
Improved Route to Linear Triblock Copolymers by Coupling with Glycidyl Ether-Activated Poly(ethylene oxide) Chains
 , ,
, ,  , ,
, ,
Abstract
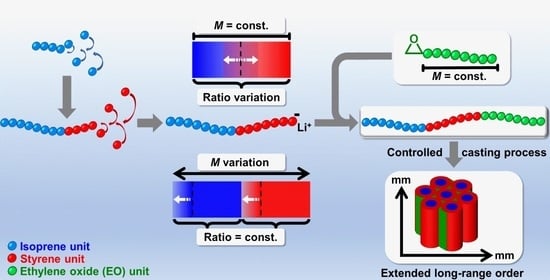
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. End Group Modification (EmPEO1.9)
2.3. Synthesis of Poly(isoprene)-b-poly(styrene)-b-alcohol Methoxy Poly(ethylene oxide) (PIxPSyAmPEO1.9)
2.4. Sample Preparation of PIxPSyAmPEO1.9 Membranes for Morphological Characterization
2.5. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.6. Gel Permeation Chromatography (GPC)
2.7. Differential Scanning Calorimetry (DSC)
2.8. Thermogravimetric Analysis (TGA)
2.9. Small-Angle X-ray Scattering (SAXS)
2.10. Cryo-Ultramicrotomy
2.11. Scanning Electron Microscope (SEM)
2.12. Transmission Electron Microscope (TEM)
3. Results and Discussion
3.1. Strategy
3.2. Synthesis
3.3. NMR Characterization
3.3.1. EmPEO1.9
3.3.2. PIxPSyAmPEO1.9
3.4. GPC Measurements
3.5. Thermal Analysis
3.6. Morphological Characterization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | fPIx 1 /% | fPSy 1 /% | fPEO1.9 1 /% | q* 2 /nm−1 | Phase 3 | d 4 /nm | rcylinder 5 /nm | φcylinder 6 /% | Domain Size 9 /nm |
|---|---|---|---|---|---|---|---|---|---|
| PI6.8PS17.3AmPEO1.9 | 29 | 65 | 6 | 0.32 | HEX | 22.4 | 6.1 | 29 (PI6.8) | 90 |
| PI14.6PS34.8AmPEO1.9 | 32 | 65 | 3 | 0.20 | HEX | 35.7 | 10.4 | 34 (PI14.6) | 122 |
| PI24.8PS25.0AmPEO1.9 | 52 | 45 | 3 | 0.20 | LAM | 31.7 | t 7 = 16.4 | 52 (PI24.8)LAM 8 | 86 |
| PI35.1PS14.8AmPEO1.9 | 71 | 26 | 3 | 0.19 | HEX | 39.2 | 10.9 | 31 (PS14.8AmPEO1.9) | 63 |
| PI26.1PS67.3AmPEO1.9 | 33 | 66 | 2 | 0.11 | HEX | 63.6 | 16.9 | 28 (PI26.1) | 147 |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Allen, C.; Maysinger, D.; Eisenberg, A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf. B Biointerfaces 1999, 16, 3–27. [Google Scholar] [CrossRef]
- Adams, M.L.; Lavasanifar, A.; Kwon, G.S. Amphiphilic block copolymers for drug delivery. J. Pharm. Sci. 2003, 92, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Nedelcu, M.; Lee, J.; Crossland, E.J.W.; Warren, S.C.; Orilall, M.C.; Guldin, S.; Hüttner, S.; Ducati, C.; Eder, D.; Wiesner, U.; et al. Block copolymer directed synthesis of mesoporous TiO2 for dye-sensitized solar cells. Soft Matter 2009, 5, 134–139. [Google Scholar]
- Perrin, L.; Phan, T.; Querelle, S.; Deratani, A.; Bertin, D. Polystyrene-block-poly(ethylene oxide)-block-polystyrene: A New Synthesis Method Using Nitroxide-Mediated Polymerization from Poly(ethylene oxide) Macroinitiators and Characterization of the Architecture Formed. Macromolecules 2008, 41, 6942–6951. [Google Scholar] [CrossRef]
- Bai, W.; Ross, C.A. Functional nanostructured materials based on self-assembly of block copolymers. MRS Bull. 2016, 41, 100–107. [Google Scholar] [CrossRef]
- Li, L.; Krissanasaeranee, M.; Pattinson, S.W.; Stefik, M.; Wiesner, U.; Steiner, U.; Eder, D. Enhanced photocatalytic properties in well-ordered mesoporous WO3. Chem. Commun. 2010, 46, 7620–7622. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gopinadhan, M.; Osuji, C.O. Directed self-assembly of block copolymers: A tutorial review of strategies for enabling nanotechnology with soft matter. Soft Matter 2014, 10, 3867–3889. [Google Scholar] [CrossRef]
- Dörr, T.S.; Fleischmann, S.; Zeiger, M.; Grobelsek, I.; de Oliveira, P.W.; Presser, V. Ordered Mesoporous Titania/Carbon Hybrid Monoliths for Lithium-ion Battery Anodes with High Areal and Volumetric Capacity. Chem. Eur. J. 2018, 24, 6358–6363. [Google Scholar] [CrossRef] [PubMed]
- Dörr, T.S.; Pelz, A.; Zhang, P.; Kraus, T.; Winter, M.; Wiemhöfer, H.-D. An Ambient Temperature Electrolyte with Superior Lithium Ion Conductivity based on a Self-Assembled Block Copolymer. Chem. Eur. J. 2018, 24, 8061–8065. [Google Scholar] [CrossRef] [PubMed]
- Pelz, A.; Dörr, T.S.; Zhang, P.; de Oliveira, P.W.; Winter, M.; Wiemhöfer, H.-D.; Kraus, T. Self-Assembled Block Copolymer Electrolytes: Enabling Superior Ambient Cationic Conductivity and Electrochemical Stability. Chem. Mater. 2019, 31, 277–285. [Google Scholar] [CrossRef]
- Butzelaar, A.J.; Röring, P.; Mach, T.P.; Hoffmann, M.; Jeschull, F.; Wilhelm, M.; Winter, M.; Brunklaus, G.; Théato, P. Styrene-Based Poly(ethylene oxide) Side-Chain Block Copolymers as Solid Polymer Electrolytes for High-Voltage Lithium-Metal Batteries. ACS Appl. Mater. Interfaces 2021, 13, 39257–39270. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry, 16th ed.; Cornell University Press: Ithaca, NY, USA, 1995; pp. 541–593. [Google Scholar]
- Bates, F.S. Polymer-Polymer Phase Behavior. Science 1991, 251, 898–905. [Google Scholar] [CrossRef]
- Leibler, L. Theory of microphase separation in block copolymers. Macromolecules 1980, 13, 1602–1617. [Google Scholar] [CrossRef]
- Lynd, N.A.; Meuler, A.J.; Hillmyer, M.A. Polydispersity and block copolymer self-assembly. Prog. Polym. Sci. 2008, 33, 875–893. [Google Scholar] [CrossRef]
- Hillmyer, M.A.; Bates, F.S. Synthesis and Characterization of Model Polyalkane−Poly(ethylene oxide) Block Copolymers. Macromolecules 1996, 29, 6994–7002. [Google Scholar] [CrossRef]
- Chatterjee, J.; Jain, S.; Bates, F.S. Comprehensive Phase Behavior of Poly(isoprene-b-styrene-b-ethylene oxide) Triblock Copolymers. Macromolecules 2007, 40, 2882–2896. [Google Scholar] [CrossRef]
- Bailey, T.S.; Pham, H.D.; Bates, F.S. Morphological Behavior Bridging the Symmetric AB and ABC States in the Poly(styrene-b-isoprene-b-ethylene oxide) Triblock Copolymer System. Macromolecules 2001, 34, 6994–7008. [Google Scholar] [CrossRef]
- Kimms, L.; Diddens, D.; Heuer, A. Understanding the Lithium Ion Transport in Concentrated Block–Copolymer Electrolytes on a Microscopic Level. arXiv 2020, arXiv:2010.11673. [Google Scholar]
- Kim, S.H.; Misner, M.J.; Xu, T.; Kimura, M.; Russell, T.P. Highly Oriented and Ordered Arrays from Block Copolymers via Solvent Evaporation. Adv. Mater. 2004, 16, 226–231. [Google Scholar] [CrossRef]
- Sun, Y.; Tan, R.; Ma, Z.; Zhou, D.; Li, J.; Kong, D.; Dong, X.-H. Quantify the contribution of chain length heterogeneity on block copolymer self-assembly. Giant 2020, 4, 100037. [Google Scholar] [CrossRef]
- Albert, J.N.; Epps, T.H. Self-assembly of block copolymer thin films. Mater. Today 2010, 13, 24–33. [Google Scholar] [CrossRef]
- Epps, T.H.; Cochran, E.W.; Bailey, T.S.; Waletzko, R.S.; Hardy, C.M.; Bates, F.S. Ordered Network Phases in Linear Poly(isoprene-b-styrene-b-ethylene oxide) Triblock Copolymers. Macromolecules 2004, 37, 8325–8341. [Google Scholar] [CrossRef]
- Coats, J.P.; Cochereau, R.; Dinu, I.A.; Messmer, D.; Sciortino, F.; Palivan, C.G. Trends in the Synthesis of Polymer Nano- and Microscale Materials for Bio-Related Applications. Macromol. Biosci. 2023, 2200474. [Google Scholar] [CrossRef]
- Xiang, L.; Li, Q.; Li, C.; Yang, Q.; Xu, F.; Mai, Y. Block Copolymer Self-Assembly Directed Synthesis of Porous Materials with Ordered Bicontinuous Structures and Their Potential Applications. Adv. Mater. 2023, 35, 2207684. [Google Scholar] [CrossRef]
- Qi, H.; Xie, R.; Yang, G.-W.; Zhang, Y.-Y.; Xu, C.-K.; Wang, Y.; Wu, G.-P. Rational Optimization of Bifunctional Organoboron Catalysts for Versatile Polyethers via Ring-Opening Polymerization of Epoxides. Macromolecules 2022, 55, 9081–9090. [Google Scholar] [CrossRef]
- Quirk, R.P.; Zhuo, Q.; Jang, S.H.; Lee, Y.; Lizarraga, G. Applications of Anionic Polymerization Research; Principles of Anionic Polymerization: An Introduction; American Chemical Society: Washington, DC, USA, 1998; pp. 2–27. [Google Scholar]
- Jagur-Grodzinski, J. Functional polymers by living anionic polymerization. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 2116–2133. [Google Scholar] [CrossRef]
- Merck Ethylene Oxide. Available online: https://www.sigmaaldrich.com/DE/en/product/sial/03906 (accessed on 28 March 2022).
- Förster, S.; Krämer, E. Synthesis of PB−PEO and PI−PEO Block Copolymers with Alkyllithium Initiators and the Phosphazene Base t-BuP4. Macromolecules 1999, 32, 2783–2785. [Google Scholar] [CrossRef]
- Herzberger, J.; Niederer, K.; Pohlit, H.; Seiwert, J.; Worm, M.; Wurm, F.R.; Frey, H. Polymerization of Ethylene Oxide, Propylene Oxide, and Other Alkylene Oxides: Synthesis, Novel Polymer Architectures, and Bioconjugation. Chem. Rev. 2016, 116, 2170–2243. [Google Scholar] [CrossRef] [PubMed]
- Esswein, B.; Möller, M. Polymerization of Ethylene Oxide with Alkyllithium Compounds and the Phosphazene Base “tBu-P4”. Angew. Chem. Int. Ed. Engl. 1996, 35, 623–625. [Google Scholar] [CrossRef]
- Brocas, A.-L.; Mantzaridis, C.; Tunc, D.; Carlotti, S. Polyether synthesis: From activated or metal-free anionic ring-opening polymerization of epoxides to functionalization. Prog. Polym. Sci. 2013, 38, 845–873. [Google Scholar] [CrossRef]
- Dreier, P.; Ahn, J.; Chang, T.; Frey, H. End Group Functionality of 95–99%: Epoxide Functionalization of Polystyryl-Lithium Evaluated via Solvent Gradient Interaction Chromatography. Macromol. Rapid Commun. 2022, 43, 2200560. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Xia, X.; Tajima, K.; Yamamoto, T.; Isono, T.; Satoh, T. Polyether/Polythioether Synthesis via Ring-Opening Polymerization of Epoxides and Episulfides Catalyzed by Alkali Metal Carboxylates. Macromolecules 2022, 55, 9373–9383. [Google Scholar] [CrossRef]
- Quirk, R.P.; Pickel, D.L.; Hasegawa, H. Anionic Polymerization Chemistry of Epoxides: Electron-Transfer Processes. Macromol. Symp. 2005, 226, 69–78. [Google Scholar] [CrossRef]
- Tonhauser, C.; Frey, H. A Road Less Traveled to Functional Polymers: Epoxide Termination in Living Carbanionic Polymer Synthesis. Macromol. Rapid Commun. 2010, 31, 1938–1947. [Google Scholar] [CrossRef]
- Tonhauser, C.; Golriz, A.A.; Moers, C.; Klein, R.; Butt, H.-J.; Frey, H. Stimuli-Responsive Y-Shaped Polymer Brushes Based on Junction-Point-Reactive Block Copolymers. Adv. Mater. 2012, 24, 5559–5563. [Google Scholar] [CrossRef] [PubMed]
- Natalello, A.; Alkan, A.; Friedel, A.; Lieberwirth, I.; Frey, H.; Wurm, F.R. Enlarging the Toolbox: Epoxide Termination of Polyferrocenylsilane (PFS) as a Key Step for the Synthesis of Amphiphilic PFS–Polyether Block Copolymers. ACS Macro Lett. 2013, 2, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Pointer, W.; Patias, G.; Al-Shok, L.; Hand, R.A.; Smith, T.; Haddleton, D.M. End functionalization of polyisoprene and polymyrcene obtained by anionic polymerization via one-pot ring-opening mono-addition of epoxides. Eur. Polym. J. 2023, 183, 111755. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar]
- Krause, D.; Grünebaum, M.; Wiemhöfer, H.-D.; Winter, M. Improved Synthesis for Producing Ordered Poly-Block Copolymers Having a Controllable Molecular Weight Distribution. International Patent Application No. PCT/WO 2022/008615 A1, 23 January 2023. [Google Scholar]
- Lochmann, L.; Pospíšil, J.; Lím, D. On the interaction of organolithium compounds with sodium and potassium alkoxides. A new method for the synthesis of organosodium and organopotassium compounds. Tetrahedron Lett. 1966, 7, 257–262. [Google Scholar] [CrossRef]
- Long, T.E.; Liu, H.Y.; Schell, B.A.; Teegarden, D.M.; Uerz, D.S. Determination of solution polymerization kinetics by near-infrared spectroscopy. 1. Living anionic polymerization processes. Macromolecules 1993, 26, 6237–6242. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Biehl, R. Jscatter, a program for evaluation and analysis of experimental data. PLoS ONE 2019, 14, e0218789. [Google Scholar] [CrossRef] [PubMed]
- Förster, S.; Apostol, L.; Bras, W. Scatter: Software for the analysis of nano- and mesoscale small-angle scattering. J. Appl. Crystallogr. 2010, 43, 639–646. [Google Scholar] [CrossRef]
- Graf, M. Polarmodifikation von Butadienkautschuk mit Polyethylenglykol. Ph.D. Thesis, Universität Bayreuth, Bayreuth, Germany, 28 October 2002. [Google Scholar]
- Zhang, B.; Zhang, Y.; Zhang, N.; Liu, J.; Cong, L.; Liu, J.; Sun, L.; Mauger, A.; Julien, C.M.; Xie, H.; et al. Synthesis and interface stability of polystyrene-poly(ethylene glycol)-polystyrene triblock copolymer as solid-state electrolyte for lithium-metal batteries. J. Power Sources 2019, 428, 93–104. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Wang, Y.; Lu, J.; Piñón, V.; Weck, M. Shell Cross-Linked Micelle-Based Nanoreactors for the Substrate-Selective Hydrolytic Kinetic Resolution of Epoxides. J. Am. Chem. Soc. 2011, 133, 14260–14263. [Google Scholar] [CrossRef] [PubMed]
- van Butsele, K.; Stoffelbach, F.; Jérôme, R.; Jérôme, C. Synthesis of Novel Amphiphilic and pH-Sensitive ABC Miktoarm Star Terpolymers. Macromolecules 2006, 39, 5652–5656. [Google Scholar] [CrossRef]
- Bhanu, V.A.; Kishore, K. Role of oxygen in polymerization reactions. Chem. Rev. 1991, 91, 99–117. [Google Scholar] [CrossRef]
- Wurm, F.; Wilms, D.; Klos, J.; Löwe, H.; Frey, H. Carbanions on Tap–Living Anionic Polymerization in a Microstructured Reactor. Macromol. Chem. Phys. 2008, 209, 1106–1114. [Google Scholar] [CrossRef]
- Castle, T.C. Synthesis of Block Copolymers by the Conversion of Living Anionic Polymerisation Into Living ROMP. Ph.D. Thesis, Durham University, Durham, UK, 13 June 2005. [Google Scholar]
- Chang, N.N.; Daly, S.R.; Girolami, G.S. Salt elimination reactions do not always eliminate. Mechanistic study of the reaction of NdCl3 with sodium N,N-dimethylaminodiboranate. Polyhedron 2019, 162, 52–58. [Google Scholar] [CrossRef]
- Surakit, T.; Adun, N.; Jitladda, T.S. Quantitative analysis of isoprene units in natural rubber and synthetic polyisoprene using 1H-NMR spectroscopy with an internal standard. Polym. Test. 2015, 43, 21–26. [Google Scholar]
- Toolan, D.T.W.; Isakova, A.; Hodgkinson, R.; Reeves-McLaren, N.; Hammond, O.S.; Edler, K.J.; Briscoe, W.H.; Arnold, T.; Gough, T.; Topham, P.D.; et al. Insights into the Influence of Solvent Polarity on the Crystallization of Poly(ethylene oxide) Spin-Coated Thin Films via In Situ Grazing Incidence Wide-Angle X-ray Scattering. Macromolecules 2016, 49, 4579–4586. [Google Scholar] [CrossRef]
- Butzelaar, A.J.; Röring, P.; Hoffmann, M.; Atik, J.; Paillard, E.; Wilhelm, M.; Winter, M.; Brunklaus, G.; Theato, P. Advanced Block Copolymer Design for Polymer Electrolytes: Prospects of Microphase Separation. Macromolecules 2021, 54, 11101–11112. [Google Scholar] [CrossRef]
- Sakurai, S.; Momii, T.; Taie, K.; Shibayama, M.; Nomura, S.; Hashimoto, T. Morphology transition from cylindrical to lamellar microdomains of block copolymers. Macromolecules 1993, 26, 485–491. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. Connection between Polymer Molecular Weight, Density, Chain Dimensions, and Melt Viscoelastic Properties. Macromolecules 1994, 27, 4639–4647. [Google Scholar] [CrossRef]
- Isono, T.; Kawakami, N.; Watanabe, K.; Yoshida, K.; Otsuka, I.; Mamiya, H.; Ito, H.; Yamamoto, T.; Tajima, K.; Borsali, R. Microphase separation of carbohydrate-based star-block copolymers with sub-10 nm periodicity. Polym. Chem. 2019, 10, 1119–1129. [Google Scholar] [CrossRef]
- Chintapalli, M.; Le, T.N.P.; Venkatesan, N.R.; Mackay, N.G.; Rojas, A.A.; Thelen, J.L.; Chen, X.C.; Devaux, D.; Balsara, N.P. Structure and Ionic Conductivity of Polystyrene-block-poly(ethylene oxide) Electrolytes in the High Salt Concentration Limit. Macromolecules 2016, 49, 1770–1780. [Google Scholar] [CrossRef]
- Zhu, L.; Cheng, S.Z.D.; Calhoun, B.H.; Ge, Q.; Quirk, R.P.; Thomas, E.L.; Hsiao, B.S.; Yeh, F.; Lotz, B. Phase structures and morphologies determined by self-organization, vitrification, and crystallization: Confined crystallization in an ordered lamellar phase of PEO-b-PS diblock copolymer. Polymer 2001, 42, 5829–5839. [Google Scholar] [CrossRef]
- Michler, G.H. Electron Microscopy of Polymers; Preparation of Thin Sections: (Cryo)ultramicrotomy and (Cryo)microtomy; Springer Laboratory: Berlin/Heidelberg, Germany, 2008; pp. 199–217. [Google Scholar]













| Polymer | n (sec-BuLi) /mmol | n (Isoprene) /mmol | n (Styrene) /mmol | n (EmPEO1.9) /mmol |
|---|---|---|---|---|
| PI6.8PS17.3AmPEO1.9 | 0.256 | 25.6 | 42.5 | 0.282 |
| PI14.6PS34.8AmPEO1.9 | 0.249 | 53.1 | 83.1 | 0.274 |
| PI24.8PS25.0AmPEO1.9 | 0.132 | 48.0 | 31.5 | 0.145 |
| PI35.1PS14.8AmPEO1.9 | 0.233 | 119.9 | 33.2 | 0.257 |
| PI26.1PS67.3AmPEO1.9 | 0.148 | 56.6 | 95.3 | 0.163 |
| Polymer | Mn,calc. /kg mol−1 | PIx CRUcalc. | Mn,calc.,PIx /kg mol−1 | PSy CRUcalc. | Mn,calc.,PSy /kg mol−1 | PEO1.9 CRUcalc. | Mn,calc.,PEO1.9 /kg mol−1 |
|---|---|---|---|---|---|---|---|
| PI6.8PS17.3AmPEO1.9 | 26.0 | 100 | 6.8 | 166 | 17.3 | 43 | 1.9 |
| PI14.6PS34.8AmPEO1.9 | 51.3 | 214 | 14.6 | 334 | 34.8 | 43 | 1.9 |
| PI24.8PS25.0AmPEO1.9 | 51.7 | 365 | 24.8 | 240 | 25.0 | 43 | 1.9 |
| PI35.1PS14.8AmPEO1.9 | 51.8 | 516 | 35.1 | 142 | 14.8 | 43 | 1.9 |
| PI26.1PS67.3AmPEO1.9 | 95.3 | 384 | 26.1 | 646 | 67.3 | 43 | 1.9 |
| Polymer | Mn,NMR /kg mol−1 | PIx CRUNMR | Mn,NMR,PIx /kg mol−1 | PSy CRUNMR | Mn,NMR,PSy /kg mol−1 | PEO1.9 CRUNMR | Mn,NMR,PEO1.9 /kg mol−1 |
|---|---|---|---|---|---|---|---|
| PI6.8PS17.3AmPEO1.9 | 26.8 | 101 | 6.9 | 174 | 18.1 | 42 | 1.9 |
| PI14.6PS34.8AmPEO1.9 | 49.7 | 206 | 14.0 | 324 | 33.8 | 44 | 1.9 |
| PI24.8PS25.0AmPEO1.9 | 49.7 | 346 | 23.6 | 231 | 24.1 | 45 | 2.0 |
| PI35.1PS14.8AmPEO1.9 | 49.6 | 489 | 33.3 | 137 | 14.3 | 45 | 2.0 |
| PI26.1PS67.3AmPEO1.9 | 106.2 | 460 | 31.3 | 702 | 73.1 | 41 | 1.8 |
| Polymer | Mn,calc. 1 /kg mol−1 | Mn,NMR 2 /kg mol−1 | Mn,GPC 3 /kg mol−1 | Đ 3 |
|---|---|---|---|---|
| EmPEO1.9 | 1.9 | 2.0 | 2.8 | 1.03 |
| PI6.8PS17.3AmPEO1.9 | 26.0 | 26.8 | 31.2 | 1.02 |
| PI14.6PS34.8AmPEO1.9 | 51.3 | 49.7 | 65.3 | 1.02 |
| PI24.8PS25.0AmPEO1.9 | 51.7 | 49.7 | 62.4 | 1.03 |
| PI35.1PS14.8AmPEO1.9 | 51.8 | 49.6 | 63.0 | 1.03 |
| PI26.1PS67.3AmPEO1.9 | 95.3 | 106.2 | 107.6 | 1.08 |
| Polymer | PIx 1 /wt.% | PSy 1 /wt.% | PEO1.9 1 /wt.% | ϑg,PIx 2 /°C | ΔCp,PIx 2 /J (g K)−1 | ϑg,PSy 2 /°C | ΔCp,PSy 2 /J (g K)−1 | ϑmp,PEO1.9 2 /°C | ϑd5 3 /°C |
|---|---|---|---|---|---|---|---|---|---|
| PI6.8PS17.3AmPEO1.9 | 25.6 | 67.5 | 6.9 | −63.7 | 0.13 | 74.6 | 0.12 | 50.2 | 331 |
| PI14.6PS34.8AmPEO1.9 | 28.2 | 68.0 | 3.9 | −65.9 | 0.14 | 86.8 | 0.24 | 50.6 | 331 |
| PI24.8PS25.0AmPEO1.9 | 47.5 | 48.5 | 4.0 | −60.7 | 0.27 | 80.9 | 0.14 | 49.9 | 327 |
| PI35.1PS14.8AmPEO1.9 | 67.2 | 28.8 | 4.0 | −60.1 | 0.37 | 76.1 | 0.06 | 49.9 | 324 |
| PI26.1PS67.3AmPEO1.9 | 29.5 | 68.8 | 1.7 | −67.6 | 0.14 | 95.8 | 0.21 | - | 334 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krause, D.T.; Krämer, S.; Siozios, V.; Butzelaar, A.J.; Dulle, M.; Förster, B.; Theato, P.; Mayer, J.; Winter, M.; Förster, S.; et al. Improved Route to Linear Triblock Copolymers by Coupling with Glycidyl Ether-Activated Poly(ethylene oxide) Chains. Polymers 2023, 15, 2128. https://doi.org/10.3390/polym15092128
Krause DT, Krämer S, Siozios V, Butzelaar AJ, Dulle M, Förster B, Theato P, Mayer J, Winter M, Förster S, et al. Improved Route to Linear Triblock Copolymers by Coupling with Glycidyl Ether-Activated Poly(ethylene oxide) Chains. Polymers. 2023; 15(9):2128. https://doi.org/10.3390/polym15092128
Chicago/Turabian StyleKrause, Daniel T., Susanna Krämer, Vassilios Siozios, Andreas J. Butzelaar, Martin Dulle, Beate Förster, Patrick Theato, Joachim Mayer, Martin Winter, Stephan Förster, and et al. 2023. "Improved Route to Linear Triblock Copolymers by Coupling with Glycidyl Ether-Activated Poly(ethylene oxide) Chains" Polymers 15, no. 9: 2128. https://doi.org/10.3390/polym15092128
APA StyleKrause, D. T., Krämer, S., Siozios, V., Butzelaar, A. J., Dulle, M., Förster, B., Theato, P., Mayer, J., Winter, M., Förster, S., Wiemhöfer, H.-D., & Grünebaum, M. (2023). Improved Route to Linear Triblock Copolymers by Coupling with Glycidyl Ether-Activated Poly(ethylene oxide) Chains. Polymers, 15(9), 2128. https://doi.org/10.3390/polym15092128