



Effective Endotoxin Removal from Chitosan That Preserves Chemical Structure and Improves Compatibility with Immune Cells

 , , ,
, , , 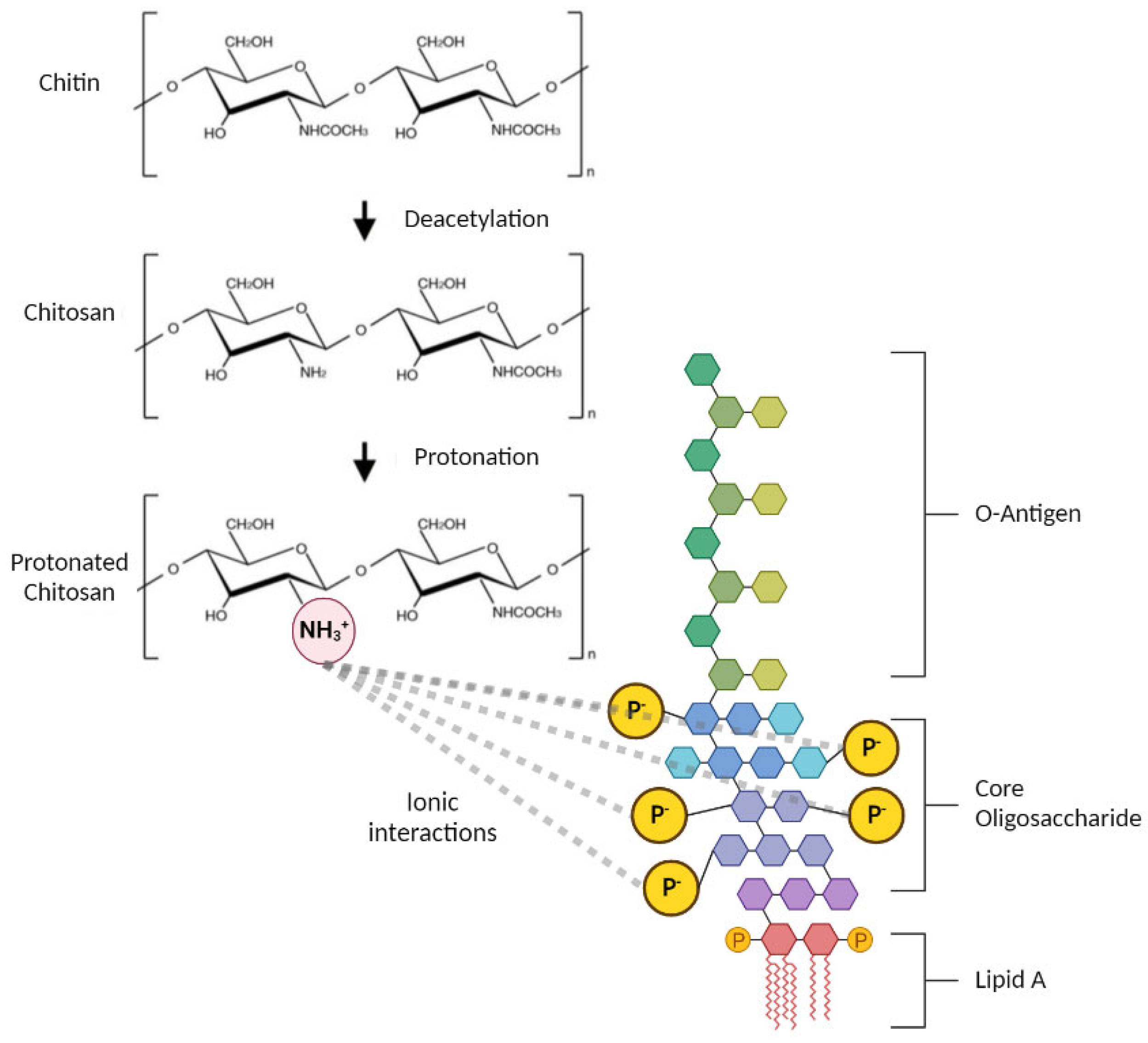{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Heat Treatment for Endotoxin Removal
2.3. NaOH Treatment for Endotoxin Removal
2.4. Preparing Chitosan Solutions
2.5. PBMC Isolation and Culture
2.5.1. CLI-095 Culture
2.5.2. TNF-α Bioassay
2.6. ELISA
2.7. Endotoxin Quantification
2.8. Synthesis of Chitosan-Genipin Hydrogel Films
2.9. Synthesis of Chitosan-Genipin Hydrogel Disks
2.10. FTIR
2.11. Lysozyme Degradation
2.12. moDC Isolation and Co-Culture Experiments
2.13. Flow Cytometry
2.14. Statistical Analyses
3. Results and Discussion
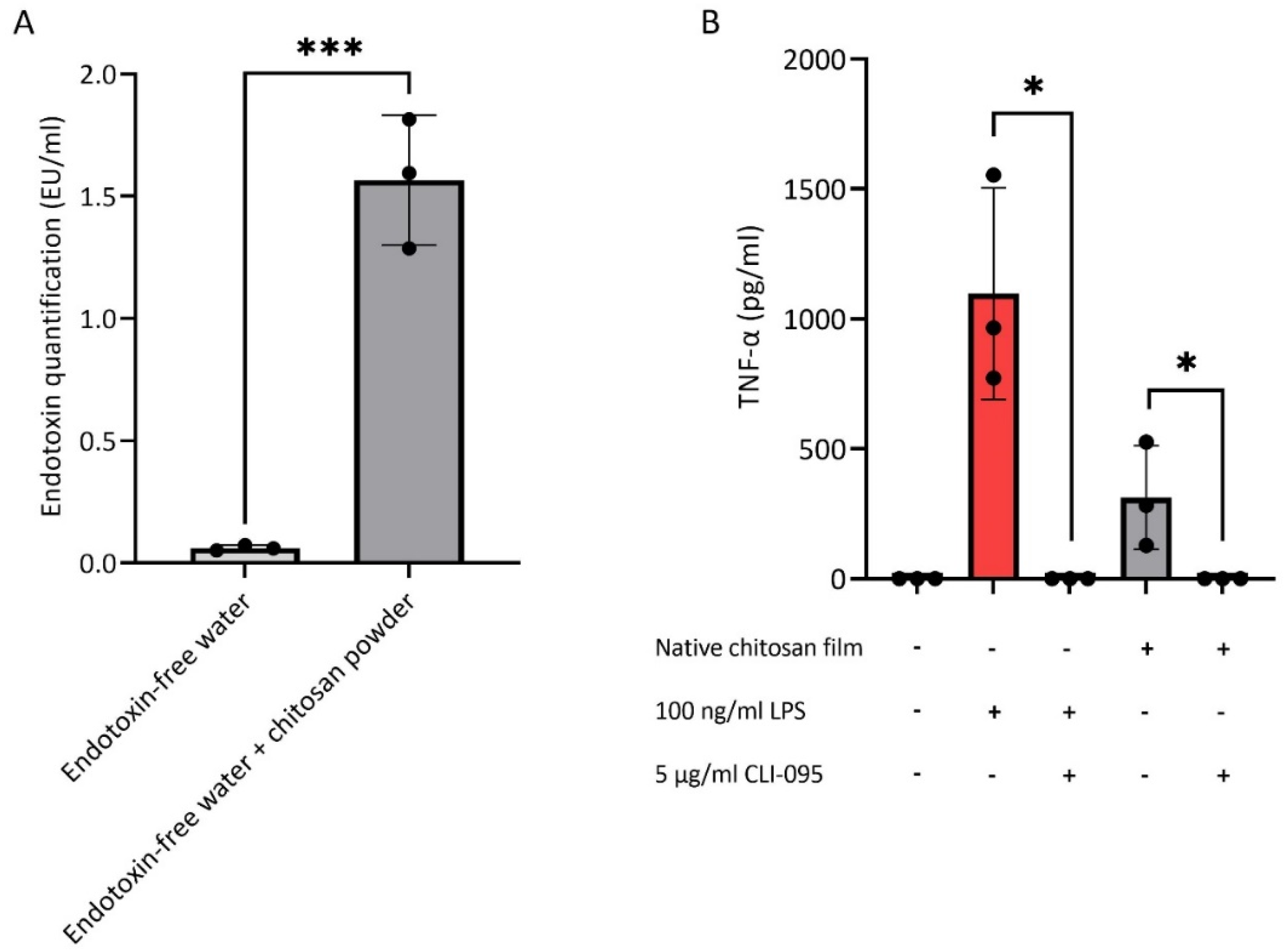
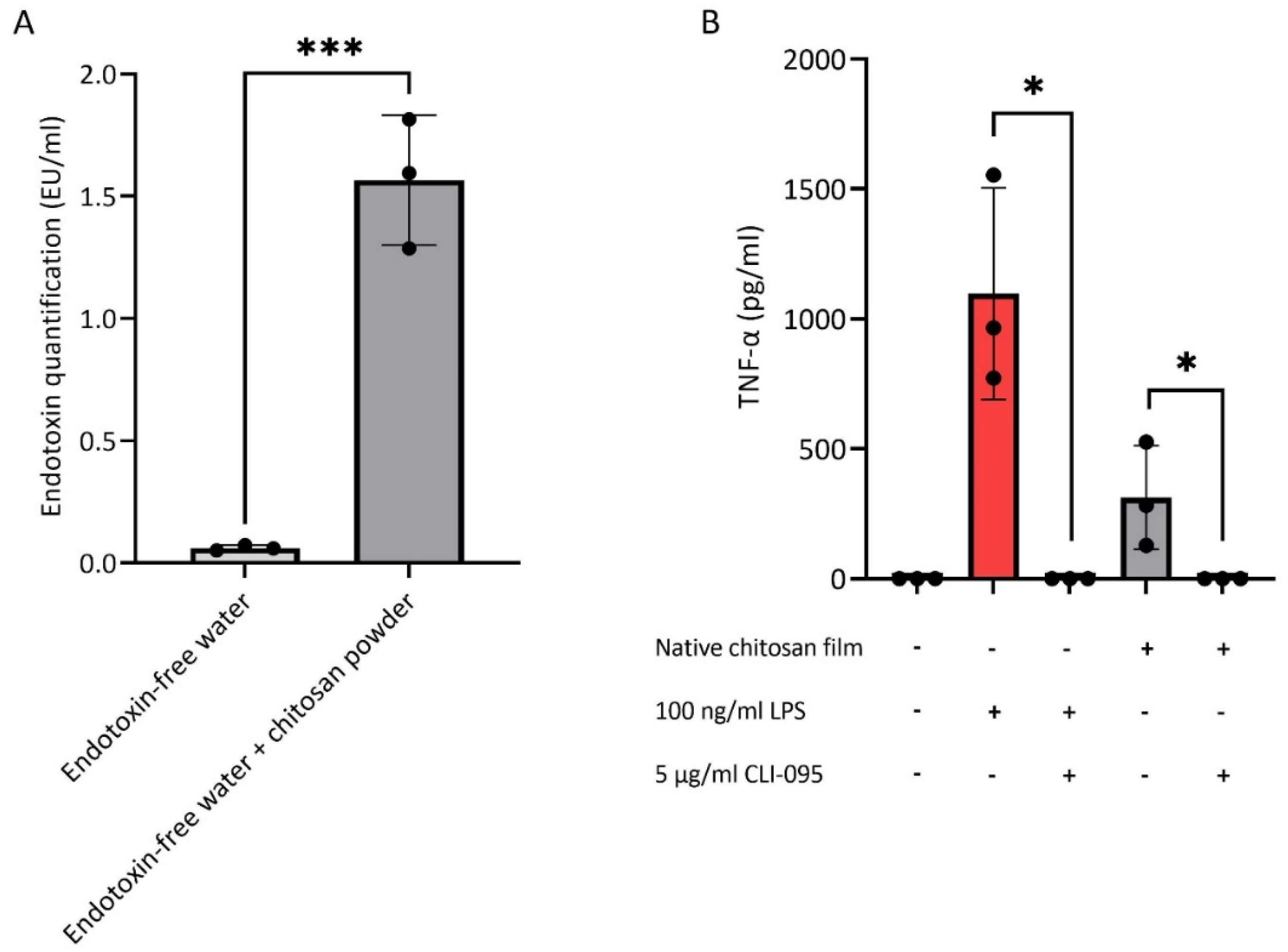
3.1. Endotoxin Contamination of Chitosan
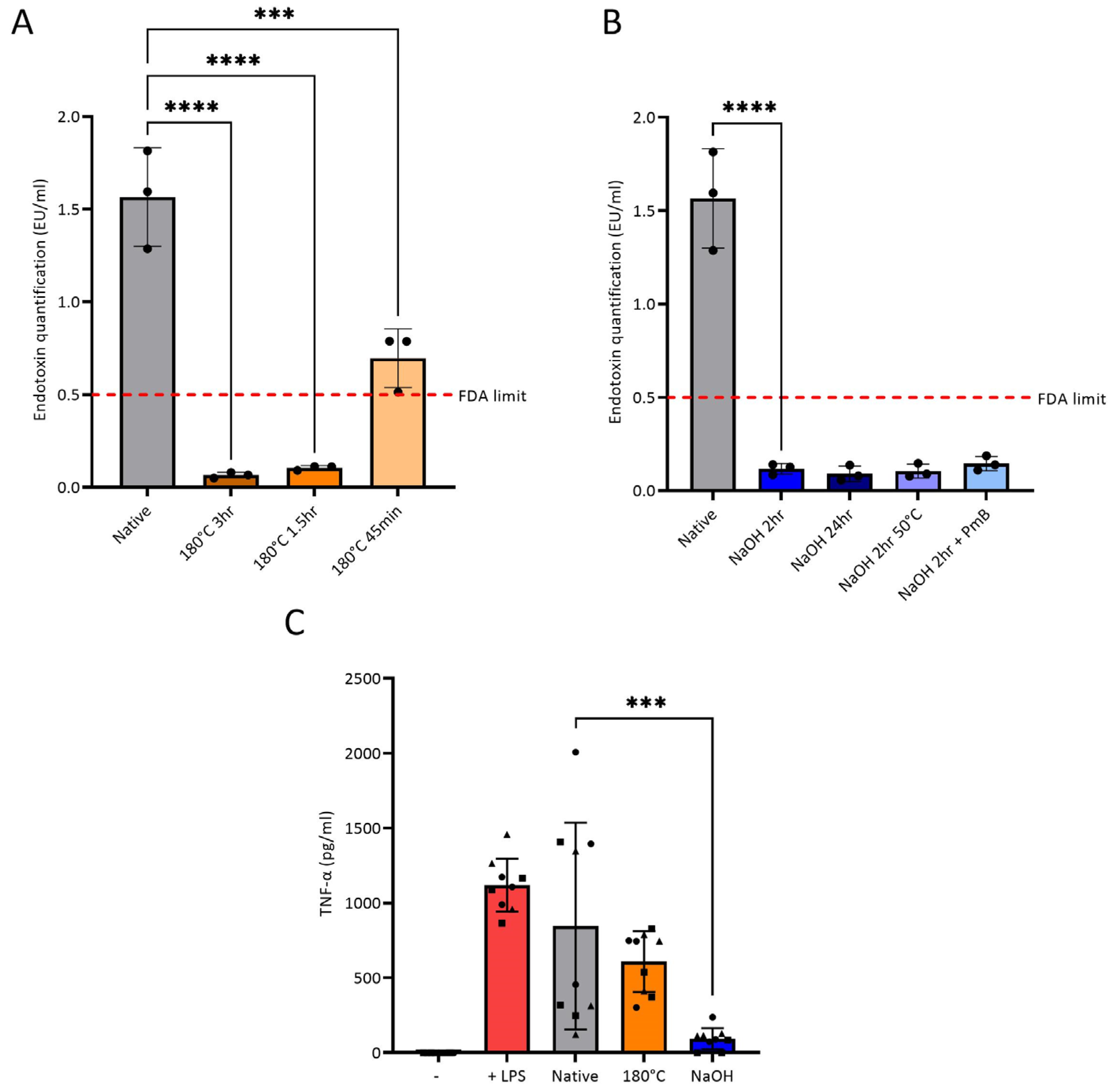
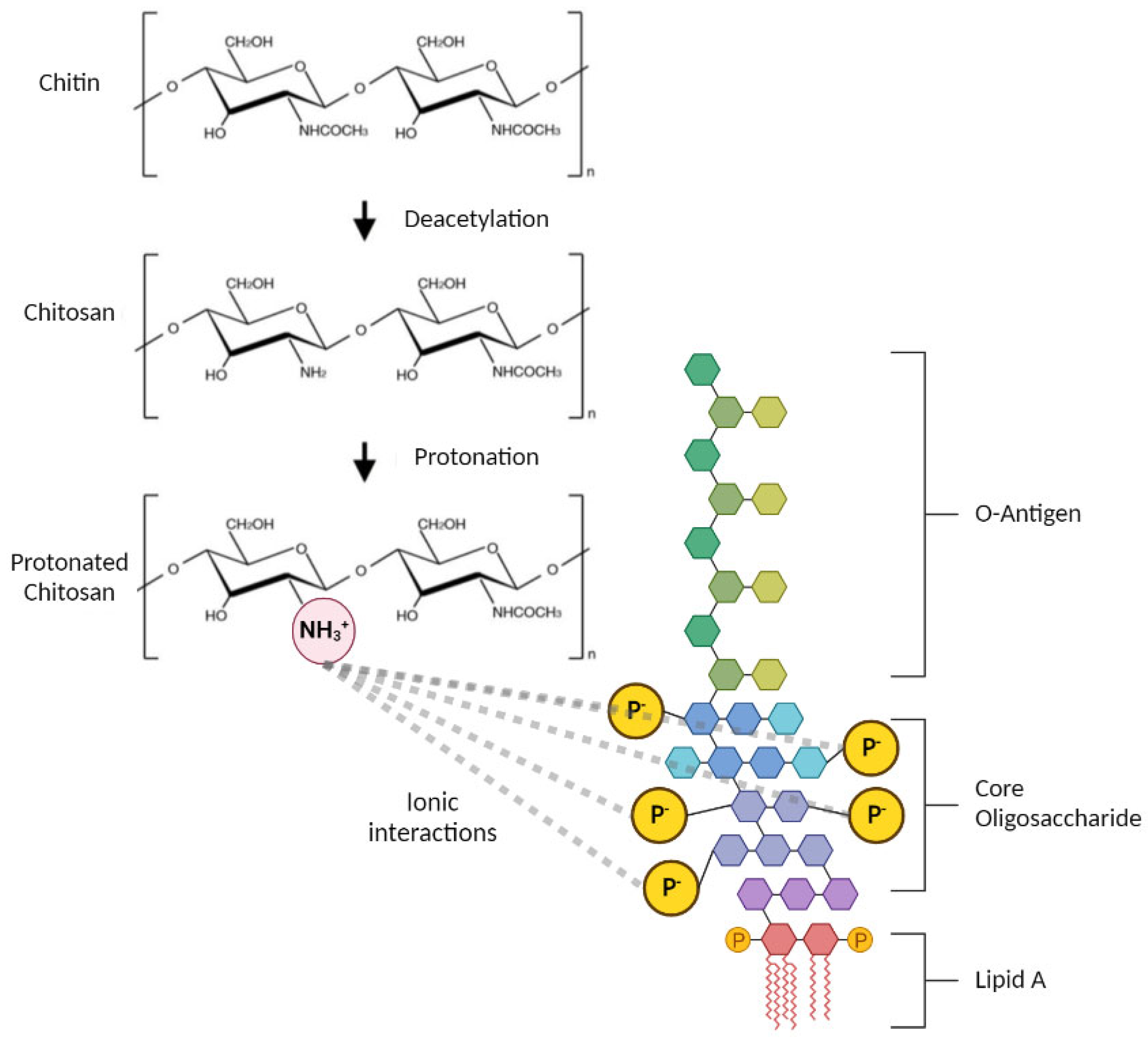
3.2. Endotoxin Removal from Chitosan
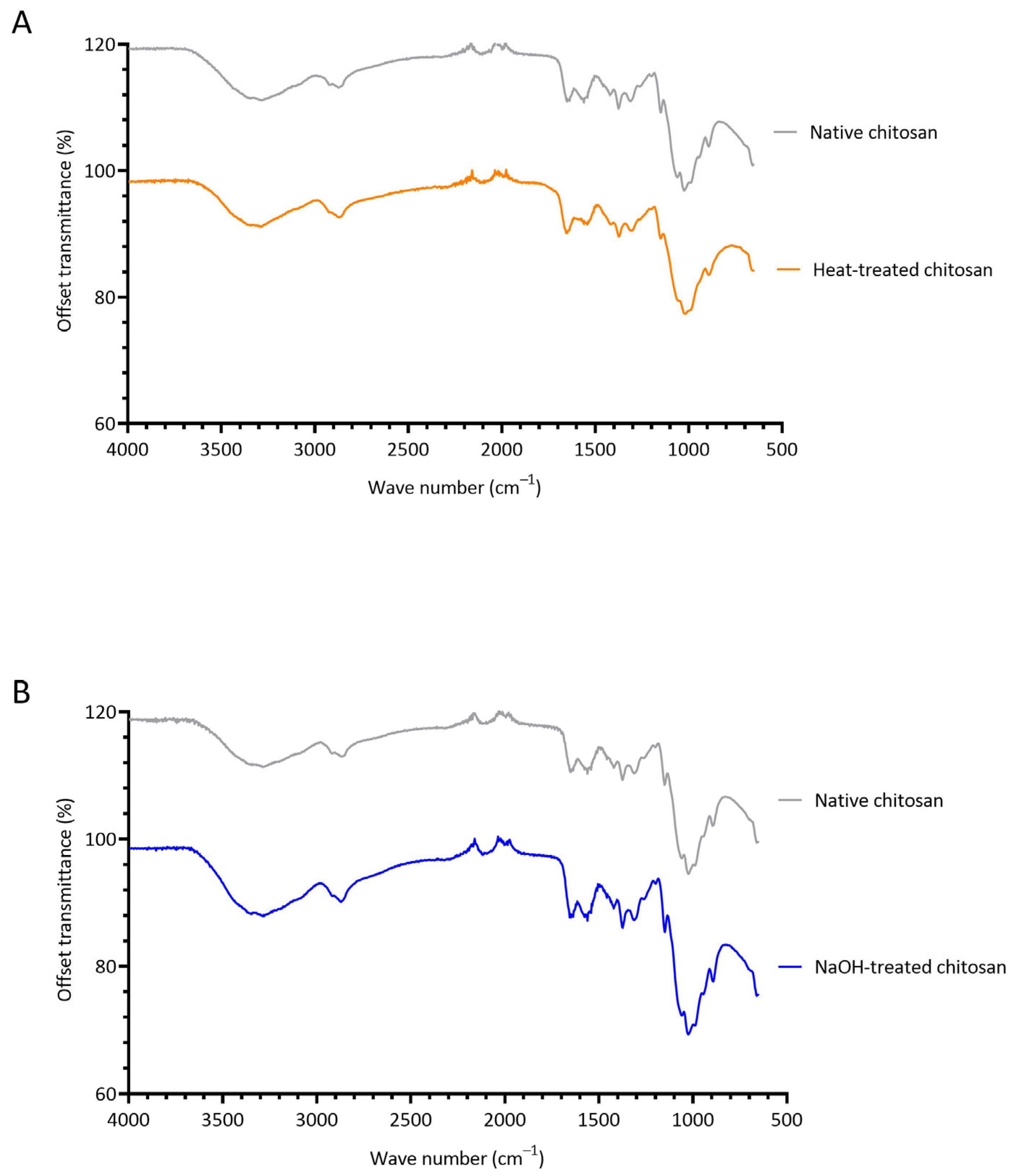
3.3. Investigating the Structural Changes of Treated Chitosan
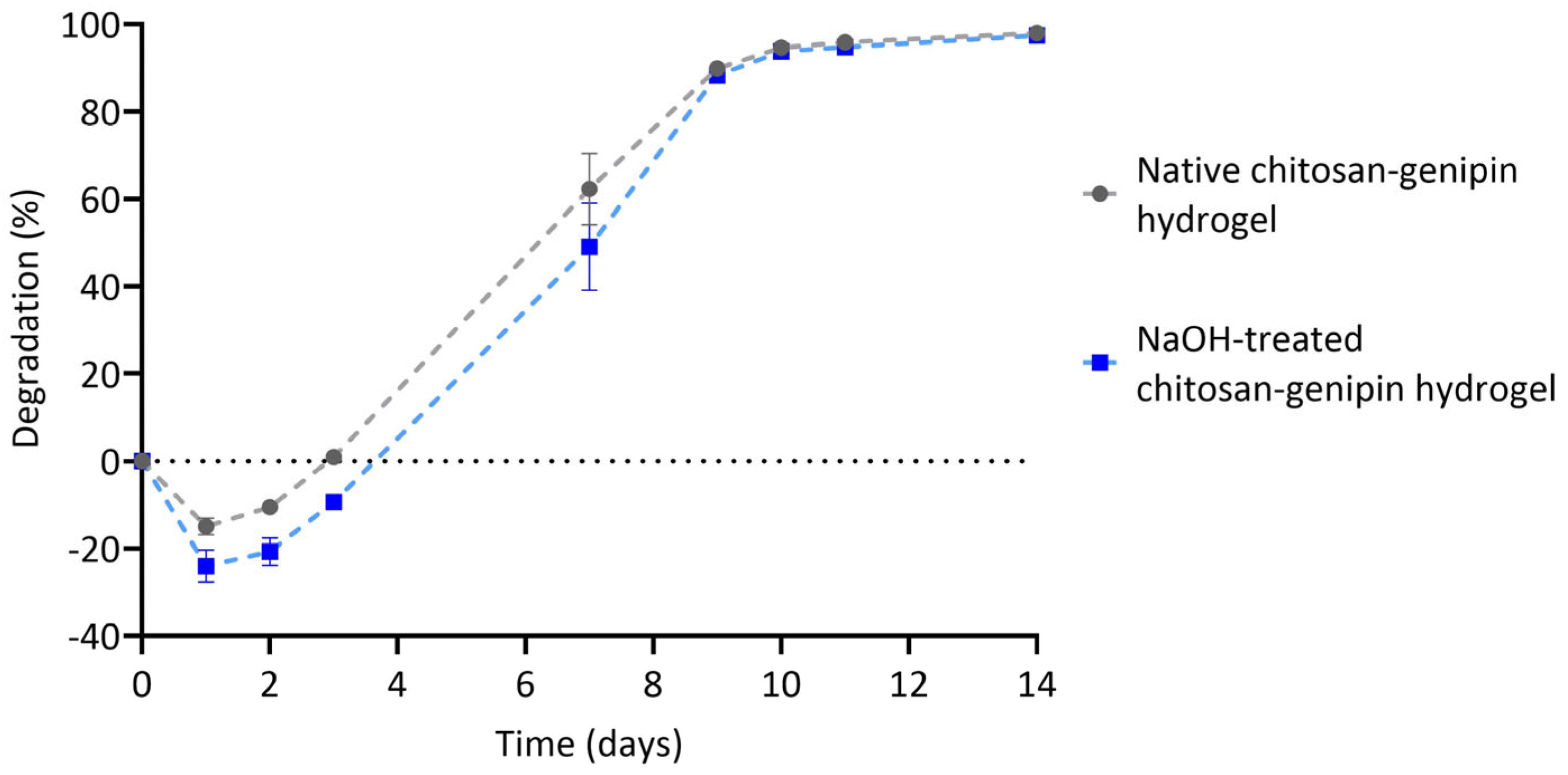
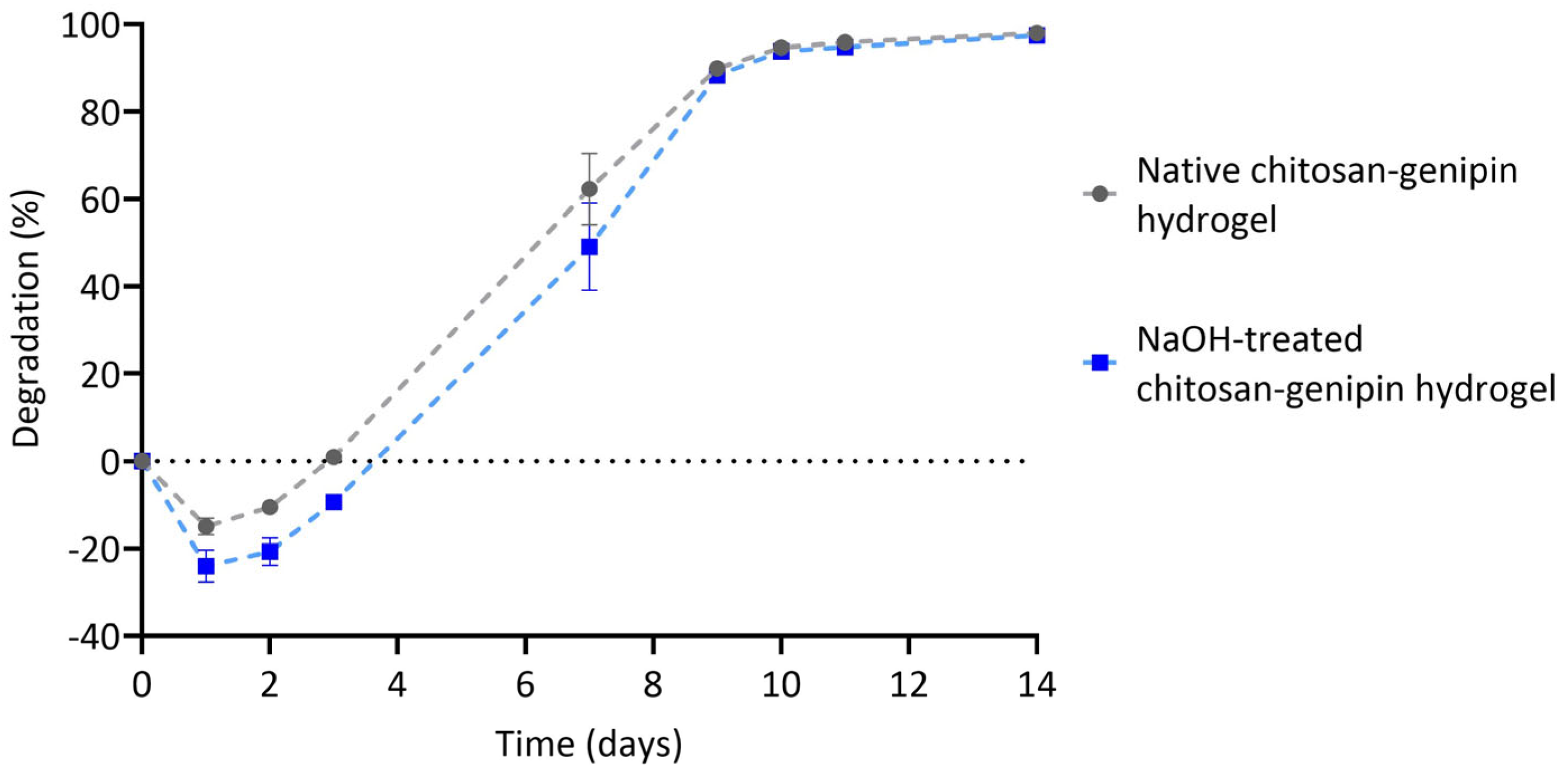
3.4. Lysozyme Degradation of Chitosan-Genipin Hydrogels Made Using NaOH Treated Chitosan
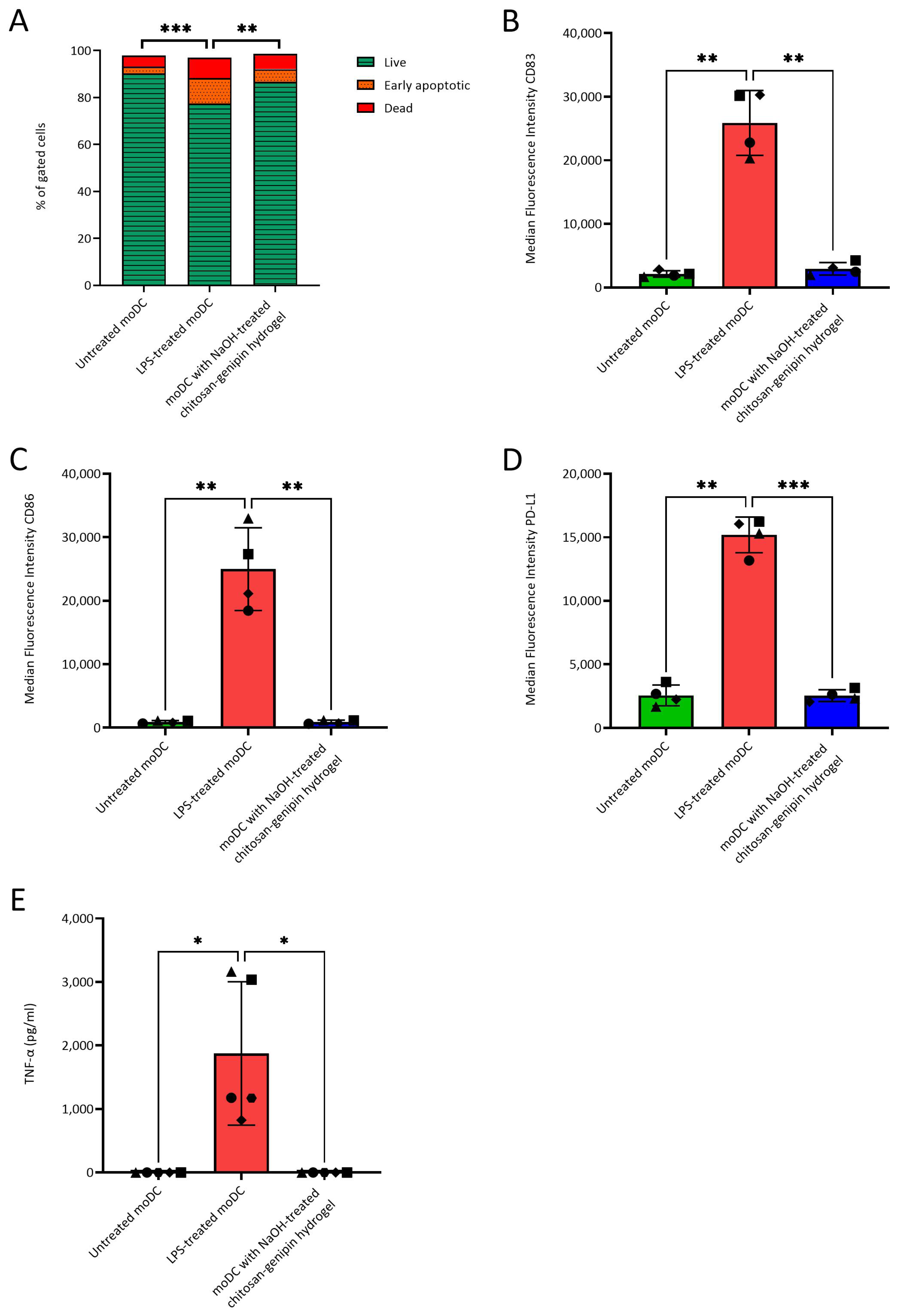
3.5. Compatibility of NaOH-Treated Chitosan-Genipin Hydrogels with moDCs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coppola, D.; Lauritano, C.; Esposito, F.P.; Riccio, G.; Rizzo, C.; de Pascale, D. Fish Waste: From Problem to Valuable Resource. Mar. Drugs 2021, 19, 116. [Google Scholar] [CrossRef]
- Singh, R.; Shitiz, K.; Singh, A. Chitin and chitosan: Biopolymers for wound management. Int. Wound J. 2017, 14, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Younes, I.; Rinaudo, M. Chitin and Chitosan Preparation from Marine Sources. Structure, Properties and Applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, L.R.; Weiser, R.S. The β-glucosaminidase activity of egg-white lysozyme. Biochim. Biophys. Acta 1957, 26, 517–521. [Google Scholar] [CrossRef]
- Kristiansen, A.; Varum, K.M.; Grasdalen, H. The interactions between highly de-N-acetylated chitosans and lysozyme from chicken egg white studied by 1H-NMR spectroscopy. JBIC J. Biol. Inorg. Chem. 2022, 251, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Reay, S.L.; Jackson, E.L.; Ferreira-Duarte, A.M.; Hilkens, C.M.U.; Novakovic, K. In vitro evaluation of the biodegradability of chitosan–genipin hydrogels. Mater. Adv. 2022, 3, 7946–7959. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Nilsen-Nygaard, J.; Strand, S.P.; Vårum, K.M.; Draget, K.I.; Nordgård, C.T. Chitosan: Gels and Interfacial Properties. Polymers 2015, 7, 552–579. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Bhuiyan, M.A.R.; Islam, M.N. Chitin and Chitosan: Structure, Properties and Applications in Biomedical Engineering. J. Polym. Environ. 2017, 25, 854–866. [Google Scholar] [CrossRef]
- FDA. Generally Regarded as Safe Notices. 2019. Available online: https://www.cfsanappsexternal.fda.gov/scripts/fdcc/index.cfm?set=GRASNotices&sort=GRN_No&order=DESC&startrow=1&type=basic&search=chitosan (accessed on 10 January 2023).
- FDA. The Global Unique Device Identification Database. 2019. Available online: https://accessgudid.nlm.nih.gov/devices/search?page=1&query=%28%22chitosan%22%29 (accessed on 10 January 2023).
- Hamedi, H.; Moradi, S.; Hudson, S.M.; Tonelli, A.E.; King, M.W. Chitosan based bioadhesives for biomedical applications: A review. Carbohydr. Polym. 2022, 282, 119100. [Google Scholar] [CrossRef]
- Ojeda-Hernández, D.D.; Canales-Aguirre, A.A.; Matias-Guiu, J.; Gomez-Pinedo, U.; Mateos-Díaz, J.C. Potential of Chitosan and Its Derivatives for Biomedical Applications in the Central Nervous System. Front. Bioeng. Biotechnol. 2020, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Cision. Global Chitosan Market Report 2022–2026 & 2031: Key Players Are Developing Innovative Technologies for Specific Applications in Film Packaging, Skin Tissue Engineering, Pharmaceutical Drugs. 2023. Available online: https://www.cision.com/?utm_medium=website&utm_source=prnewswire&utm_content=cishomepage&utm_campaign=prnewswire (accessed on 10 January 2023).
- Gorbet, M.B.; Sefton, M.V. Endotoxin: The uninvited guest. Biomaterials 2005, 26, 6811–6817. [Google Scholar] [CrossRef]
- Magalhães, P.O.; Lopes, A.M.; Mazzola, P.G.; Rangel-Yagui, C.; Penna, T.C.V.; Pessoa Júnior, A. Methods of endotoxin removal from biological preparations: A review. J. Pharm. Pharm. Sci. 2007, 10, 388–404. [Google Scholar]
- Heinrich, M.A.; Mangia, M.; Prakash, J. Impact of endotoxins on bioengineered tissues and models. Trends Biotechnol. 2021, 40, 532–534. [Google Scholar] [CrossRef] [PubMed]
- Clifton, L.A.; Ciesielski, F.; Skoda, M.W.A.; Paracini, N.; Holt, S.A.; Lakey, J.H. The Effect of Lipopolysaccharide Core Oligosaccharide Size on the Electrostatic Binding of Antimicrobial Proteins to Models of the Gram Negative Bacterial Outer Membrane. Langmuir 2016, 32, 3485–3494. [Google Scholar] [CrossRef] [Green Version]
- Naberezhnykh, G.; Gorbach, V.; Kalmykova, E.; Solov’Eva, T. Determination of the parameters of binding between lipopolysaccharide and chitosan and its N-acetylated derivative using a gravimetric piezoquartz biosensor. Biophys. Chem. 2015, 198, 9–13. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Salthouse, D.; Novakovic, K.; Hilkens, C.M.; Ferreira, A.M. Interplay between biomaterials and the immune system: Challenges and opportunities in regenerative medicine. Acta Biomater. 2023, 155, 1–18. [Google Scholar] [CrossRef]
- Schwarz, H.; Schmittner, M.; Duschl, A.; Horejs-Hoeck, J. Residual Endotoxin Contaminations in Recombinant Proteins Are Sufficient to Activate Human CD1c+ Dendritic Cells. PLoS ONE 2014, 9, e113840. [Google Scholar] [CrossRef] [Green Version]
- FDA. Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers. 2012. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-pyrogen-and-endotoxins-testing-questions-and-answers (accessed on 10 January 2023).
- Yang, T.-L. Chitin-based Materials in Tissue Engineering: Applications in Soft Tissue and Epithelial Organ. Int. J. Mol. Sci. 2011, 12, 1936–1963. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Murai, T.; Hasegawa, C.; Hirata, M.; Tsuchiya, T.; Yagami, T.; Haishima, Y. Endotoxin contamination in wound dressings made of natural biomaterials. J. Biomed. Mater. Res. 2003, 66B, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Korntner, S.; Olijve, J.; Mullen, A.; Zeugolis, D. The Influence of Bloom Index, Endotoxin Levels and Polyethylene Glycol Succinimidyl Glutarate Crosslinking on the Physicochemical and Biological Properties of Gelatin Biomaterials. Biomolecules 2021, 11, 1003. [Google Scholar] [CrossRef]
- Lieder, R.; Gaware, V.S.; Thormodsson, F.; Einarsson, J.M.; Ng, C.-H.; Gislason, J.; Masson, M.; Petersen, P.H.; Sigurjonsson, O.E. Endotoxins affect bioactivity of chitosan derivatives in cultures of bone marrow-derived human mesenchymal stem cells. Acta Biomater. 2013, 9, 4771–4778. [Google Scholar] [CrossRef]
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An Update on Potential Biomedical and Pharmaceutical Applications. Mar. Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef] [PubMed]
- Vasiliev, Y. Chitosan-based vaccine adjuvants: Incomplete characterization complicates preclinical and clinical evaluation. Expert Rev. Vaccines 2015, 14, 37–53. [Google Scholar] [CrossRef]
- Smith, A.; Perelman, M.; Hinchcliffe, M. Chitosan. Hum. Vaccines Immunother. 2014, 10, 797–807. [Google Scholar] [CrossRef]
- Dmour, I.; Islam, N. Recent advances on chitosan as an adjuvant for vaccine delivery. Int. J. Biol. Macromol. 2022, 200, 498–519. [Google Scholar] [CrossRef]
- Parmaksız, S.; Şenel, S. An Overview on Chitosan-Based Adjuvant/Vaccine Delivery Systems. In Chitosan for Biomaterials IV; Springer: Cham, Switzerland, 2021; pp. 293–379. [Google Scholar] [CrossRef]
- Fong, D.; Hoemann, C.D.; Vrana, N.E.; Jennings, J.A.; Wells, C.M.; McGraw, G.S.; Pulgarin, D.A.V.; Whitaker, M.D.; Pruitt, R.L.; Bumgardner, J.D.; et al. Chitosan immunomodulatory properties: Perspectives on the impact of structural properties and dosage. Futur. Sci. OA 2017, 4, FSO225. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, D.P.; Fonseca, A.C.; Costa, M.; Amaral, I.F.; Barbosa, M.A.; Águas, A.P.; Barbosa, J.N. Macrophage polarization following chitosan implantation. Biomaterials 2013, 34, 9952–9959. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.; Santos, S.; Torres, A.; Barbosa, M. Chitosan drives anti-inflammatory macrophage polarisation and pro-inflammatory dendritic cell stimulation. Eur. Cells Mater. 2012, 24, 136–153. [Google Scholar] [CrossRef]
- Guan, G.; Azad, M.A.K.; Lin, Y.; Kim, S.W.; Tian, Y.; Liu, G.; Wang, H. Biological Effects and Applications of Chitosan and Chito-Oligosaccharides. Front. Physiol. 2019, 10, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.; Kolbe, H.; Duncan, R. Potential of low molecular mass chitosan as a DNA delivery system: Biocompatibility, body distribution and ability to complex and protect DNA. Int. J. Pharm. 1999, 178, 231–243. [Google Scholar] [CrossRef]
- Carreño-Gómez, B.; Duncan, R. Evaluation of the biological properties of soluble chitosan and chitosan microspheres. Int. J. Pharm. 1997, 148, 231–240. [Google Scholar] [CrossRef]
- Park, J.; Babensee, J.E. Differential functional effects of biomaterials on dendritic cell maturation. Acta Biomater. 2012, 8, 3606–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, H.; John, A.; Shafarin, J. Potentiation of LPS-Induced Apoptotic Cell Death in Human Hepatoma HepG2 Cells by Aspirin via ROS and Mitochondrial Dysfunction: Protection by N-Acetyl Cysteine. PLoS ONE 2016, 11, e0159750. [Google Scholar] [CrossRef] [Green Version]
- Lebre, F.; Lavelle, E.C.; Borges, O. Easy and effective method to generate endotoxin-free chitosan particles for immunotoxicology and immunopharmacology studies. J. Pharm. Pharmacol. 2019, 71, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Merck. Cell Culture FAQs: Bacterial Endotoxin Contamination. 2022. Available online: https://www.sigmaaldrich.com/GB/en/technical-documents/technical-article/microbiological-testing/pyrogen-testing/what-is-endotoxin (accessed on 10 January 2023).
- Harm, S.; Schildböck, C.; Strobl, K.; Hartmann, J. An In vitro study on factors affecting endotoxin neutralization in human plasma using the Limulus Amebocyte Lysate test. Sci. Rep. 2021, 11, 4192. [Google Scholar] [CrossRef]
- Ongkudon, C.M.; Chew, J.H.; Liu, B.; Danquah, M.K. Chromatographic Removal of Endotoxins: A Bioprocess Engineer’s Perspective. ISRN Chromatogr. 2012, 2012, 649746. [Google Scholar] [CrossRef] [Green Version]
- Dimida, S.; Demitri, C.; De Benedictis, V.M.; Scalera, F.; Gervaso, F.; Sannino, A. Genipin-cross-linked chitosan-based hydrogels: Reaction kinetics and structure-related characteristics. J. Appl. Polym. Sci. 2015, 132, 1–8. [Google Scholar] [CrossRef]
- Neto, C.; Giacometti, J.; Job, A.; Ferreira, F.; Fonseca, J.; Pereira, M. Thermal Analysis of Chitosan Based Networks. Carbohydr. Polym. 2005, 62, 97–103. [Google Scholar] [CrossRef]
- Jana, S.; Trivedi, M.K.; Tallapragada, R.M. Characterization of Physicochemical and Thermal Properties of Chitosan and Sodium Alginate after Biofield Treatment. Pharm. Anal. Acta 2015, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.-K.; Hung, C.-C.; Lin, M.-F. A kinetic study of thermal degradations of chitosan/polycaprolactam blends. Macromol. Res. 2004, 12, 466–473. [Google Scholar] [CrossRef]
- Sandle, T. A Comparative Study of Different Methods for Endotoxin Destruction, American Pharmaceutical Review. 2013. Available online: https://www.americanpharmaceuticalreview.com/Featured-Articles/148858-A-Comparative-Study-of-Different-Methods-for-Endotoxin-Destruction/ (accessed on 20 March 2023).
- Cytiva. Use of Sodium Hydroxide for Cleaning and Sanitization of Chromatography Resins and Systems. 2020. Available online: https://cdn.cytivalifesciences.com/api/public/content/digi-20986-original#:~:text=Sodium%20hydroxide%20has%20shown%20to,yeasts%2C%20fungi%2C%20and%20endotoxins (accessed on 10 January 2023).
- Cardoso, L.S.; Araujo, M.I.; Góes, A.M.; Pacífico, L.G.; Oliveira, R.R.; Oliveira, S.C. Polymyxin B as inhibitor of LPS contamination of Schistosoma mansoni recombinant proteins in human cytokine analysis. Microb. Cell Factories 2007, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Tynan, G.A.; McNaughton, A.; Jarnicki, A.; Tsuji, T.; Lavelle, E.C. Polymyxin B Inadequately Quenches the Effects of Contaminating Lipopolysaccharide on Murine Dendritic Cells. PLoS ONE 2012, 7, e37261. [Google Scholar] [CrossRef] [Green Version]
- Vo, N.T.N.; Huang, L.; Lemos, H.; Mellor, A.; Novakovic, K. Poly(ethylene glycol)-interpenetrated genipin-crosslinked chitosan hydrogels: Structure, pH responsiveness, gelation kinetics, and rheology. J. Appl. Polym. Sci. 2020, 137, 1–16. [Google Scholar] [CrossRef]
- Vukajlovic, D.; Parker, J.; Bretcanu, O.; Novakovic, K. Chitosan based polymer/bioglass composites for tissue engineering applications. Mater. Sci. Eng. C 2019, 96, 955–967. [Google Scholar] [CrossRef]
- Matcham, S.; Novakovic, K. Fluorescence Imaging in Genipin Crosslinked Chitosan–Poly(vinyl pyrrolidone) Hydrogels. Polymers 2016, 8, 385. [Google Scholar] [CrossRef] [Green Version]
- Fatima, B. Quantitative Analysis by IR: Determination of Chitin/Chitosan DD. In Modern Spectroscopic Techniques and Applications; Books on Demand: Norderstedt, Germany, 2020. [Google Scholar] [CrossRef] [Green Version]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [Green Version]
- Schildhauer, T.A.; Peter, E.; Muhr, G.; Köller, M. Activation of human leukocytes on tantalum trabecular metal in comparison to commonly used orthopedic metal implant materials. J. Biomed. Mater. Res. Part A 2009, 88A, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, F.A.; Young, S.A.; Kasapis, S. Swelling behaviour and glass transition in genipin-crosslinked chitosan systems. Int. J. Biol. Macromol. 2020, 164, 3075–3083. [Google Scholar] [CrossRef] [PubMed]
- Lončarević, A.; Ivanković, M.; Rogina, A. Lysozyme-Induced Degradation of Chitosan: The Characterisation of Degraded Chitosan Scaffolds. J. Tissue Repair Regen. 2017, 1, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Fernandes Queiroz, M.; Melo, K.R.T.; Sabry, D.A.; Sassaki, G.L.; Rocha, H.A.O. Does the Use of Chitosan Contribute to Oxalate Kidney Stone Formation? Mar. Drugs 2014, 13, 141–158. [Google Scholar] [CrossRef]
- Mekahlia, S.; Bouzid, B. Chitosan-Copper (II) complex as antibacterial agent: Synthesis, characterization and coordinating bond- activity correlation study. Phys. Procedia 2009, 2, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Hemmler, D.; Roullier-Gall, C.; Marshall, J.W.; Rychlik, M.; Taylor, A.J.; Taylor, P. Schmitt-Kopplin, Insights into the Chemistry of Non-Enzymatic Browning Reactions in Different Ribose-Amino Acid Model Systems. Sci. Rep.-Uk 2018, 8, 16879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Lu, X.; Li, N.; Zheng, Z.; Qiao, X. Characterization, Variables, and Antioxidant Activity of the Maillard Reaction in a Fructose–Histidine Model System. Molecules 2019, 24, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.M.; Zhao, Y.H.; Liu, X.H.; Ding, F.; Gu, X.S. The effect of different sterilization procedures on chitosan dried powder. Appl. Polym. Sci. 2007, 104, 1968–1972. [Google Scholar] [CrossRef]
- Lim, L.; Khor, E.; Koo, O. γ Irradiation of chitosan. J. Biomed. Mater. Res. 1998, 43, 282–290. [Google Scholar] [CrossRef]
- Lund, M.N.; Ray, C.A. Control of Maillard Reactions in Foods: Strategies and Chemical Mechanisms. J. Agr. Food Chem. 2017, 65, 4537–4552. [Google Scholar] [CrossRef] [Green Version]
- Ajandouz, E.H.; Tchiakpe, L.S.; Ore, F.D.; Benajiba, A.; Puigserver, A. Effects of pH on Caramelization and Maillard Reaction Kinetics in Fructose-Lysine Model Systems. J. Food Sci. 2001, 66, 926–931. [Google Scholar] [CrossRef]
- Aalaei, K.; Rayner, M.; Sjöholm, I. Chemical methods and techniques to monitor early Maillard reaction in milk products; A review. Crit. Rev. Food Sci. 2019, 59, 1829–1839. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ouyang, W.; Lawuyi, B.; Martoni, C.; Prakash, S. Reaction of chitosan with genipin and its fluorogenic attributes for potential microcapsule membrane characterization. J. Biomed. Mater. Res. A 2005, 75A, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, R.A.A.; Mehtedi, M.E.; Bottegoni, C.; Aquili, A.; Gigante, A. Genipin-Crosslinked Chitosan Gels and Scaffolds for Tissue Engineering and Regeneration of Cartilage and Bone. Mar. Drugs 2015, 13, 7314–7338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, D.A.; Bent, G.-A. Reducing Acrylamide Exposure: A Review of the Application of Sulfur-Containing Compounds—A Caribbean Outlook. Eur. J. Nutr. Food Saf. 2019, 9, 192–209. [Google Scholar] [CrossRef]
- Merck. IR Spectrum Table & Chart. 2022. Available online: https://www.sigmaaldrich.com/GB/en/technical-documents/technical-article/analytical-chemistry/photometry-and-reflectometry/ir-spectrum-table (accessed on 10 January 2023).
- Shirvan, A.R.; Shakeri, M.; Bashari, A. Recent advances in application of chitosan and its derivatives in functional finishing of textiles. In The Impact and Prospects of Green Chemistry for Textile Technology; Woodhead Publishing: Sawston, UK, 2019; pp. 107–133. [Google Scholar] [CrossRef]
- Chen, X.; Yang, H.; Zhong, Z.; Yan, N. Base-catalysed, one-step mechanochemical conversion of chitin and shrimp shells into low molecular weight chitosan. Green Chem. 2017, 19, 2783–2792. [Google Scholar] [CrossRef] [Green Version]
- Galed, G.; Diaz, E.; Goycoolea, F.M.; Heras, A. Influence of N-Deacetylation Conditions on Chitosan Production from α-Chitin. Nat. Prod. Commun. 2008, 3, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Pires, C.T.; Vilela, J.A.; Airoldi, C. The Effect of Chitin Alkaline Deacetylation at Different Condition on Particle Properties. Procedia Chem. 2014, 9, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Danarto, Y.; Distantina, S. Optimizing deacetylation process for chitosan production from green mussel (perna viridis) shell. AIP Publishing LLC 2016, 1710, 030028. [Google Scholar] [CrossRef]
- Novikov, V.Y.; Konovalova, I.N.; Dolgopyatova, N.V. The Mechanism of Chitin and Chitosan Deacetylation during Long-Term Alkaline Treatment. Appl. Biochem. Microbiol. 2022, 58, 309–314. [Google Scholar] [CrossRef]
- No, H.K.; Cho, Y.I.; Kim, H.R.; Meyers, S.P. Effective Deacetylation of Chitin under Conditions of 15 psi/121 °C. J. Agric. Food Chem. 2000, 48, 2625–2627. [Google Scholar] [CrossRef]
- Pangburn, S.; Trescony, P.; Heller, J. Lysozyme degradation of partially deacetylated chitin, its films and hydrogels. Biomaterials 1982, 3, 105–108. [Google Scholar] [CrossRef]
- Kim, S.-H.; Jung, H.-H.; Lee, C.-K. Generation, Characteristics and Clinical Trials of Ex Vivo Generated Tolerogenic Dendritic Cells. Yonsei Med. J. 2018, 59, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.T.N.; Huang, L.; Lemos, H.; Mellor, A.L.; Novakovic, K. Genipin-crosslinked chitosan hydrogels: Preliminary evaluation of the In vitro biocompatibility and biodegradation. J. Appl. Polym. Sci. 2021, 138, 49259. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reay, S.L.; Jackson, E.L.; Salthouse, D.; Ferreira, A.M.; Hilkens, C.M.U.; Novakovic, K. Effective Endotoxin Removal from Chitosan That Preserves Chemical Structure and Improves Compatibility with Immune Cells. Polymers 2023, 15, 1592. https://doi.org/10.3390/polym15071592
Reay SL, Jackson EL, Salthouse D, Ferreira AM, Hilkens CMU, Novakovic K. Effective Endotoxin Removal from Chitosan That Preserves Chemical Structure and Improves Compatibility with Immune Cells. Polymers. 2023; 15(7):1592. https://doi.org/10.3390/polym15071592
Chicago/Turabian StyleReay, Sophie L., Emma L. Jackson, Daniel Salthouse, Ana Marina Ferreira, Catharien M. U. Hilkens, and Katarina Novakovic. 2023. "Effective Endotoxin Removal from Chitosan That Preserves Chemical Structure and Improves Compatibility with Immune Cells" Polymers 15, no. 7: 1592. https://doi.org/10.3390/polym15071592
APA StyleReay, S. L., Jackson, E. L., Salthouse, D., Ferreira, A. M., Hilkens, C. M. U., & Novakovic, K. (2023). Effective Endotoxin Removal from Chitosan That Preserves Chemical Structure and Improves Compatibility with Immune Cells. Polymers, 15(7), 1592. https://doi.org/10.3390/polym15071592