
Facile Synthesis of Light-Switchable Polymers with Diazocine Units in the Main Chain



Abstract
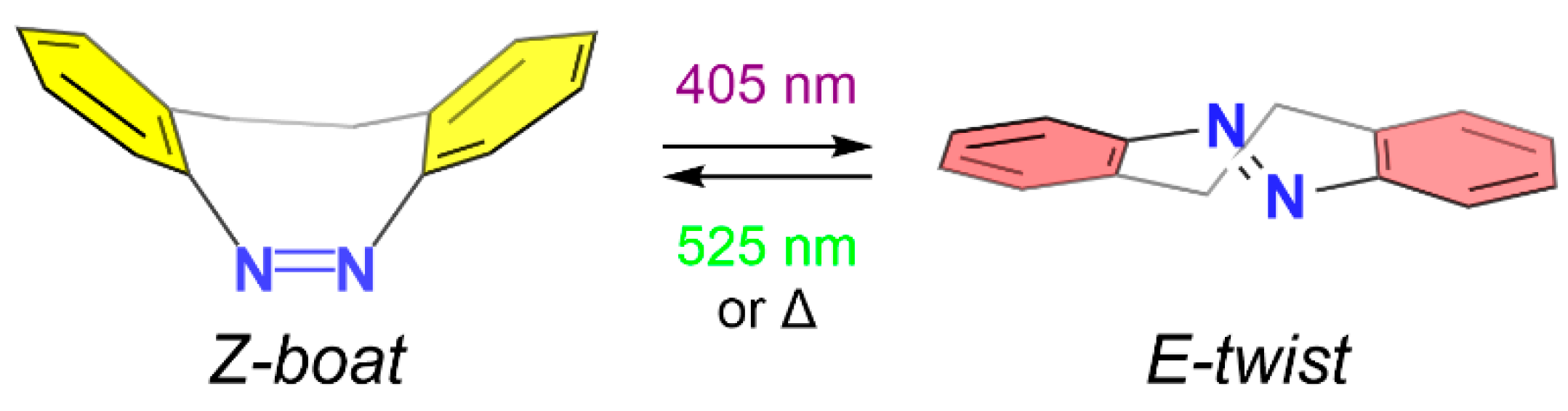
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
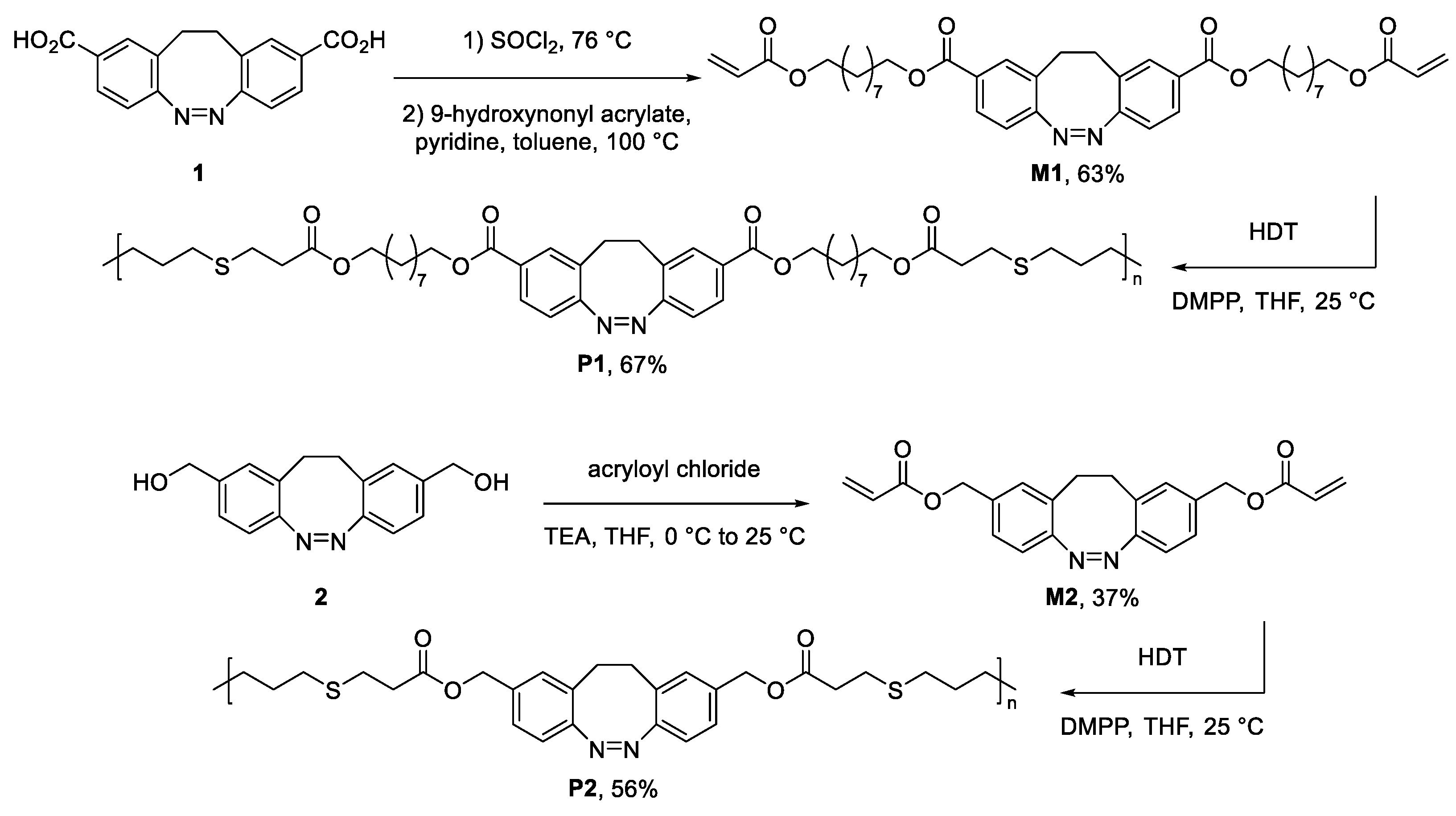
2.3. Synthetic Procedures





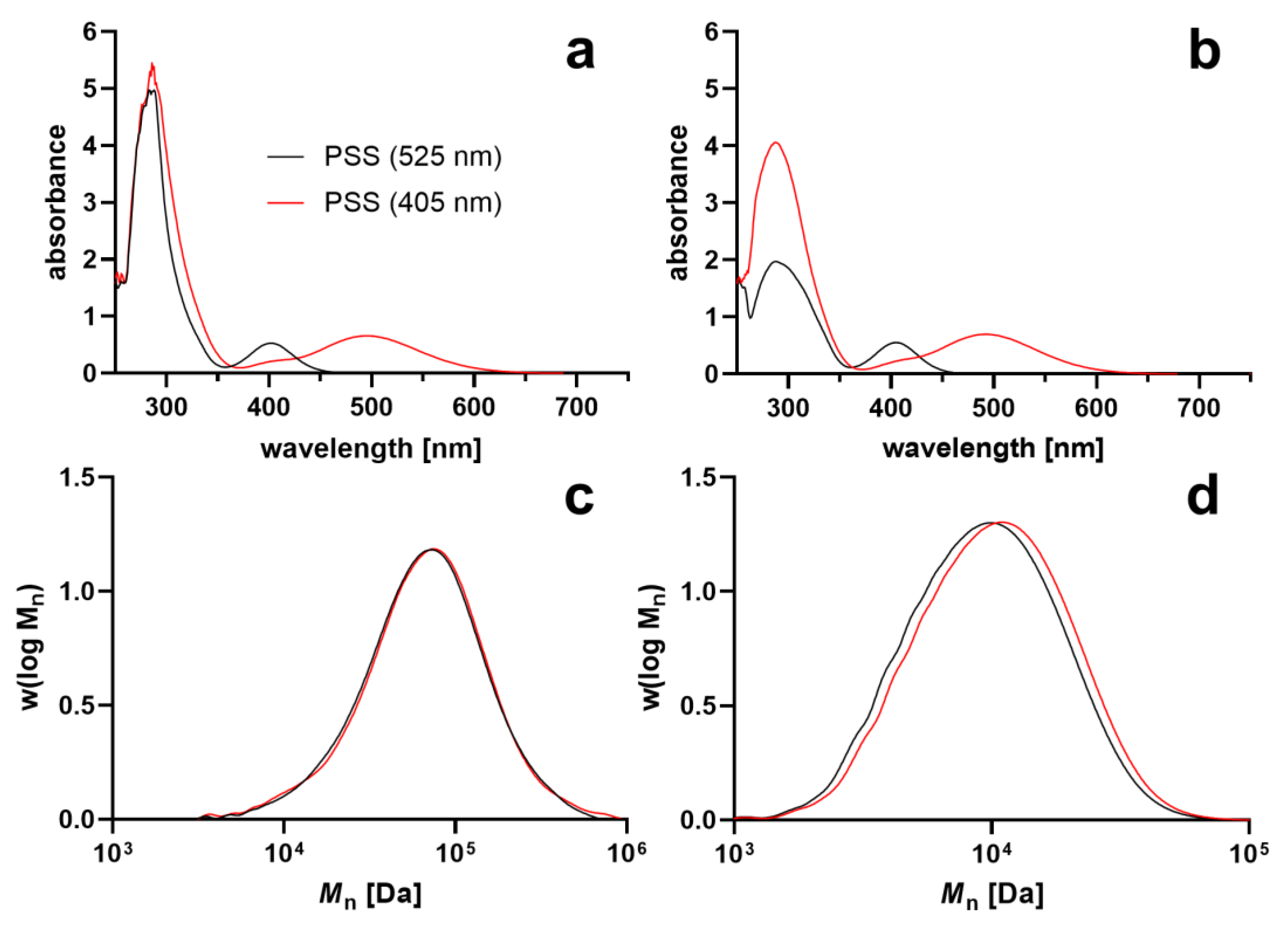
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pianowski, Z.L. (Ed.) Molecular Photoswitches; Wiley: Hoboken, NJ, USA, 2022; ISBN 9783527351046. [Google Scholar]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in Different Classes of Azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Kizilkan, E.; Strueben, J.; Staubitz, A.; Gorb, S.N. Bioinspired Photocontrollable Microstructured Transport Device. Sci. Robot. 2017, 2, 9454. [Google Scholar] [CrossRef] [PubMed]
- Oscurato, S.L.; Salvatore, M.; Maddalena, P.; Ambrosio, A. From Nanoscopic to Macroscopic Photo-Driven Motion in Azobenzene-Containing Materials. Nanophotonics 2018, 7, 1387–1422. [Google Scholar] [CrossRef]
- Erbas-Cakmak, S.; Leigh, D.A.; McTernan, C.T.; Nussbaumer, A.L. Artificial Molecular Machines. Chem. Rev. 2015, 115, 10081–10206. [Google Scholar] [CrossRef]
- Beharry, A.A.; Woolley, G.A. Azobenzene Photoswitches for Biomolecules. Chem. Soc. Rev. 2011, 40, 4422. [Google Scholar] [CrossRef] [PubMed]
- Dattler, D.; Fuks, G.; Heiser, J.; Moulin, E.; Perrot, A.; Yao, X.; Giuseppone, N. Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors. Chem. Rev. 2020, 120, 310–433. [Google Scholar] [CrossRef]
- Bléger, D.; Liebig, T.; Thiermann, R.; Maskos, M.; Rabe, J.P.; Hecht, S. Light-Orchestrated Macromolecular “Accordions”: Reversible Photoinduced Shrinking of Rigid-Rod Polymers. Angew. Chemie Int. Ed. 2011, 50, 12559–12563. [Google Scholar] [CrossRef]
- Sogawa, H.; Shiotsuki, M.; Sanda, F. Synthesis and Photoresponse of Helically Folded Poly(Phenyleneethynylene)s Bearing Azobenzene Moieties in the Main Chains. Macromolecules 2013, 46, 4378–4387. [Google Scholar] [CrossRef]
- Yu, Z.; Hecht, S. Reversible and Quantitative Denaturation of Amphiphilic Oligo(Azobenzene) Foldamers. Angew. Chem. Int. Ed. 2011, 50, 1640–1643. [Google Scholar] [CrossRef] [PubMed]
- Tie, C.; Gallucci, J.C.; Parquette, J.R. Helical Conformational Dynamics and Photoisomerism of Alternating Pyridinedicarboxamide/m-(Phenylazo)Azobenzene Oligomers. J. Am. Chem. Soc. 2006, 128, 1162–1171. [Google Scholar] [CrossRef]
- Izumi, A.; Nomura, R.; Masuda, T. Design and Synthesis of Stimuli-Responsive Conjugated Polymers Having Azobenzene Units in the Main Chain. Macromolecules 2001, 34, 4342–4347. [Google Scholar] [CrossRef]
- Zhong, H.-Y.; Chen, L.; Yang, R.; Meng, Z.-Y.; Ding, X.-M.; Liu, X.-F.; Wang, Y.-Z. Azobenzene-Containing Liquid Crystalline Polyester with π–π Interactions: Diverse Thermo- and Photo-Responsive Behaviours. J. Mater. Chem. C 2017, 5, 3306–3314. [Google Scholar] [CrossRef]
- Appiah, C.; Woltersdorf, G.; Pérez-Camargo, R.A.; Müller, A.J.; Binder, W.H. Crystallization Behavior of Precision Polymers Containing Azobenzene Defects. Eur. Polym. J. 2017, 97, 299–307. [Google Scholar] [CrossRef]
- Kuenstler, A.S.; Clark, K.D.; Read de Alaniz, J.; Hayward, R.C. Reversible Actuation via Photoisomerization-Induced Melting of a Semicrystalline Poly(Azobenzene). ACS Macro Lett. 2020, 9, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Yao, Y.; Zhuang, Q.; Lin, S. Mainchain Alternating Azopolymers with Fast Photo-Induced Reversible Transition Behavior. Macromolecules 2021, 54, 10040–10048. [Google Scholar] [CrossRef]
- Carstensen, N.O. QM/MM Surface-Hopping Dynamics of a Bridged Azobenzene Derivative. Phys. Chem. Chem. Phys. 2013, 15, 15017–15026. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Wang, D.H.; Koerner, H.; Vaia, R.A.; Tan, L.-S.; White, T.J. Enhancement of Photogenerated Mechanical Force in Azobenzene-Functionalized Polyimides. Angew. Chem. Int. Ed. 2012, 51, 4117–4121. [Google Scholar] [CrossRef]
- Fang, L.; Zhang, H.; Li, Z.; Zhang, Y.; Zhang, Y.; Zhang, H. Synthesis of Reactive Azobenzene Main-Chain Liquid Crystalline Polymers via Michael Addition Polymerization and Photomechanical Effects of Their Supramolecular Hydrogen-Bonded Fibers. Macromolecules 2013, 46, 7650–7660. [Google Scholar] [CrossRef]
- Wang, D.H.; Lee, K.M.; Lee, D.H.; Baczkowski, M.; Park, H.; McConney, M.E.; Tan, L.-S. Role of Alicyclic Conformation-Isomerization in the Photomechanical Performance of Azobenzene-Functionalized Cross-Linked Polyimides Containing Tetra-Substituted Cyclohexane Moieties. ACS Macro Lett. 2021, 10, 278–283. [Google Scholar] [CrossRef]
- Xue, X.; Zhu, J.; Zhang, Z.; Zhou, N.; Tu, Y.; Zhu, X. Soluble Main-Chain Azobenzene Polymers via Thermal 1,3-Dipolar Cycloaddition: Preparation and Photoresponsive Behavior. Macromolecules 2010, 43, 2704–2712. [Google Scholar] [CrossRef]
- Wu, Y.; Natansohn, A.; Rochon, P. Photoinduced Birefringence and Surface Relief Gratings in Polyurethane Elastomers with Azobenzene Chromophore in the Hard Segment. Macromolecules 2004, 37, 6090–6095. [Google Scholar] [CrossRef]
- Siewertsen, R.; Neumann, H.; Buchheim-Stehn, B.; Herges, R.; Näther, C.; Renth, F.; Temps, F. Highly Efficient Reversible Z−E Photoisomerization of a Bridged Azobenzene with Visible Light through Resolved S 1 (Nπ*) Absorption Bands. J. Am. Chem. Soc. 2009, 131, 15594–15595. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jing, X.; Zhang, J.; Yang, F.; Duan, C. Modifying Electron Injection Kinetics for Selective Photoreduction of Nitroarenes into Cyclic and Asymmetric Azo Compounds. Nat. Commun. 2022, 13, 1940. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.S.; Hüll, K.; Reynders, M.; Matsuura, B.S.; Leippe, P.; Ko, T.; Schäffer, L.; Trauner, D. Oxidative Approach Enables Efficient Access to Cyclic Azobenzenes. J. Am. Chem. Soc. 2019, 141, 17295–17304. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Eleya, N.; Staubitz, A. Cross-Coupling Strategy for the Synthesis of Diazocines. Org. Lett. 2020, 22, 1624–1627. [Google Scholar] [CrossRef] [PubMed]
- Carstensen, O.; Sielk, J.; Schönborn, J.B.; Granucci, G.; Hartke, B. Unusual Photochemical Dynamics of a Bridged Azobenzene Derivative. J. Chem. Phys. 2010, 133, 124305. [Google Scholar] [CrossRef]
- Sell, H.; Näther, C.; Herges, R. Amino-Substituted Diazocines as Pincer-Type Photochromic Switches. Beilstein J. Org. Chem. 2013, 9, 1–7. [Google Scholar] [CrossRef]
- Samanta, S.; Qin, C.; Lough, A.J.; Woolley, G.A. Bidirectional Photocontrol of Peptide Conformation with a Bridged Azobenzene Derivative. Angew. Chemie Int. Ed. 2012, 51, 6452–6455. [Google Scholar] [CrossRef]
- Wang, G.A.; Xu, J.; Traynor, S.M.; Chen, H.; Eljabu, F.; Wu, X.; Yan, H.; Li, F. DNA Balance for Native Characterization of Chemically Modified DNA. J. Am. Chem. Soc. 2021, 143, 13655–13663. [Google Scholar] [CrossRef]
- Li, S.; Han, G.; Zhang, W. Concise Synthesis of Photoresponsive Polyureas Containing Bridged Azobenzenes as Visible-Light-Driven Actuators and Reversible Photopatterning. Macromolecules 2018, 51, 4290–4297. [Google Scholar] [CrossRef]
- Dowds, M.; Bank, D.; Strueben, J.; Soto, D.P.; Sönnichsen, F.D.; Renth, F.; Temps, F.; Staubitz, A. Efficient Reversible Photoisomerisation with Large Solvodynamic Size-Switching of a Main Chain Poly(Azobenzene-Alt-Trisiloxane). J. Mater. Chem. C 2020, 8, 1835–1845. [Google Scholar] [CrossRef]
- Izumi, A.; Teraguchi, M.; Nomura, R.; Masuda, T. Synthesis of Poly(p-Phenylene)-Based Photoresponsive Conjugated Polymers Having Azobenzene Units in the Main Chain. Macromolecules 2000, 33, 5347–5352. [Google Scholar] [CrossRef]
- Li, S.; Colaco, R.; Staubitz, A. ARGET ATRP of Methyl Acrylate and Methyl Methacrylate with Diazocine-Derived Initiators. ACS Appl. Polym. Mater. 2022, 4, 6825–6833. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Wang, G.; Tong, X.; Zhao, Y. Preparation of Azobenzene-Containing Amphiphilic Diblock Copolymers for Light-Responsive Micellar Aggregates. Macromolecules 2004, 37, 8911–8917. [Google Scholar] [CrossRef]
- Coghill, A.M.; Garson, L.R.; American Chemical Society (Eds.) The ACS Style Guide: Effective Communication of Scientific Information, 3rd ed.; American Chemical Society; Oxford University Press: Washington, DC, USA; Oxford, UK; New York, NY, USA, 2006; ISBN 9780841239999/9780841274006. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PSS (405 nm) | PSS (525 nm) | |||||||
|---|---|---|---|---|---|---|---|---|
| ΓZ→E (%) E | λmax(E) (nm) | D (10−6 cm2 s−1) 1 | ΓE→Z (%) Z | λmax(Z) (nm) | D (10−6 cm2 s−1)1 | k (E→Z) (10−3 min−1) | t1/2 (min) | |
| M1 | 60% | 491 | 7.45 ± 0.14 (Z) 7.39 ± 0.07 (E) | >99% | 399 | 7.46 ± 0.11 | 26.73 ± 0.84 | 26 ± 1 |
| P1 | 60% | 496 | 1.90 ± 0.03 (Z) 1.91 ± 0.02 (E) | >99% | 402 | 1.96 ± 0.05 | 28.56 ± 0.37 | 24 ± 0 |
| M2 | 64% | 488 | 10.4 ± 0.16 (Z) 11.0 ± 0.26 (E) | >99% | 402 | 10.6 ± 0.09 | 1.90 ± 0.04 | 366 ± 7 |
| P2 | 64% | 492 | 2.72 ± 0.21 (Z) 2.75 ± 0.07 (E) | >99% | 405 | 2.84 ± 0.08 | 1.90 ± 0.05 | 366 ± 9 |
| Mn,GPC (kDa) | Đ | Tg (°C) | ||
|---|---|---|---|---|
| P1 | PSS (525 nm) | 43 | 2.5 | −11.3 |
| PSS (405 nm) | 43 | 2.5 | −10.9 | |
| P2 | PSS (525 nm) | 7.4 | 1.6 | 12.3 |
| PSS (405 nm) | 8.1 | 1.6 | 10.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Bamberg, K.; Lu, Y.; Sönnichsen, F.D.; Staubitz, A. Facile Synthesis of Light-Switchable Polymers with Diazocine Units in the Main Chain. Polymers 2023, 15, 1306. https://doi.org/10.3390/polym15051306
Li S, Bamberg K, Lu Y, Sönnichsen FD, Staubitz A. Facile Synthesis of Light-Switchable Polymers with Diazocine Units in the Main Chain. Polymers. 2023; 15(5):1306. https://doi.org/10.3390/polym15051306
Chicago/Turabian StyleLi, Shuo, Katrin Bamberg, Yuzhou Lu, Frank D. Sönnichsen, and Anne Staubitz. 2023. "Facile Synthesis of Light-Switchable Polymers with Diazocine Units in the Main Chain" Polymers 15, no. 5: 1306. https://doi.org/10.3390/polym15051306
APA StyleLi, S., Bamberg, K., Lu, Y., Sönnichsen, F. D., & Staubitz, A. (2023). Facile Synthesis of Light-Switchable Polymers with Diazocine Units in the Main Chain. Polymers, 15(5), 1306. https://doi.org/10.3390/polym15051306