

Plasma-Treated Electrospun PLGA Nanofiber Scaffold Supports Limbal Stem Cells
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation
2.3. Target Drum Design
2.4. Plasma Treatment
2.5. Sterilization
2.5.1. Gamma (γ) Irradiation
2.5.2. Alcohol Disinfection Followed by UV Radiation
2.6. Characterization
2.6.1. Scanning Electron Microscopy
2.6.2. Porosity
2.6.3. Mechanical Strength
2.6.4. Thermal Analysis
2.6.5. In Vitro Degradation
2.6.6. Wettability
2.7. Biological Testing
2.7.1. Limbal Stem Cells Isolation
2.7.2. Cell Attachment
2.7.3. Cytotoxicity Assay
2.8. Statistical Analysis
3. Results
3.1. Sterilization
3.2. Characterization
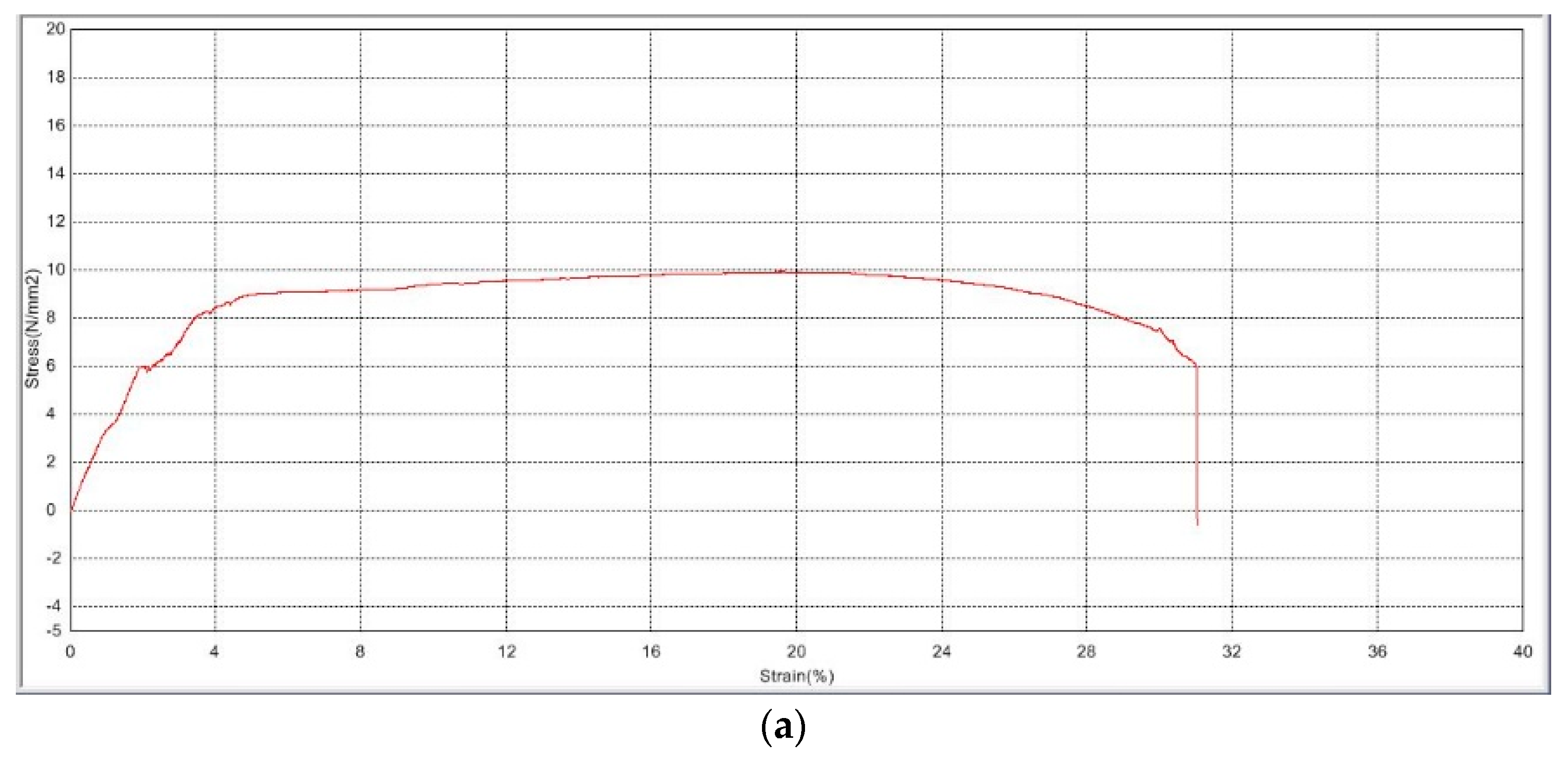
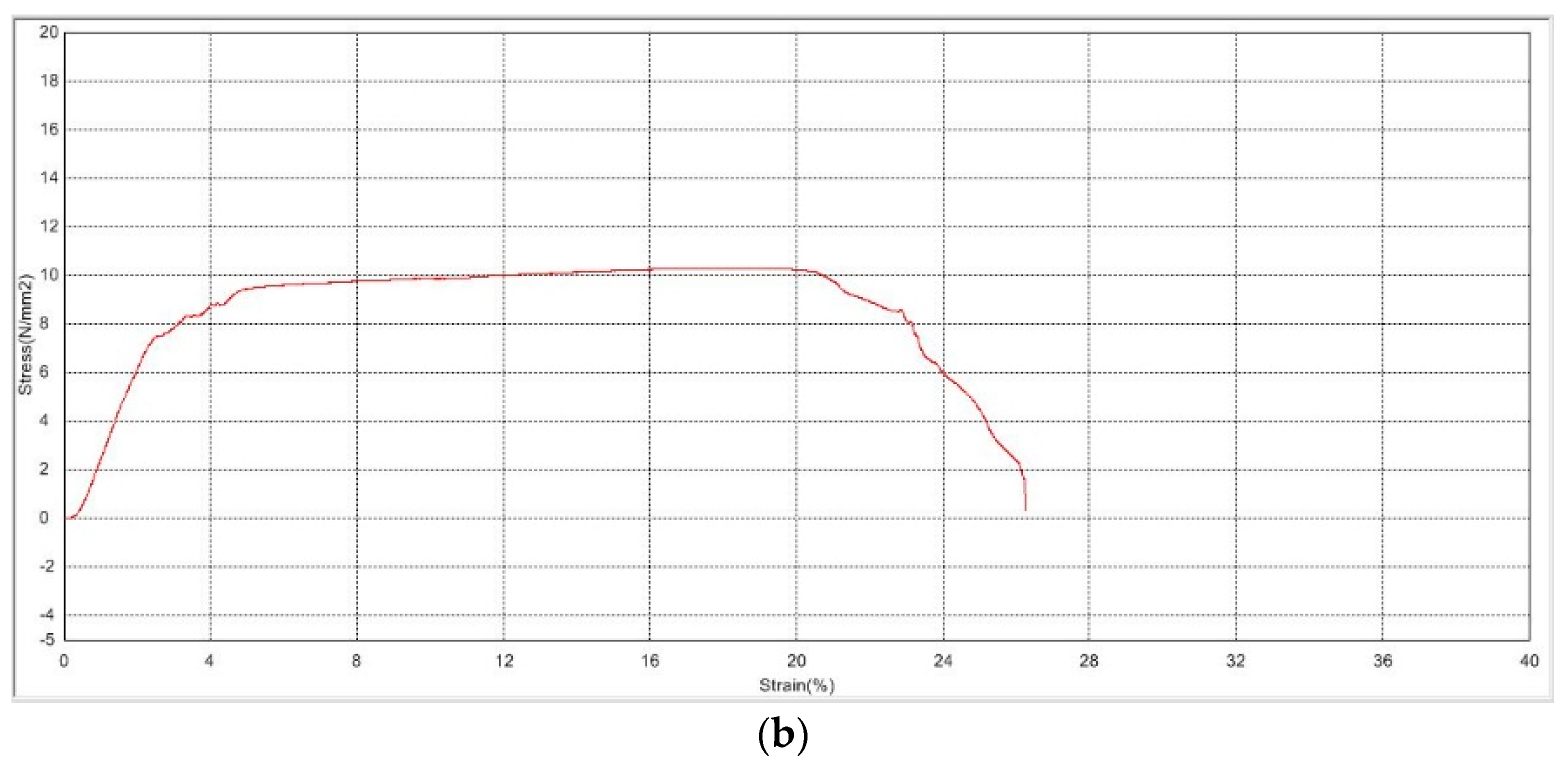
3.2.1. Mechanical Strength
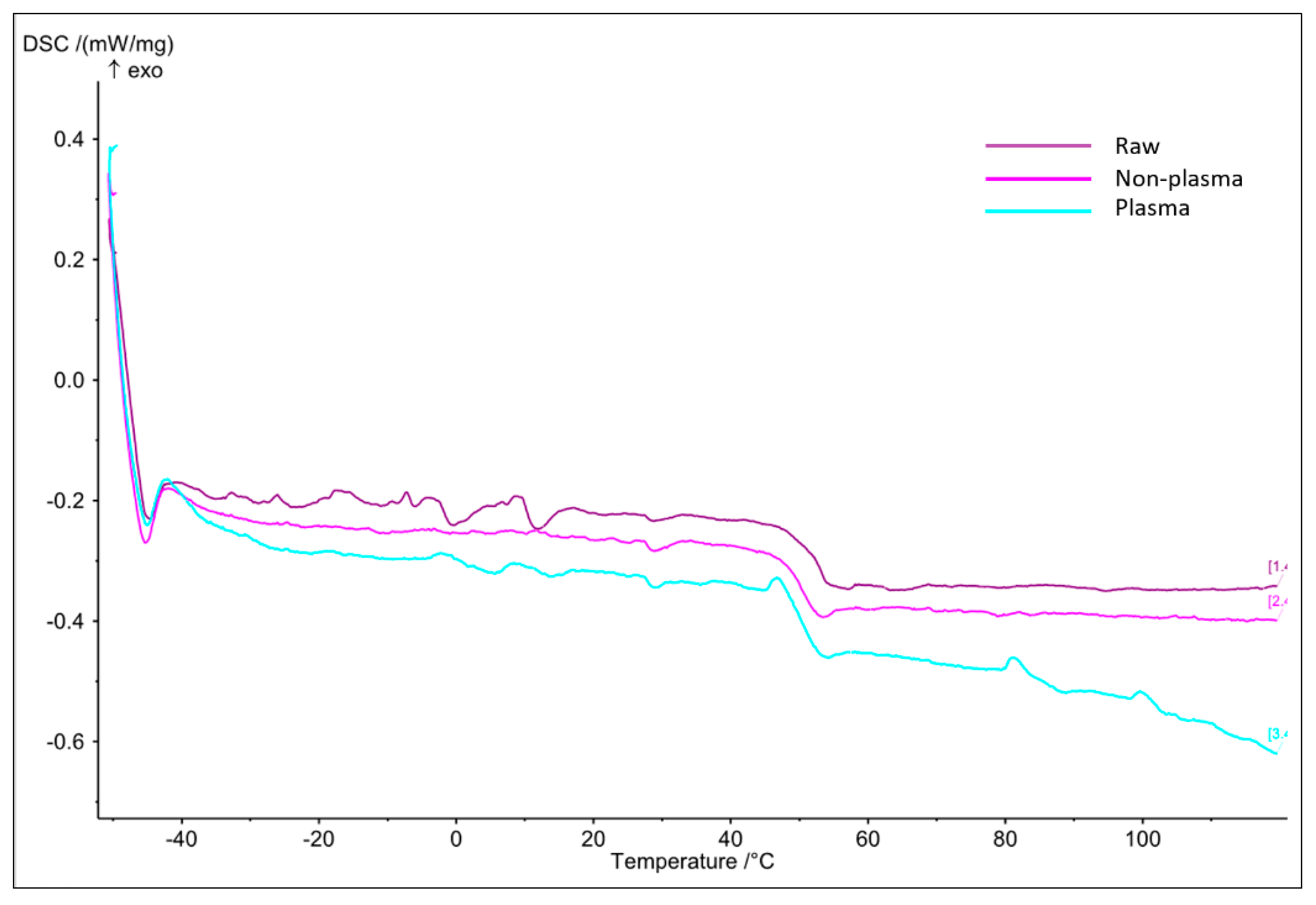
3.2.2. Thermal Analysis
- DSC
- b.
- TGA
3.2.3. Degradation
- Morphology
- b.
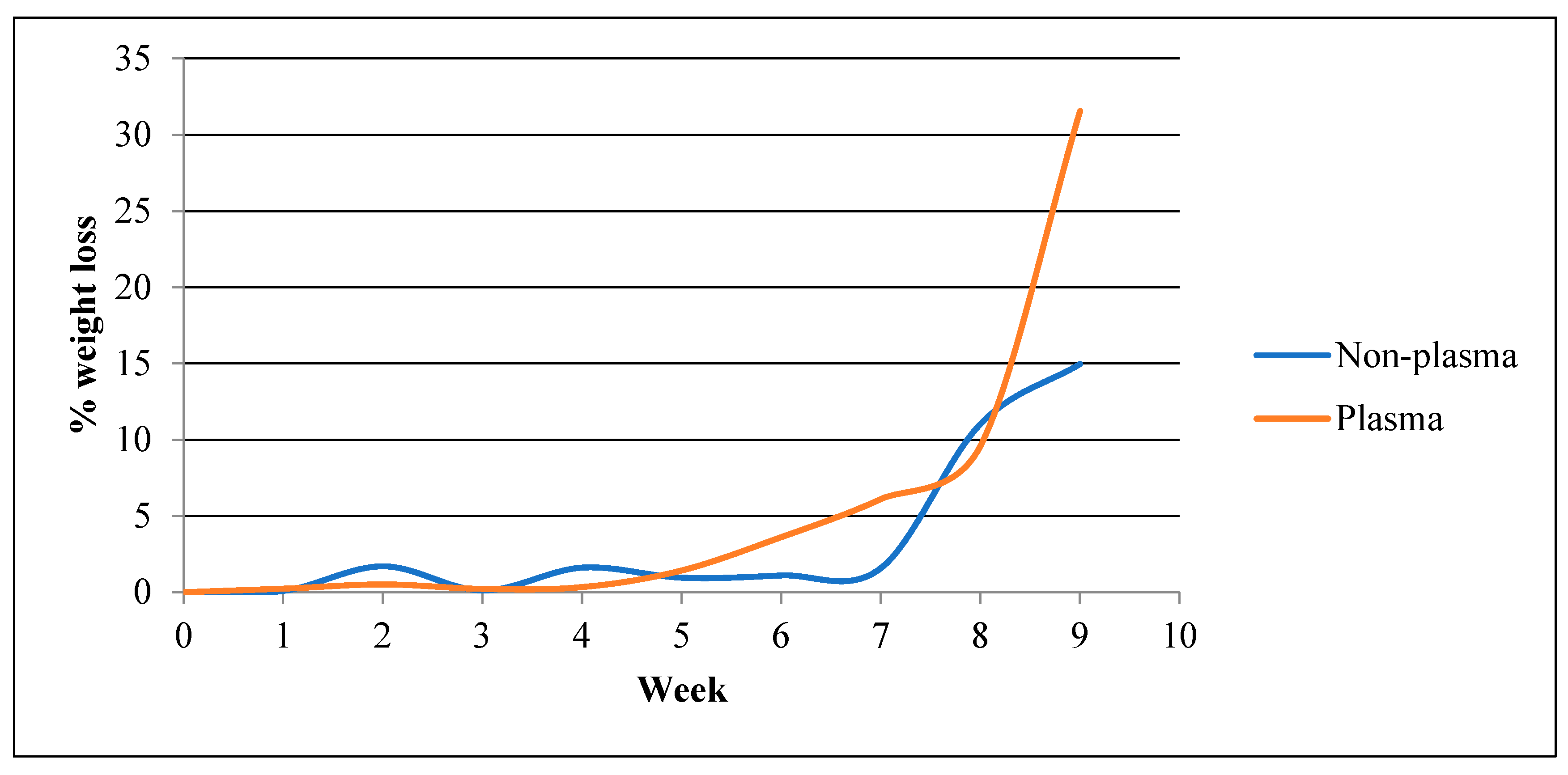
- Weight Loss
- c.
- pH changes
3.2.4. Wettability
- Water Uptake
- b.
- Contact Angle
3.3. Scanning Electron Microscopy
3.4. Biological Testing
3.4.1. Cell Attachment
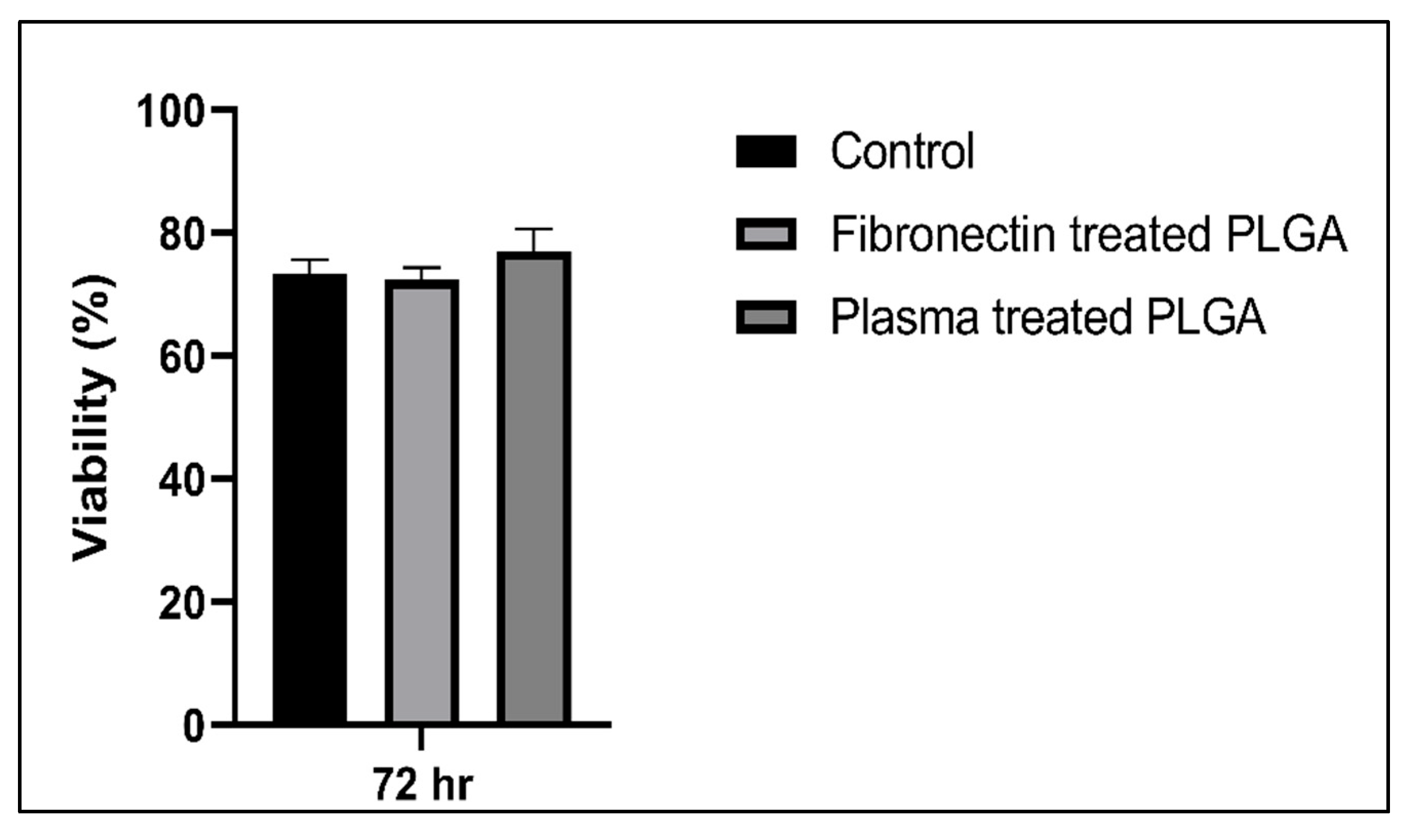
3.4.2. Cytotoxic Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Navaratnam, J.; Utheim, T.P.; Rajasekhar, V.K.; Shahdadfar, A. Substrates for Expansion of Corneal Endothelial Cells towards Bioengineering of Human Corneal Endothelium. J. Funct. Biomater. 2015, 6, 917–945. [Google Scholar] [CrossRef] [PubMed]
- Nowell, C.S.; Radtke, F. Corneal Epithelial Stem Cells and Their Niche at a Glance. J. Cell Sci. 2017, 130, 1021–1025. [Google Scholar]
- Deng, S.X.; Borderie, V.; Chan, C.C.; Dana, R.; Figueiredo, F.C.; Gomes, J.A.P.; Pellegrini, G.; Shimmura, S.; Kruse, F.E.; The International Limbal Stem Cell Deficiency Working Group. Global Consensus on Definition, Classification, Diagnosis, and Staging of Limbal Stem Cell Deficiency. Cornea 2019, 38, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Eom, Y.W.; Oh, J.-E.; Lee, J.I.; Baik, S.K.; Rhee, K.-J.; Shin, H.C.; Kim, Y.M.; Ahn, C.M.; Kong, J.H.; Kim, H.S. The Role of Growth Factors in Maintenance of Stemness in Bone Marrow-Derived Mesenchymal Stem Cells. Biochem. Biophys. Res. Commun. 2014, 445, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.N.; Bobba, S.; Richardson, A.; Park, M.; Watson, S.L.; Wakefield, D.; Di Girolamo, N. Native and Synthetic Scaffolds for Limbal Epithelial Stem Cell Transplantation. Acta Biomater. 2018, 65, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, H.; Shimmura, S.; Kobayashi, H.; Taguchi, T.; Asano-Kato, N.; Uchino, Y.; Kato, M.; Shimazaki, J.; Tanaka, J.; Tsubota, K. Collagen-immobilized Poly (Vinyl Alcohol) as an Artificial Cornea Scaffold That Supports a Stratified Corneal Epithelium. J. Biomed. Mater. Res. Part B Appl. Biomater. Off. J. Soc. Biomater. 2006, 76, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Sheardown, H. Dendrimer Crosslinked Collagen as a Corneal Tissue Engineering Scaffold: Mechanical Properties and Corneal Epithelial Cell Interactions. Biomaterials 2006, 27, 4608–4617. [Google Scholar] [CrossRef]
- Orwin, E.J.; Hubel, A. In Vitro Culture Characteristics of Corneal Epithelial, Endothelial, and Keratocyte Cells in a Native Collagen Matrix. Tissue Eng. 2000, 6, 307–319. [Google Scholar] [CrossRef]
- Sharma, S.; Mohanty, S.; Gupta, D.; Jassal, M.; Agrawal, A.K.; Tandon, R. Cellular Response of Limbal Epithelial Cells on Electrospun Poly-ε-Caprolactone Nanofibrous Scaffolds for Ocular Surface Bioengineering: A Preliminary in Vitro Study. Mol. Vis. 2011, 17, 2898. [Google Scholar]
- Baradaran-Rafii, A.; Biazar, E.; Heidari-Keshel, S. Cellular Response of Limbal Stem Cells on Polycaprolactone Nanofibrous Scaffolds for Ocular Epithelial Regeneration. Curr. Eye Res. 2016, 41, 326–333. [Google Scholar] [CrossRef]
- Roointan, A.; Kianpour, S.; Memari, F.; Gandomani, M.; Gheibi Hayat, S.M.; Mohammadi-Samani, S. Poly (Lactic-Co-Glycolic Acid): The Most Ardent and Flexible Candidate in Biomedicine! Int. J. Polym. Mater. Polym. Biomater. 2018, 67, 1028–1049. [Google Scholar] [CrossRef]
- Sefat, F.; McKean, R.; Deshpande, P.; Ramachandran, C.; Hill, C.J.; Sangwan, V.S.; Ryan, A.J.; MacNeil, S. Production, Sterilisation and Storage of Biodegradable Electrospun PLGA Membranes for Delivery of Limbal Stem Cells to the Cornea. Procedia Eng. 2013, 59, 101–116. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-Based Nanoparticles: An Overview of Biomedical Applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.; McKean, R.; Blackwood, K.A.; Senior, R.A.; Ogunbanjo, A.; Ryan, A.J.; MacNeil, S. Using Poly (Lactide-Co-Glycolide) Electrospun Scaffolds to Deliver Cultured Epithelial Cells to the Cornea. Regen. Med. 2010, 5, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Ortega, Í.; Ryan, A.J.; Deshpande, P.; MacNeil, S.; Claeyssens, F. Combined Microfabrication and Electrospinning to Produce 3-D Architectures for Corneal Repair. Acta Biomater. 2013, 9, 5511–5520. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, F.A.; Ju, H.W.; Moon, B.M.; Lee, O.J.; Kim, J.; Park, H.J.; Kim, D.W.; Kim, D.; Jang, J.E.; Khang, G. Hybrid Scaffolds Based on PLGA and Silk for Bone Tissue Engineering. J. Tissue Eng. Regen. Med. 2016, 10, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Feng, Y.; Guo, J.; Wang, H.; Li, Q.; Yang, J.; Hao, X.; Lv, J.; Ma, N.; Li, W. Surface Modification and Endothelialization of Biomaterials as Potential Scaffolds for Vascular Tissue Engineering Applications. Chem. Soc. Rev. 2015, 44, 5680–5742. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.O.; Liu, X.H.; Smith, L.A.; Ma, P.X. Nanostructured Polymer Scaffolds for Tissue Engineering and Regenerative Medicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 226–236. [Google Scholar] [CrossRef]
- Mann, B.K.; West, J.L. Cell Adhesion Peptides Alter Smooth Muscle Cell Adhesion, Proliferation, Migration, and Matrix Protein Synthesis on Modified Surfaces and in Polymer Scaffolds. J. Biomed. Mater. Res. 2002, 60, 86–93. [Google Scholar] [CrossRef]
- Niemczyk-soczynska, B.; Gradys, A.; Sajkiewicz, P. Hydrophilic Surface Functionalization of Electrospun. Polymers 2020, 12, 2636. [Google Scholar] [CrossRef]
- Intranuovo, F.; Gristina, R.; Fracassi, L.; Lacitignola, L.; Crovace, A.; Favia, P. Plasma Processing of Scaffolds for Tissue Engineering and Regenerative Medicine. Plasma Chem. Plasma Process. 2016, 36, 269–280. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Qi, N. Controllable Structure, Properties, and Degradation of the Electrospun PLGA/PLA-blended Nanofibrous Scaffolds. J. Appl. Polym. Sci. 2012, 125, E468–E476. [Google Scholar] [CrossRef]
- Pellegrini, G.; Golisano, O.; Paterna, P.; Lambiase, A.; Bonini, S.; Rama, P.; De Luca, M. Location and Clonal Analysis of Stem Cells and Their Differentiated Progeny in the Human Ocular Surface. J. Cell Biol. 1999, 145, 769–782. [Google Scholar] [CrossRef] [PubMed]
- López-Santos, C.; Terriza, A.; Portolés, J.; Yubero, F.; González-Elipe, A.R. Physiological Degradation Mechanisms of PLGA Membrane Films under Oxygen Plasma Treatment. J. Phys. Chem. C 2015, 119, 20446–20452. [Google Scholar] [CrossRef]
- Park, H.; Lee, K.Y.; Lee, S.J.; Park, K.E.; Park, W.H. Plasma-Treated Poly (Lactic-Co-Glycolic Acid) Nanofibers for Tissue Engineering. Macromol. Res. 2007, 15, 238–243. [Google Scholar] [CrossRef]
- Fu, K.; Pack, D.W.; Klibanov, A.M.; Langer, R. Visual Evidence of Acidic Environment within Degrading Poly(Lactic-Co-Glycolic Acid) (PLGA) Microspheres. Pharm. Res. 2000, 17, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Park, K.E.; Lee, K.Y.; Lee, S.J.; Park, W.H. Surface Characteristics of Plasma-treated PLGA Nanofibers. In Proceedings of the Macromolecular Symposia; Wiley Online Library: Hoboken, NJ, USA, 2007; Volume 249, pp. 103–108. [Google Scholar]
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol. 2016, 134, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Porth, J.M.; Deiotte, E.; Dunn, M.; Bashshur, R. A Review of the Literature on the Global Epidemiology of Corneal Blindness. Cornea 2019, 38, 1602–1609. [Google Scholar] [CrossRef] [PubMed]
- Gorna, K.; Gogolewski, S. The Effect of Gamma Radiation on Molecular Stability and Mechanical Properties of Biodegradable Polyurethanes for Medical Applications. Polym. Degrad. Stab. 2003, 79, 465–474. [Google Scholar] [CrossRef]
- Dua, H.S.; Gomes, J.A.P.; King, A.J.; Maharajan, V.S. The Amniotic Membrane in Ophthalmology. Surv. Ophthalmol. 2004, 49, 51–77. [Google Scholar] [CrossRef]
- Tosi, G.M.; Massaro-Giordano, M.; Caporossi, A.; Toti, P. Amniotic Membrane Transplantation in Ocular Surface Disorders. J. Cell Physiol. 2005, 202, 849–851. [Google Scholar] [CrossRef] [PubMed]
- Fatimah, S.S.; Ng, S.L.; Chua, K.H.; Hayati, A.R.; Tan, A.E.; Tan, G.C. Value of Human Amniotic Epithelial Cells in Tissue Engineering for Cornea. Hum. Cell 2010, 23, 141–151. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | 2 h | 24 h |
|---|---|---|
| Plasma average (%) | 0.6 ± 0.07 ↑ | 0.10 ± 0.03 ↑ |
| Non-plasma average (%) | 2.71 ± 0.12 ↓ | 4.0 ± 0.15 ↓ |
| p value | 0.32 | 0.16 |
| Scaffold | Average Angle |
|---|---|
| Before plasma treatment | 143.2 ± 1.0° |
| After plasma treatment | 130.9 ± 1.4° |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jafar, H.; Ahmed, K.; Rayyan, R.; Sotari, S.; Buqain, R.; Ali, D.; Al Bdour, M.; Awidi, A. Plasma-Treated Electrospun PLGA Nanofiber Scaffold Supports Limbal Stem Cells. Polymers 2023, 15, 4244. https://doi.org/10.3390/polym15214244
Jafar H, Ahmed K, Rayyan R, Sotari S, Buqain R, Ali D, Al Bdour M, Awidi A. Plasma-Treated Electrospun PLGA Nanofiber Scaffold Supports Limbal Stem Cells. Polymers. 2023; 15(21):4244. https://doi.org/10.3390/polym15214244
Chicago/Turabian StyleJafar, Hanan, Khalid Ahmed, Rama Rayyan, Shorouq Sotari, Rula Buqain, Dema Ali, Muawyah Al Bdour, and Abdalla Awidi. 2023. "Plasma-Treated Electrospun PLGA Nanofiber Scaffold Supports Limbal Stem Cells" Polymers 15, no. 21: 4244. https://doi.org/10.3390/polym15214244
APA StyleJafar, H., Ahmed, K., Rayyan, R., Sotari, S., Buqain, R., Ali, D., Al Bdour, M., & Awidi, A. (2023). Plasma-Treated Electrospun PLGA Nanofiber Scaffold Supports Limbal Stem Cells. Polymers, 15(21), 4244. https://doi.org/10.3390/polym15214244