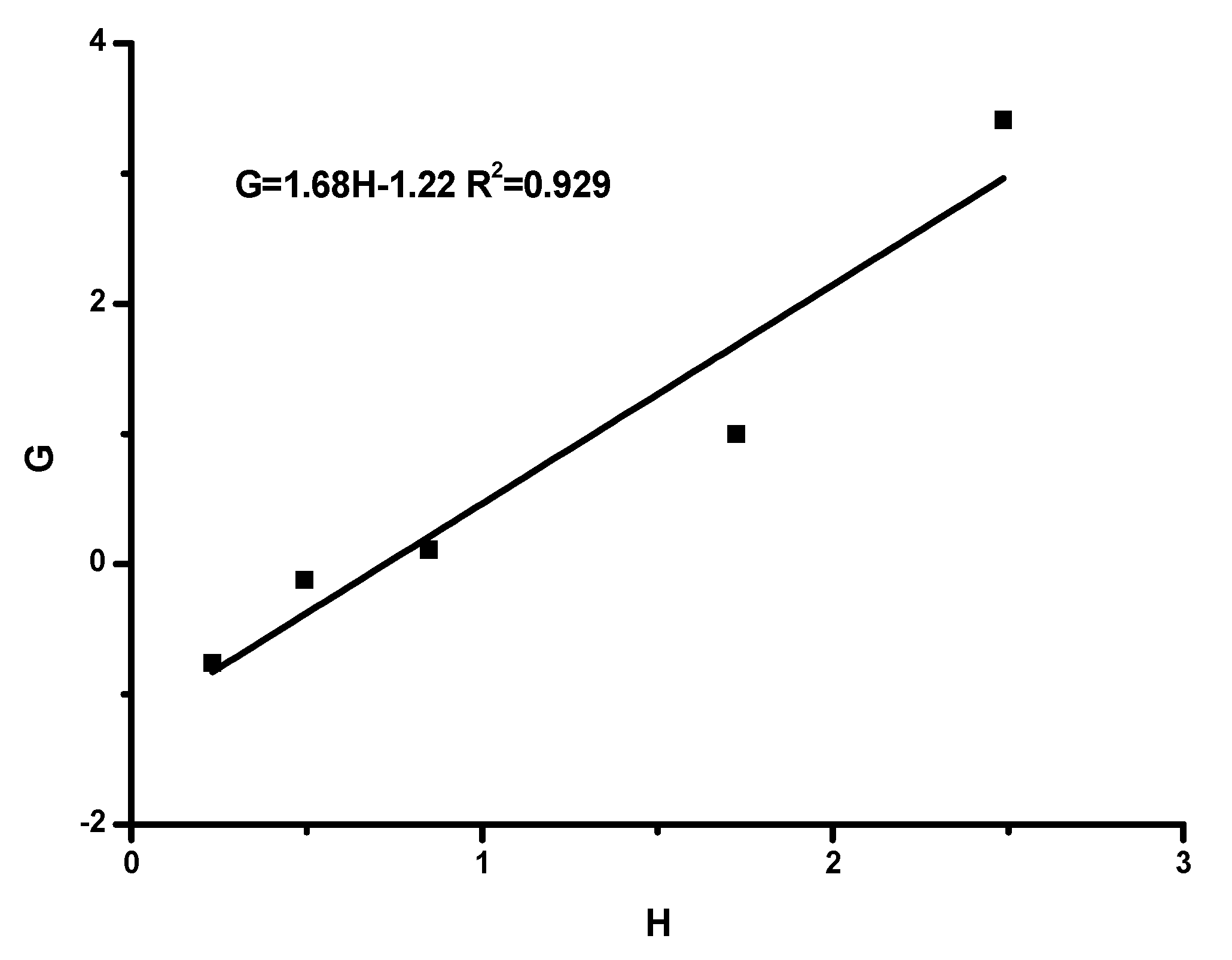
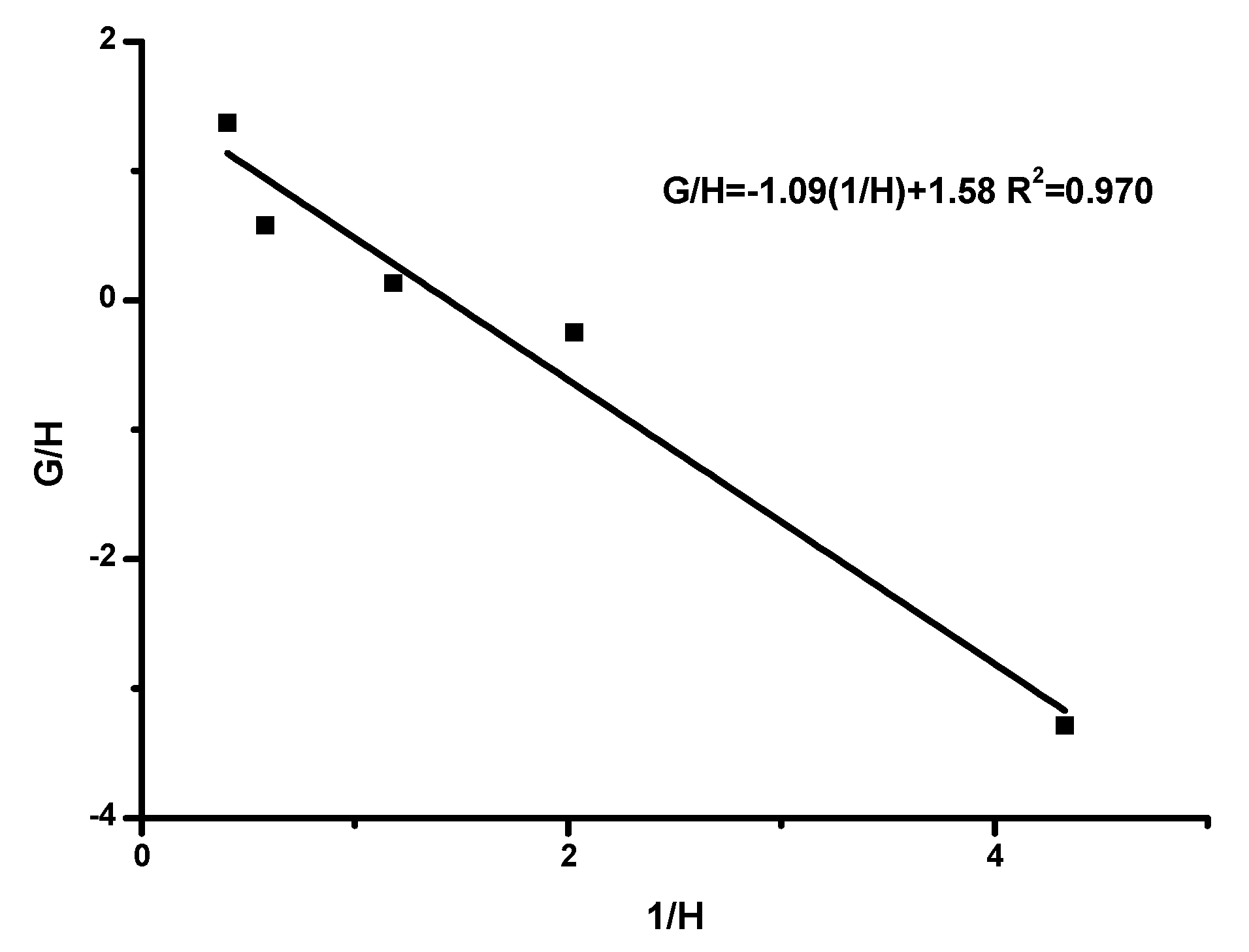
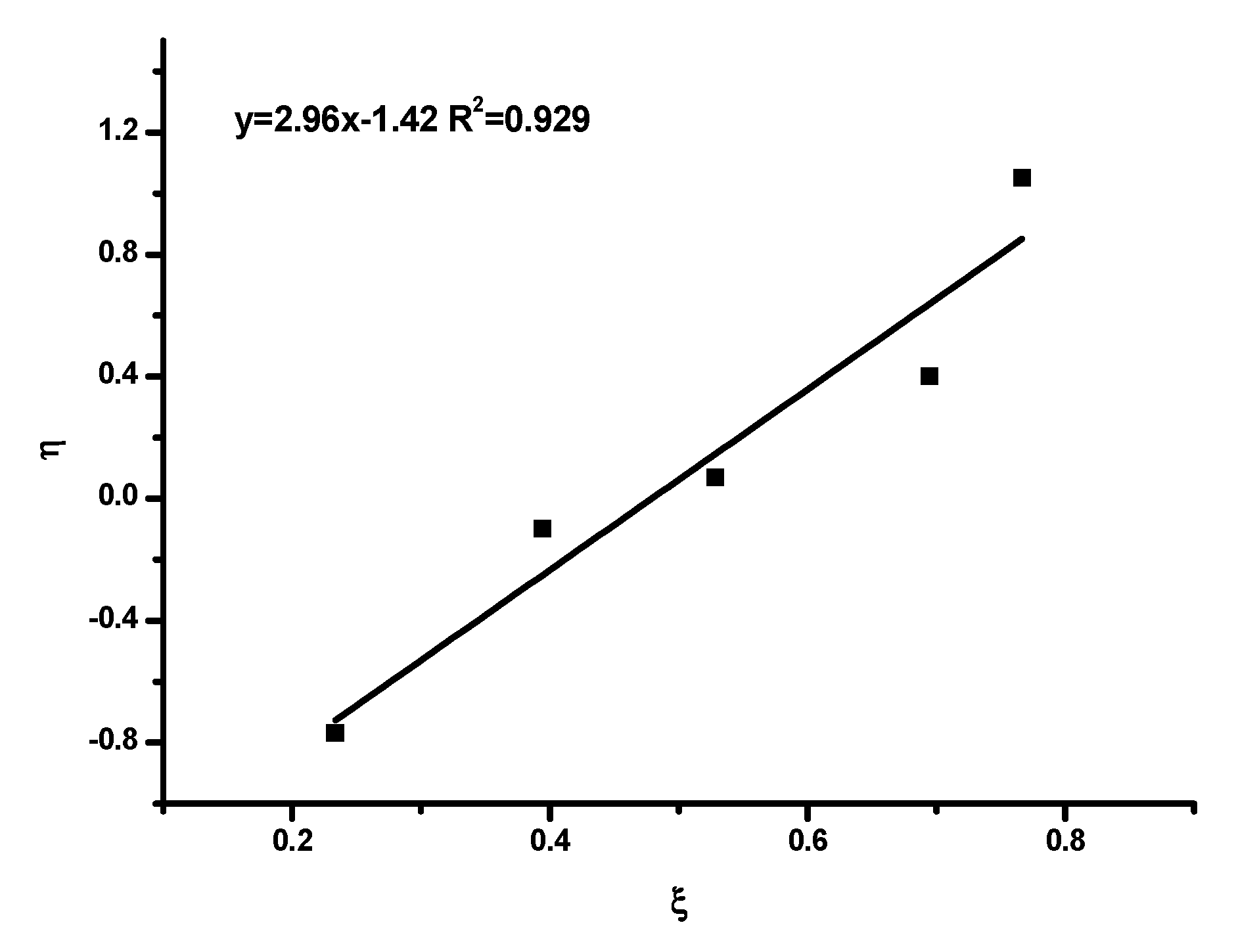
Reactivity Ratios
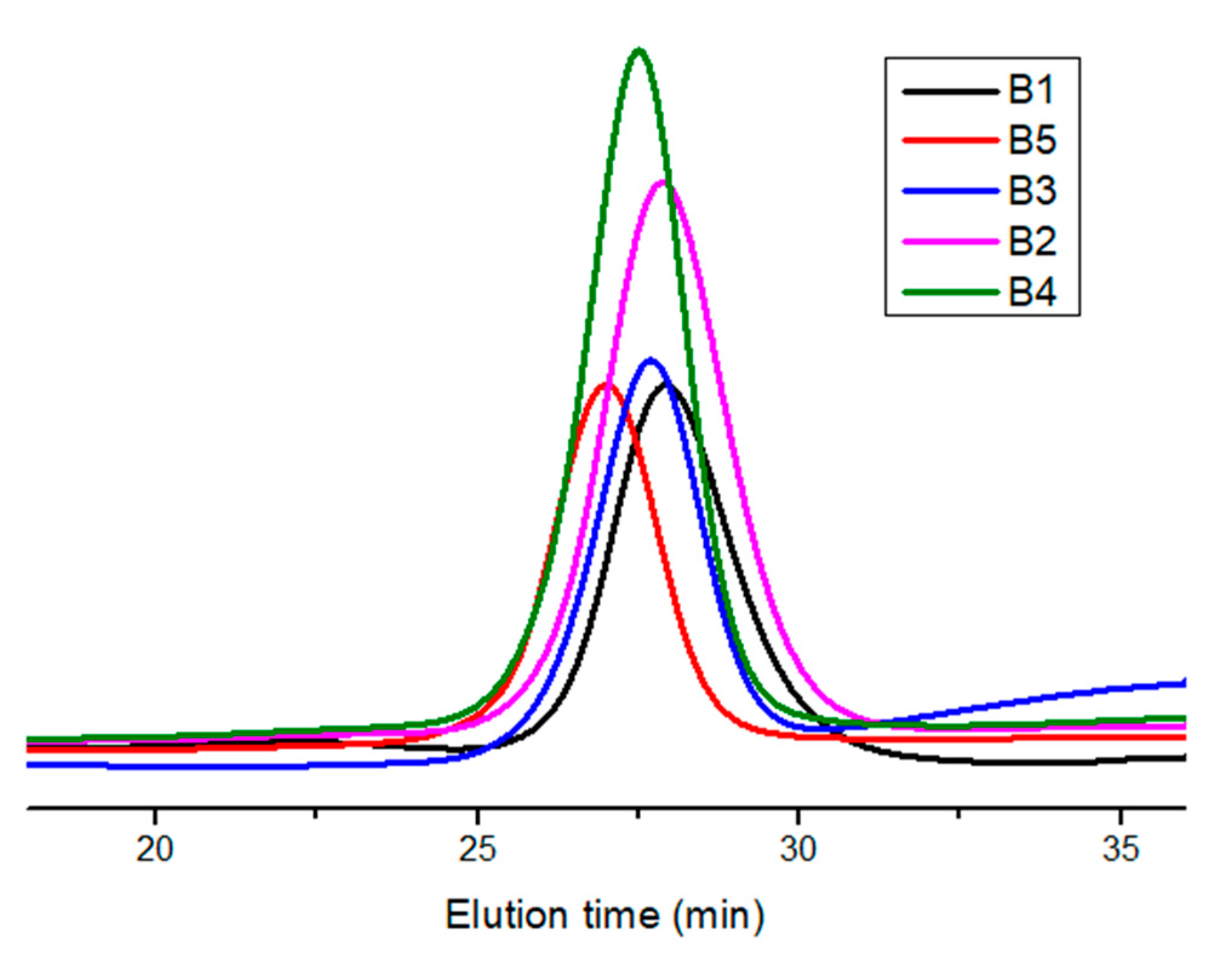
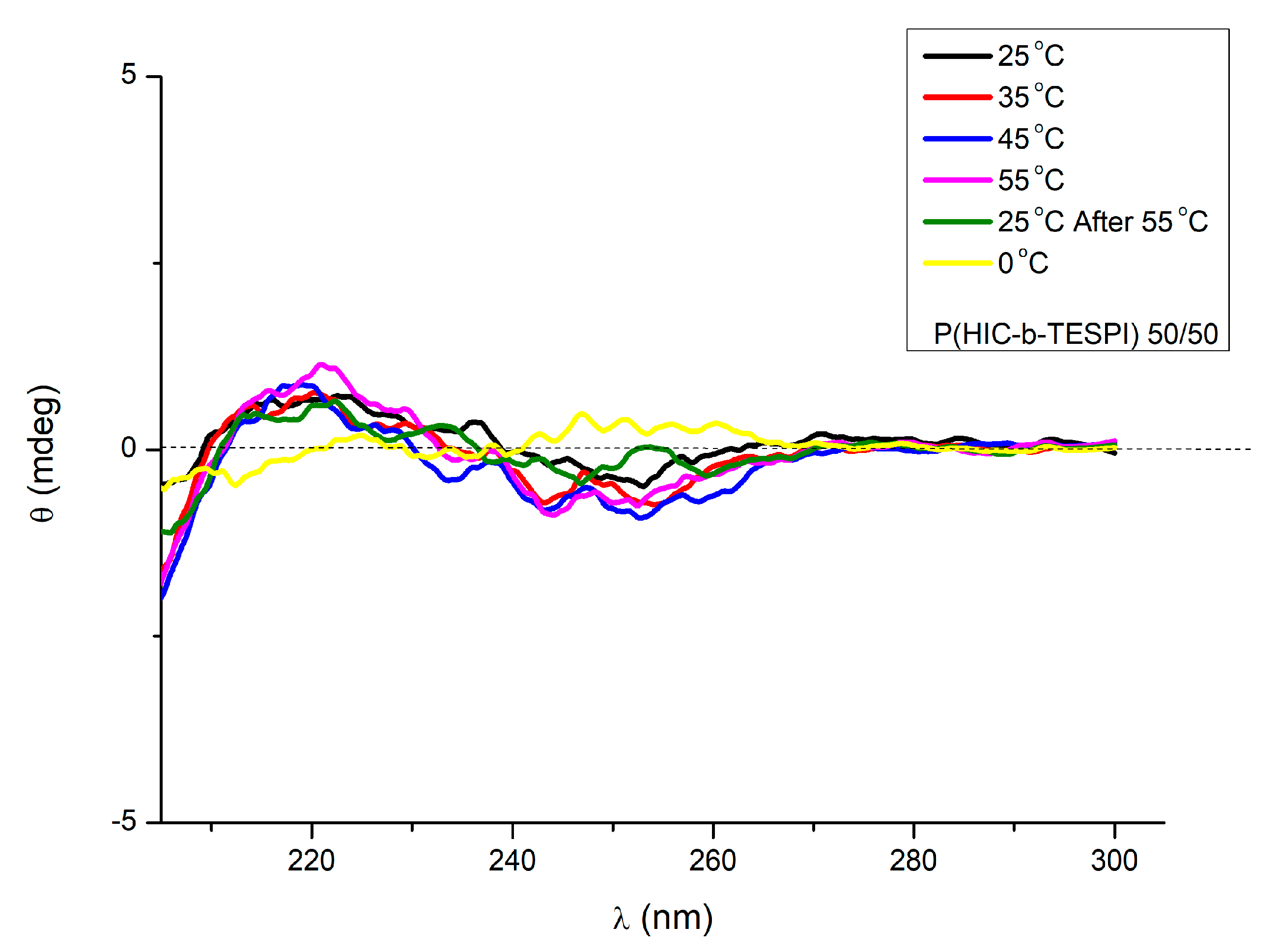
3.3. Block Copolymers
3.4. Thermal Decomposition Studies
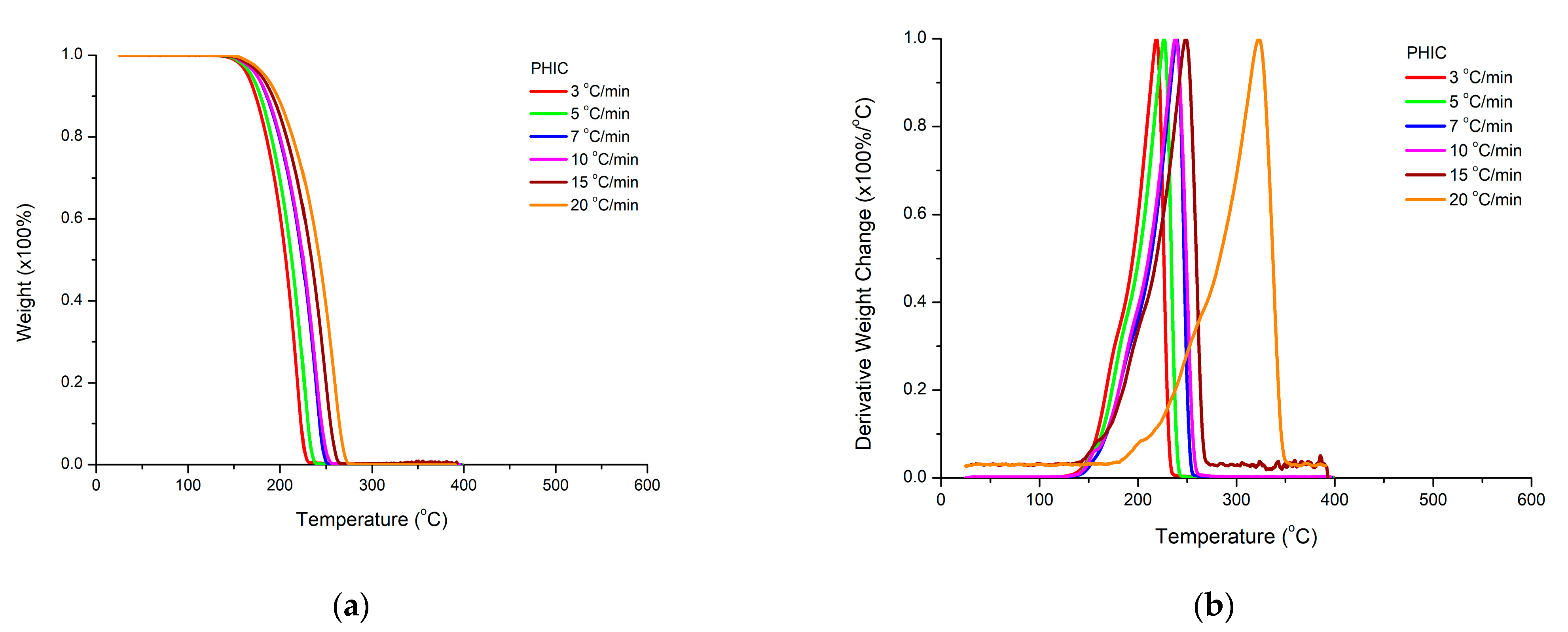
3.4.1. Homopolymers
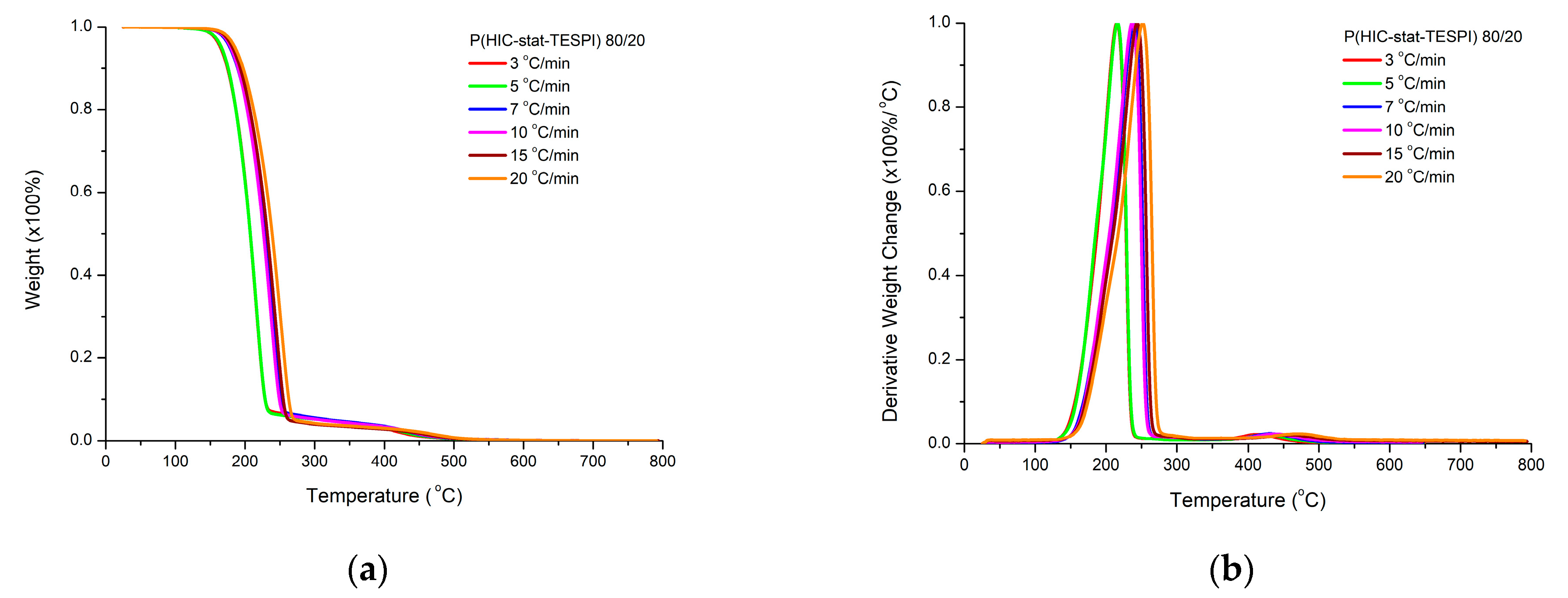
3.4.2. Statistical Copolymers
3.4.3. Block Copolymers
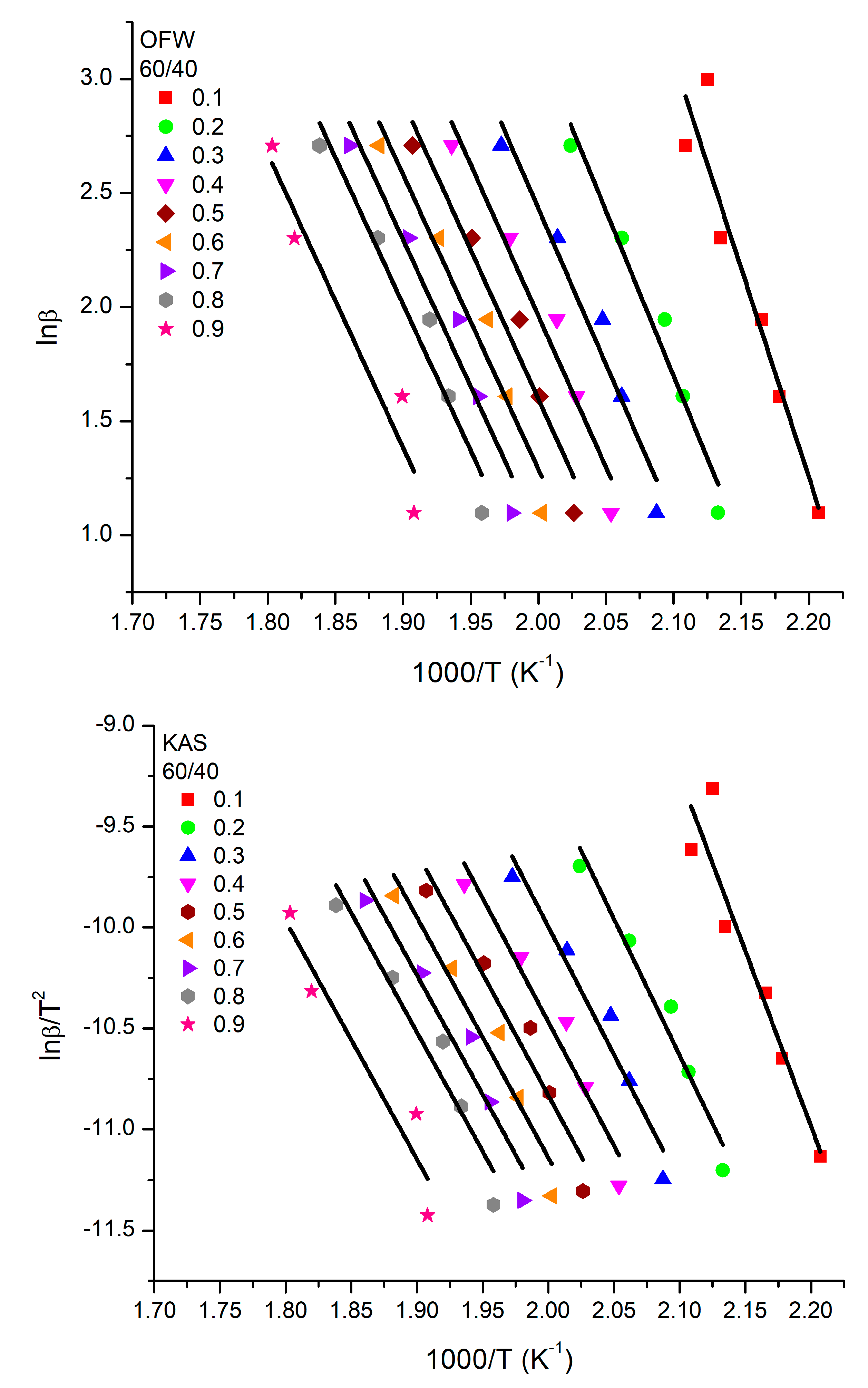
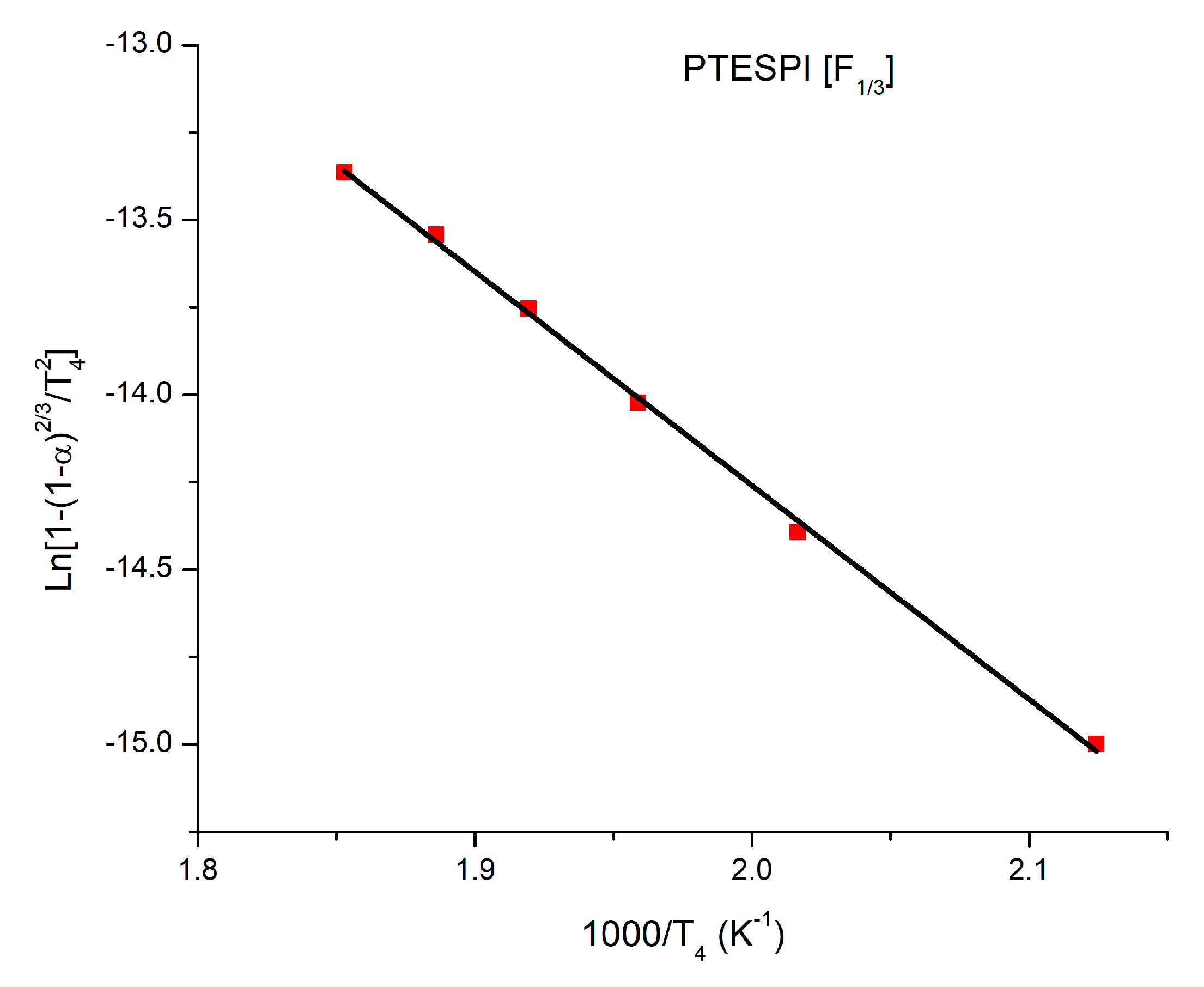
3.5. Kinetics of the Thermal Decomposition of the Homopolymers and the Statistical Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Βur, A.J.; Fetters, L.J. The Chain Structure, Polymerization and Conformation of Polyisocyanates. Chem. Rev. 1976, 76, 727–745. [Google Scholar] [CrossRef]
- Schneider, N.S.; Furusaki, S.; Lenz, R.W. Chain stiffness in polyisocyanates. J. Polym. Sci. Part A Gen. Pap. 1965, 3, 933–948. [Google Scholar] [CrossRef]
- Zhao, W.; Kloczkowski, A.; Mark, J.E.; Erman, B.; Bahar, I. Main-Chain Lyotropic Liquid-Crystalline Elastomers. 1. Syntheses of Cross-Linked Polyisocyanate Gels Acquiring Liquid-Crystalline Behavior in the Swollen State. Macromolecules 1996, 29, 2796–2804. [Google Scholar] [CrossRef]
- Gu, H.; Sato, T.; Teramoto, A.; Varichon, L.; Green, M.M. Molecular Mechanisms for the Optical Activities of Polyisocyanates Induced by Intramolecular Chiral Perturbations. Polym. J. 1997, 29, 77–84. [Google Scholar] [CrossRef]
- Shashoua, V.E.; Sweeny, W.; Tietz, R.F. The Homopolymerization of Monoisocyanates. J. Am. Chem. Soc. 1960, 82, 866–873. [Google Scholar] [CrossRef]
- Tonelli, A.E. Conformational Characteristics of the Poly(n-alkyl isocyanates). Macromolecules 1974, 7, 628–631. [Google Scholar] [CrossRef]
- Shashoua, V.E. The Homopolymerization of Monoisocyanates. J. Am. Chem. Soc. 1959, 81, 3156. [Google Scholar] [CrossRef]
- Lecomte, L.; Desreux, V. Dielectric properties of poly(4-methylphenylisocyanate) and poly(4-methoxyphenylisocyanate) in solution. Eur. Polym. J. 1976, 12, 741–747. [Google Scholar] [CrossRef]
- Chen, J.T.; Thomas, E.L.; Ober, C.K.; Mao, G.-P. Self-Assembled Smectic Phases in Rod-Coil Block Copolymers. Science 1996, 273, 343–346. [Google Scholar] [CrossRef]
- Chen, J.T.; Thomas, E.L.; Ober, C.K.; Hwang, S.S. Zigzag Morphology of a Poly(styrene-b-hexyl isocyanate) Rod-Coil Block Copolymer. Macromolecules 1995, 28, 1688–1697. [Google Scholar] [CrossRef]
- Vazaios, A.; Touris, A.; Echeverria, M.; Zorba, G.; Pitsikalis, M. Micellization Behaviour of Linear and Nonlinear Block Copolymers Based on Poly(n-hexyl isocyanate) in Selective Solvents. Polymers 2020, 12, 1678. [Google Scholar] [CrossRef] [PubMed]
- Green, M.M.; Peterson, N.C.; Sato, T.; Teramoto, A.; Cook, R.; Lifson, S. A Helical Polymer with a Cooperative Response to Chiral Information. Science 1995, 268, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Matsuda, M.; Nakano, T.; Yashima, E. Asymmetric Polymerization of Isocyanates with Optically Active Anionic Initiators. Polym. J. 1993, 4, 391–396. [Google Scholar] [CrossRef]
- Μayer, S.; Zentel, R. A new chiral polyisocyanate: An optical switch triggered by a small amount of photochromic side groups, Macromol. Chem. Phys. 1998, 199, 1675–1682. [Google Scholar]
- Baudis, S.; Ligon, S.C.; Seidler, K.; Weigel, G.; Grasl, C.; Bergmeister, H.; Schima, H.; Liska, R. Hard-block degradable thermoplastic urethane-elastomers for electrospun vascular prostheses. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1272–1280. [Google Scholar] [CrossRef]
- Godfrey, R.A.; Miller, G.W. Block polymers of isocyanates and vinyl monomers by homogeneous anionic polymerization. J. Polym. Sci. Part A-1 Polym. Chem. 1969, 7, 2387–2404. [Google Scholar] [CrossRef]
- East, G.; Furukawa, H. Initiation of the anionic polymerization of isocyanates with sodium naphthalane. Polymer 1979, 20, 659–661. [Google Scholar] [CrossRef]
- Berger, M.N. Addition Polymers of Monofunctional Isocyanates. J. Macromol. Sci. Part C Polym. Rev. 1973, 9, 269–303. [Google Scholar] [CrossRef]
- Chae, C.-G.; Seo, H.-B.; Lee, J.-S. Living anionic polymerization of isocyanates. In Anionic Polymerization: Priciples, Practice, Strength, Consequences and Applications; Hadjichristidis, N., Hirao, A., Eds.; Springer: Tokyo, Japan, 2015; pp. 339–386. [Google Scholar]
- Shin, Y.-D.; Ahn, J.-H.; Lee, J.-S. Anionic polymerization of isocyanates with optical functionalities. Polymer 2001, 42, 7979–7985. [Google Scholar] [CrossRef]
- Shin, Y.D.; Kim, S.Y.; Ahn, J.H.; Lee, J.S. Synthesis of Poly(n-hexyl isocyanate) by Controlled Anionic Polymerization in the Presence of NaBPh4. Macromolecules 2001, 34, 2408–2410. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Shin, Y.-D.; Kim, S.-Y.; Lee, J.-S. Synthesis of well-defined block copolymers of n-hexyl isocyanate with isoprene by living anionic polymerization. Polymer 2003, 44, 3847–3854. [Google Scholar] [CrossRef]
- Lee, J.-S.; Ryu, S.-W. Anionic living polymerization of 3-(triethoxysilyl)propyl isicyanate. Macromolecules 1999, 32, 2085–2087. [Google Scholar] [CrossRef]
- Kang, N.-G.; Kang, B.-G.; Koh, H.-D.; Changez, M.; Lee, J.-S. Block copolymers containing pyridine moieties: Precise synthesis and applications. React. Funct. Polym. 2009, 69, 470–479. [Google Scholar] [CrossRef]
- Vazaios, A.; Pitsikalis, M.; Hadjichristidis, N. Triblock copolymers and penatblock terpolymers of n-hexyl isocyanate with styrene and isoprene: Synthesis, characterization and thermal properties. J. Polym. Sci. Polym. Chem. Ed. 2003, 41, 3094–3102. [Google Scholar] [CrossRef]
- Zorba, G.; Vazaios, A.; Pitsikalis, M.; Hadjichristidis, N. Anionic polymerization of n-hexyl isocyanate with monofunctional initiators. Application in the synthesis of diblock copolymers with styrene and isoprene. J. Polym. Sci. Polym. Chem. Ed. 2005, 43, 3533–3542. [Google Scholar] [CrossRef]
- Min, J.; Yoo, H.-S.; Shah, P.N.; Chae, C.-G.; Lee, J.-S. Enolate anionic initiator, sodium deoxybenzoin, for leading living natures by formation of aggregates at the growth chain ends. J. Polym. Sci. Polym. Chem. Ed. 2013, 51, 1742–1748. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Shin, Y.-D.; Nath, G.Y.; Park, S.-Y.; Rahman, M.S.; Samal, S.; Lee, J.-S. Unprecedented Control over Polymerization of n-Hexyl Isocyanate using an Anionic Initiator Having Synchronized Function of Chain-End Protection. J. Am. Chem. Soc. 2005, 127, 4132–4133. [Google Scholar] [CrossRef]
- Rahman, M.S.; Yoo, H.-S.; Changez, M.; Lee, J.-S. Living Anionic Polymerization of Isocyanate Containing a Reactive Carbamate Group. Macromolecules 2009, 42, 3927–3932. [Google Scholar] [CrossRef]
- Zorba, G.; Pitsikalis, M.; Hadjichristidis, N. Novel well-defined star homopolymers and star-block copolymers of poly(n-hexyl isocyanate) by anionic polymerization. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2387–2399. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. Organotitanium(IV) compounds as catalysts for the polymerization of isocyanates: The polymerization of isocyanates with functionalized side chains. Macromolecules 1993, 26, 436–439. [Google Scholar] [CrossRef]
- Mourmouris, S.; Kostakis, K.; Pitsikalis, M.; Hadjichristidis, N. Polymerization of n-hexyl isocyanate with CpTiCl2(OR) (R = functional group or macromolecular chain): A route to ω-functionalized and block copolymers and terpolymers of n-hexyl isocyanate. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 6503–6514. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. “Living” titanium(IV) catalyzed coordination polymerizations of isocyanates. J. Am. Chem. Soc. 1991, 113, 5065–5066. [Google Scholar] [CrossRef]
- Wu, J.; Pearce, E.M.; Kwei, T.K. A Novel Rod−Coil Block Copolymer and Its Compatible Blends. Macromolecules 2001, 34, 1828–1836. [Google Scholar] [CrossRef]
- Liu, X.; Deng, J.; Wu, Y.; Zhang, L. Amphiphilic triblock terpolymers consisting of poly(n-hexyl isocyanate) and poly(ethylene glycol): Preparation and characterization. Polymer 2012, 53, 5717–5722. [Google Scholar] [CrossRef]
- Hoff, S.M.; Novak, B.M. Complex architectures through living polymerizations. The synthesis of “once-broken worms” and triblock copolymers using bimetallic initiators. Macromolecules 1993, 26, 4067–4069. [Google Scholar] [CrossRef]
- Touris, A.; Kostakis, K.; Mourmouris, S.; Kotzabasakis, V.; Pitsikalis, M.; Hadjichristidis, N. Complex Macromolecular Architectures Based on n-Hexyl Isocyanate and ϵ-Caprolactone Using Titanium-Mediated Coordination Polymerization. Macromolecules 2008, 41, 2426–2438. [Google Scholar] [CrossRef]
- Hoff, S.M.; Novak, B.M. Synthesis and Characterization of Wormlike Three-Arm Poly(n-hexyl isocyanate) Star Polymers. Macromolecules 2001, 34, 3849–3855. [Google Scholar]
- Choinopoulos, I.; Koinis, S.; Pitsikalis, M. Synthesis and characterization of chiral poly(l-lactide-b-hexyl isocyanate) macromonomers with norbornenyl end groups and their homopolymerization through ring opening metathesis polymerization to afford polymer brushes. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1102–1112. [Google Scholar] [CrossRef]
- Choinopoulos, I.; Patias, G.; Koinis, S.; Pitsikalis, M. Synthesis and characterization of brush diblock and triblock copolymers bearing polynorbornene backbone and poly(l-lactide) and/or poly(hexyl isocyanate) side chains by a combination of coordination and ring opening metathesis polymerization. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3455–3465. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Du, J.; Rainbolt, E.A.; Pathiranage, T.M.S.K.; Huang, P.; Reuther, J.F.; Novak, B.M.; Biewer, M.C.; Stefan, M.C. A semiconducting liquid crystalline regioregular poly(3-hexylthiophene) and nematic poly(n-hexyl isocyanate) and its application in bulk heterojunction solar cells. J. Mater. Chem. A 2014, 2, 16148–16156. [Google Scholar] [CrossRef]
- Miyake, G.M.; Weitekamp, R.A.; Piunova, V.A.; Grubbs, R.H. Grubbs, Synthesis of isocyanate-based brush block copolymers and their rapid self-assembly to infrared-reflecting photonic crystals. J. Am. Chem. Soc. 2012, 134, 14249–14254. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Nishikawa, N.; Kawato, D.; Suemasa, D.; Jung, S.; Kim, Y.Y.; Ree, M.; Kakuchi, T. Precise synthesis of a rod-coil miktoarm star copolymer containing poly(n-hexyl isocyanate) and aliphatic ester. Polym. Chem. 2014, 5, 588–599. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Mihara, T.; Kikuchi, M.; Lien, L.T.N.; Nagai, K. Synthesis of Methacrylate-Ended Poly(n-hexyl isocyanate) Rodlike Macromonomers and Their Radical Copolymerization Behavior. Macromolecules 2007, 40, 950–958. [Google Scholar] [CrossRef]
- Deike, S.; Binder, W.H. Induction of Chirality in β-Turn Mimetic Polymer Conjugates via Postpolymerization “Click” Coupling. Macromolecules 2017, 50, 2637–2644. [Google Scholar] [CrossRef]
- Lee, J.S.; Hirao, A.; Nakahama, S. Polymerization of monomers containing fynctional silyl groups. 5. Synthesis of new porous membranes with functional groups. Macromolecules 1988, 21, 274–276. [Google Scholar] [CrossRef]
- Lee, J.S.; Hirao, A.; Nakahama, S. Polymerization of monomers containing fynctional silyl groups. 7. Synthesis of new porous membranes with fcontrolled microstructures. Macromolecules 1989, 22, 2602–2606. [Google Scholar] [CrossRef]
- Yu, K.; Yang, L.; Wang, J.; Zhu, Z.; Wang, T.-J. Modification of nanosilica particles with hydrophobic modifier bis[3-(triethoxysilyl)propyl]tetrasulfide by using micro-injection in aqueous solutions. Colloids Surf. A 2020, 599, 124852. [Google Scholar] [CrossRef]
- Sayyadian, M.; Jamshidi, M.; Ghamarpoor, R.; Razavizadeh, M. Silanization of functionalized PET fabric to improve PET-nitrile rubber (NBR) adhesion; effects of functionalization type and silane concentration. Arab. J. Chem. 2023, 16, 105098. [Google Scholar] [CrossRef]
- Tian, Q.; Tang, Y.; Ding, T.; Li, X.; Zhang, Z. Effect of nano-sioica surface-capped with bis[3-(triethoxysilyl)propyl]tetrasulfide on the mechanical properties of styrene-butadiene rubber/butadiene rubber nanocomposites. Comp. Commun. 2018, 10, 190–193. [Google Scholar]
- Borase, T.; Ninjbadgar, T.; Kapetanakis, A.; Roche, S.; Connor, R.O.; Kerskens, C.; Heise, A.; Brougham, D.F. Stable aqueous dispersions of glycopeptide-grafted selectably functionalized magnetic nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 3164–3167. [Google Scholar] [CrossRef]
- Choinopoulos, I.; Koinis, S.; Pitsikalis, M. Synthesis and characterization of chiral poly(alkyl isocyanates) by coordination polymerization using a chiral half-titanocene complex. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 2141–2151. [Google Scholar] [CrossRef]
- Ratkanthwar, K.; Zhao, J.; Zhang, H.; Hadjichristidis, N.; Mays, J.W. Schlenk techniques for anioinic polymerization. In Anionic Polymerization: Principles, Practice, Strength, Consequences and Applications; Hadjichristidis, N., Hirao, A., Eds.; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Hagiopol, C. Copolymerization: Toward a Systematic Approach; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Kelen, T.; Tüdos, F. Analysis of the Linear Methods for Determining Copolymerization Reactivity Ratios. I. A New Improved Linear Graphic Method. J. Macromol. Sci. Part A Chem. 1975, 9, 1–27. [Google Scholar] [CrossRef]
- Beginn, U. COPOINT—A simple computer program to determine copolymerization parameters by numerical integration. e-Polymers 2005, 5, 759–773. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 155–270. [Google Scholar]
- Igarashi, S. Representation of composition and blockiness of the copolymer by a triangular coordinate system. J. Polym. Sci. Part B Polym. Lett. 1963, 1, 359–363. [Google Scholar] [CrossRef]
- Shah, F.N.; Min, J.; Chae, C.G.; Nishikawa, N.; Suemasa, D.; Kakuchi, T.; Satoh, T.; Lee, J.S. “Helicity Inversion”: Linkage Effects of Chiral Poly(n-hexyl isocyanate)s. Macromolecules 2012, 45, 8961–8969. [Google Scholar] [CrossRef]
- Mantzara, D.; Katara, A.; Panteli, M.; Choinopoulos, I.; Pitsikalis, M. Synthesis and characterization of statistical and block copolymers of n-hexyl isocyanate and 2-chloroethyl isocyanate via coordination polymerization. J. Polym. Sci. 2023; submitted. [Google Scholar]
- Katara, A.; Mantzara, D.; Panteli, M.; Choinopoulos, I.; Pitsikalis, M. Statistical and Block Copolymers of n-Hexyl Isocyanate and 2–Phenylethyl Isocyanate via Coordination Polymerization. Synthesis, Characterization and Thermal Properties. Eur. Polym. J. 2023, 199, 112441. [Google Scholar] [CrossRef]
- Galukhin, A.; Liavitskaya, T.; Vyazovkin, S. Kinetic and Mechanistic Insights into Thermally Initiated Polymerization of Cyanate Esters with Different Bridging Groups. Macromol. Chem. Phys. 2019, 220, 1900141. [Google Scholar] [CrossRef]
- Ozawa, T. A New Method of Analyzing Thermogravimetric Data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. A quick, direct method for the determination of activation energy from thermogravimetric data. J. Polym. Sci. Part B Polym. Lett. 1966, 4, 323–328. [Google Scholar] [CrossRef]
- Ozawa, T. Kinetic analysis of derivative curves in thermal analysis. J. Therm. Anal. Calorim. 1970, 2, 301–324. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Aharoni, S.M. Rigid backbone polymers. X. Transitions in bulk polyisocyanates. J. Polym. Sci. Polym. Phys. Ed. 1980, 18, 1303–1310. [Google Scholar] [CrossRef]
- Durairaj, B.; Dimock, A.W.; Samulski, E.T.; Shaw, M.T. Investigation of the thermal degradation of alkyl isocyanate polymers by direct pyrolysis mass spectrometry. J. Polym. Sci. Polym. Chem. Ed. 1989, 27, 3211–3225. [Google Scholar] [CrossRef]
- Trache, D.; Abdelaziz, A.; Siouani, B. A simple and linear isoconversional method to determine the pre-exponential factors and the mathematical reaction mechanism functions. J. Therm. Anal. Calorim. 2017, 128, 335–348. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ratio | 80/20 | 60/40 | 50/50 | 40/60 | 20/80 |
|---|---|---|---|---|---|
| [CpTiCl2(O-(S)-2-Bu)] | 0.0528 g, (0.20 mmol) | 0.0564 g, (0.21 mmol) | 0.0568 g, (0.22 mmol) | 0.0544 g, (0.21 mmol) | 0.0529 g, (0.21 mmol) |
| TESPI | 1.6 mL, (6.46 mmol) | 2.8 mL, (11.31 mmol) | 3.3 mL, (13.32 mmol) | 3.7 mL, (14.94 mmol) | 4.4 mL, (17.77 mmol) |
| HIC | 3.8 mL, (26.08 mmol) | 2.5 mL, (17.16 mmol) | 1.9 mL, (13.04 mmol) | 1.4 mL, (9,61 mmol) | 0.6 mL, (4.11 mmol) |
| Yield | 38% | 39% | 47% | 41% | 44% |
| Sample | B1 | B2 | B3 | B4 | B5 |
|---|---|---|---|---|---|
| [CpTiCl2(O-(S)-2-Bu)] | 0.0345 g (0.13 mmol) | 0.0293 g (0.11 mmol) | 0.0349 g (0.14 mmol) | 0.0273 g (0.11 mmol) | 0.0285 g (0.12 mmol) |
| TESPI | 0.5 mL (2.01 mmol) | 0.8 mL (3.23 mmol) | 1.0 mL (4.03 mmol) | 1.1 mL (4.44 mmol) | 1.30 mL (5.25 mmol) |
| HIC | 1.0 mL (6.86 mmol) | 0.7 mL (4.80 mmol) | 0.5 mL (3.43 mmol) | 0.4 mL (2.74 mmol) | 0.2 mL (1.37 mmol) |
| Molar Monomer Ratio in Feed (HIC/TESPI) | a Mw × 10−3 | a Ð | b Mol Composition (HIC/TESPI) | c Yield |
|---|---|---|---|---|
| 80/20 | 22.1 | 1.17 | 87/13 | 38% |
| 60/40 | 25.0 | 1.25 | 68/32 | 39% |
| 50/50 | 23.6 | 1.26 | 53/47 | 47% |
| 40/60 | 22.0 | 1.28 | 45/54 | 41% |
| 20/80 | 11.8 | 1.36 | 19/81 | 44% |
| Method | rHIC | rTESPI |
|---|---|---|
| F-R | 1.68 | 1.22 |
| IF-R | 1.58 | 1.09 |
| K-T | 1.54 | 1.07 |
| ext K-T | 1.69 | 1.05 |
| COPOINT | 1.49 a | 0.95 b |
| SAMPLE | M(HIC)–M(HIC) | M(TESPI)–M(TESPI) | M(HIC)–M(TESPI) | μ(HIC) | μ(TESPI) |
|---|---|---|---|---|---|
| 20/80 | 0.04496 | 0.66496 | 0.29009 | 1.35 | 5.09 |
| 40/60 | 0.22981 | 0.31721 | 0.45299 | 1.96 | 2.48 |
| 50/50 | 0.30276 | 0.24256 | 0.45467 | 2.46 | 1.97 |
| 60/40 | 0.47609 | 0.12109 | 0.40283 | 3.84 | 1.50 |
| 80/20 | 0.75697 | 0.02277 | 0.22026 | 7.02 | 1.24 |
| Sample | a Mw × 10−3 | a Ð | b Mole% Composition (HIC/TESPI) |
|---|---|---|---|
| B1 | 11.1 | 1.19 | 84/16 |
| B2 | 12.2 | 1.19 | 65/35 |
| B3 | 13.5 | 1.12 | 28/72 |
| B4 | 14.7 | 1.14 | 26/74 |
| B5 | 17.3 | 1.16 | 14/86 |
| Conversion | Εa (kJ/mol) PHIC | Εa (kJ/mol) PTESPI | Εa (kJ/mol) 50/50 | Εa (kJ/mol) 60/40 | Εa (kJ/mol) 80/20 |
|---|---|---|---|---|---|
| 0.1 | 133.78 | 73.58 | 68.11 | 144.34 | 117.54 |
| 0.2 | 116.64 | 67.09 | 98.78 | 123.32 | 106.75 |
| 0.3 | 107.00 | 62.55 | 54.31 | 109.91 | 100.04 |
| 0.4 | 95.58 | 60.48 | 88.30 | 104.05 | 96.75 |
| 0.5 | 100.54 | 58.65 | 86.52 | 101.26 | 94.86 |
| 0.6 | 97.39 | 48.19 | 85.52 | 99.82 | 93.74 |
| 0.7 | 96.06 | 61.86 | 85.28 | 99.30 | 85.23 |
| 0.8 | 94.71 | 57.05 | 85.35 | 99.02 | 94.16 |
| 0.9 | 93.42 | 27.77 | 86.19 | 78.53 | 101.64 |
| Conversion | Εa (kJ/mol) PHIC | Εa (kJ/mol) PTESPI | Εa (kJ/mol) 50/50 | Εa (kJ/mol) 60/40 | Εa (kJ/mol) 80/20 |
|---|---|---|---|---|---|
| 0.1 | 134.46 | 81.39 | 75.25 | 152.06 | 125.16 |
| 0.2 | 118.38 | 75.29 | 106.76 | 131.56 | 114.58 |
| 0.3 | 109.39 | 71.04 | 62.69 | 118.10 | 108.03 |
| 0.4 | 98.64 | 69.19 | 96.81 | 112.38 | 104.87 |
| 0.5 | 103.47 | 67.55 | 95.19 | 109.72 | 103.08 |
| 0.6 | 100.57 | 57.52 | 94.22 | 108.38 | 102.05 |
| 0.7 | 99.38 | 62.17 | 94.43 | 107.96 | 93.59 |
| 0.8 | 98.15 | 69.27 | 95.25 | 107.78 | 102.64 |
| 0.9 | 96.99 | 42.13 | 75.25 | 87.49 | 110.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panteli, M.; Mantzara, D.; Katara, A.; Choinopoulos, I.; Pitsikalis, M. Synthesis and Characterization of Statistical and Block Copolymers of n-Hexyl Isocyanate and 3-(Triethoxysilyl) Propyl Isocyanate via Coordination Polymerization. Polymers 2023, 15, 4113. https://doi.org/10.3390/polym15204113
Panteli M, Mantzara D, Katara A, Choinopoulos I, Pitsikalis M. Synthesis and Characterization of Statistical and Block Copolymers of n-Hexyl Isocyanate and 3-(Triethoxysilyl) Propyl Isocyanate via Coordination Polymerization. Polymers. 2023; 15(20):4113. https://doi.org/10.3390/polym15204113
Chicago/Turabian StylePanteli, Maria, Dimitra Mantzara, Aikaterini Katara, Ioannis Choinopoulos, and Marinos Pitsikalis. 2023. "Synthesis and Characterization of Statistical and Block Copolymers of n-Hexyl Isocyanate and 3-(Triethoxysilyl) Propyl Isocyanate via Coordination Polymerization" Polymers 15, no. 20: 4113. https://doi.org/10.3390/polym15204113