
Catalytic Ring-Opening Polymerisation of Cyclic Ethylene Carbonate: Importance of Elementary Steps for Determining Polymer Properties Revealed via DFT-MTD Simulations Validated Using Kinetic Measurements


Abstract
:1. Introduction
2. Materials and Methods
2.1. Computational Details
2.2. Chemicals Used and Suppliers
2.3. Experimental Method
3. Results and Discussion
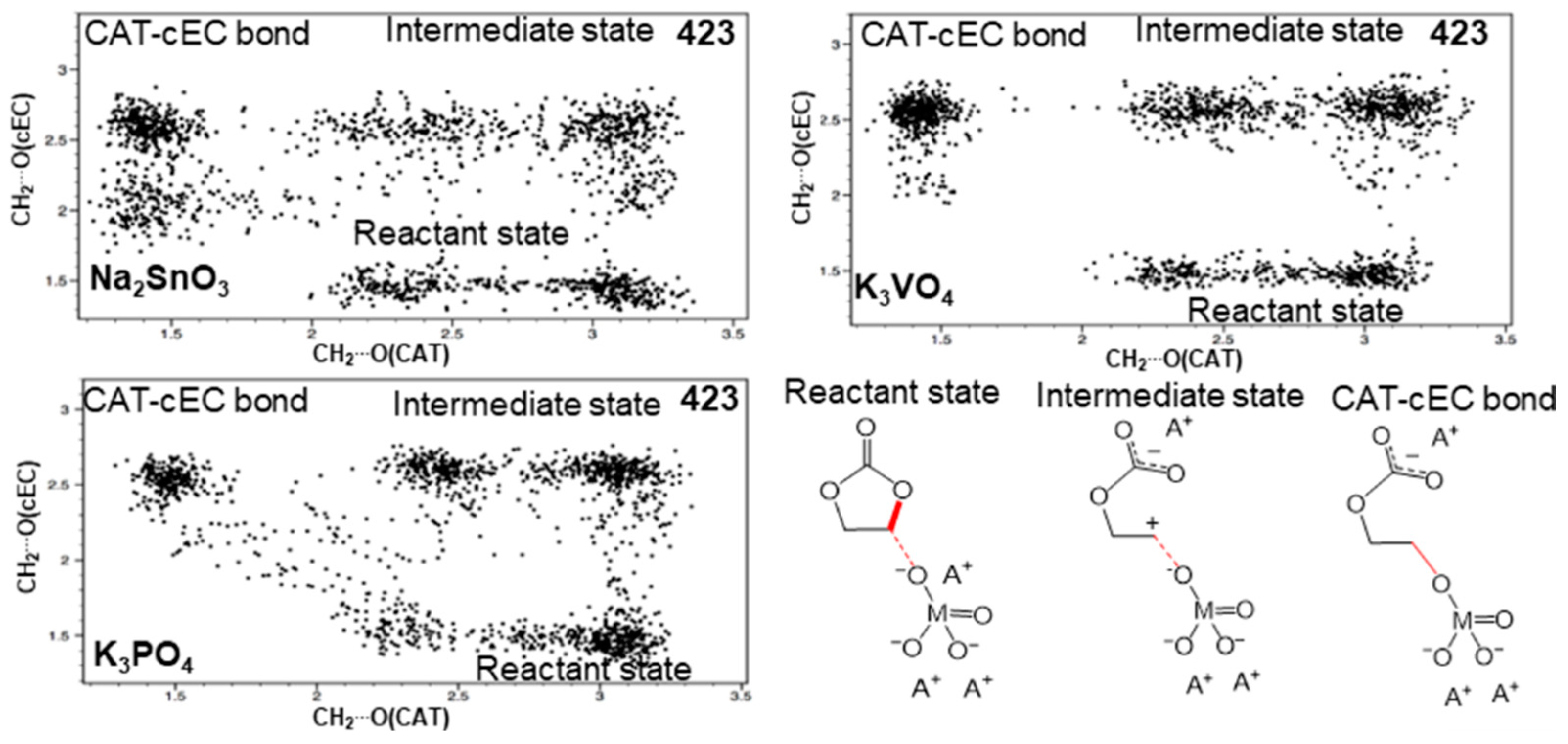
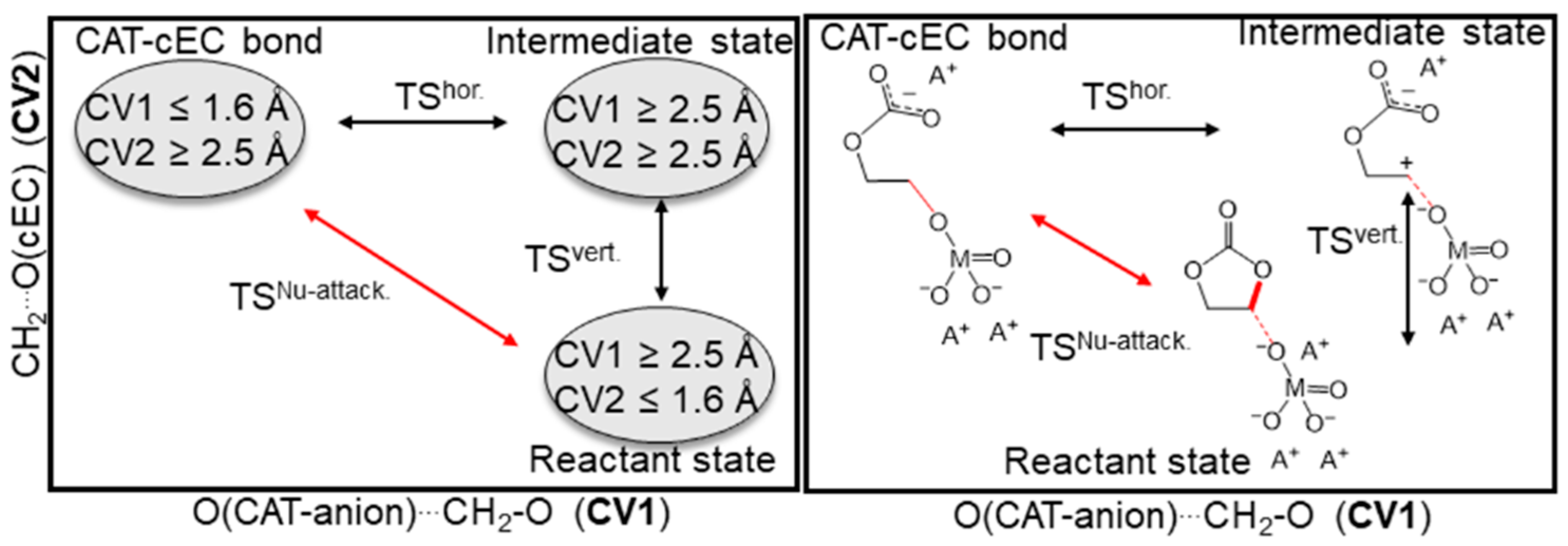
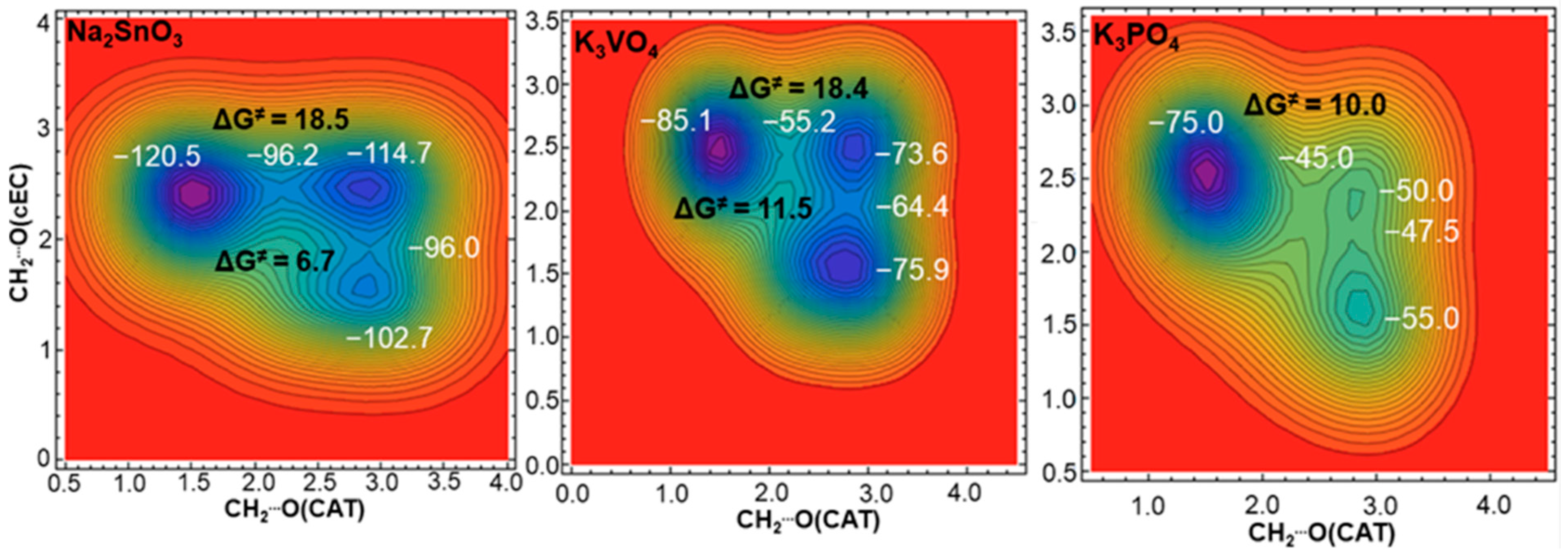
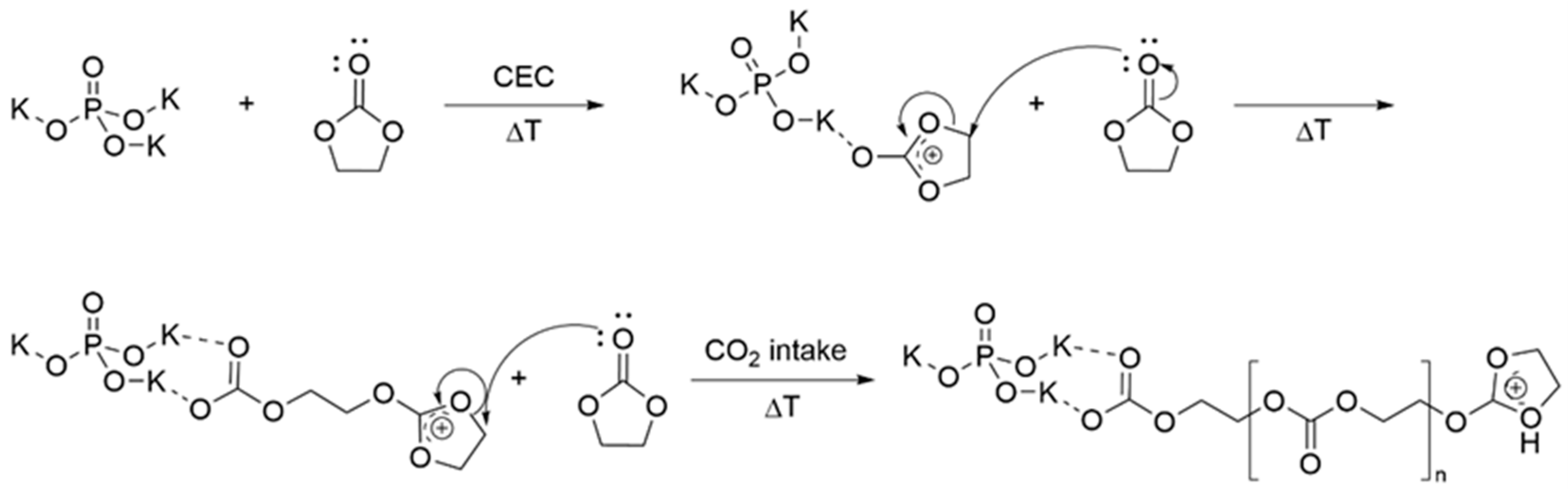
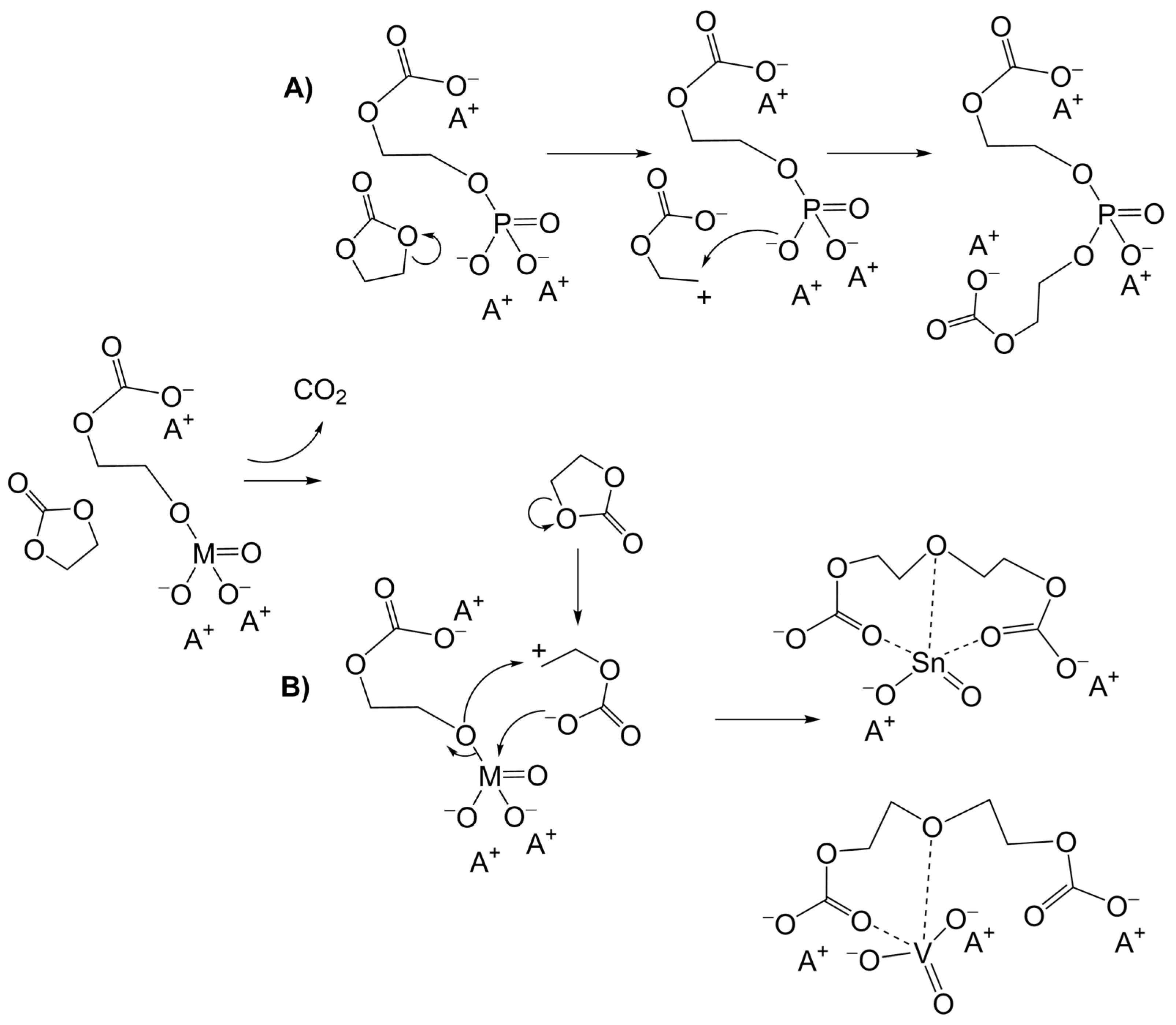
3.1. Simulation of the Catalyst–cEC Attachment Path
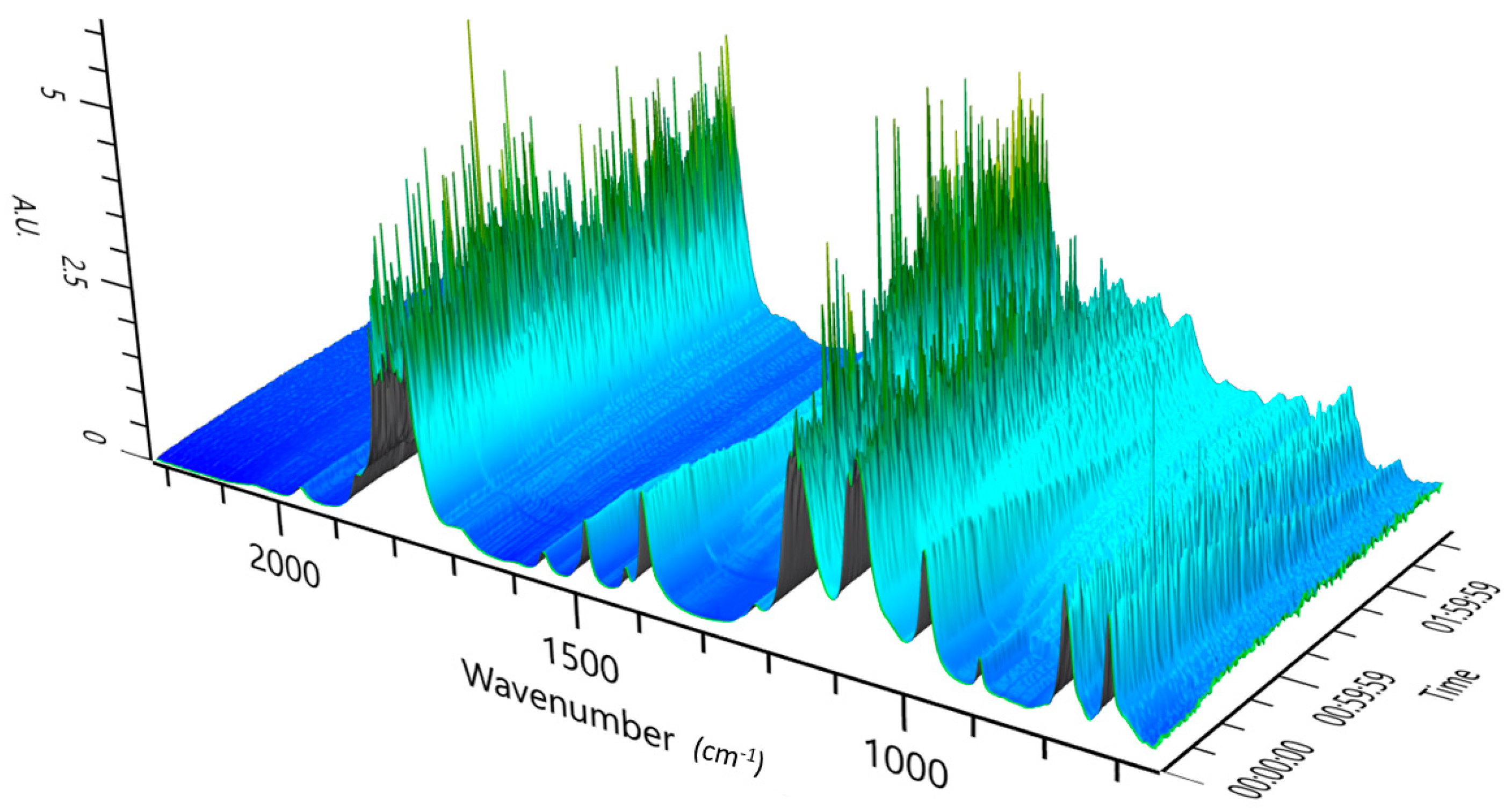
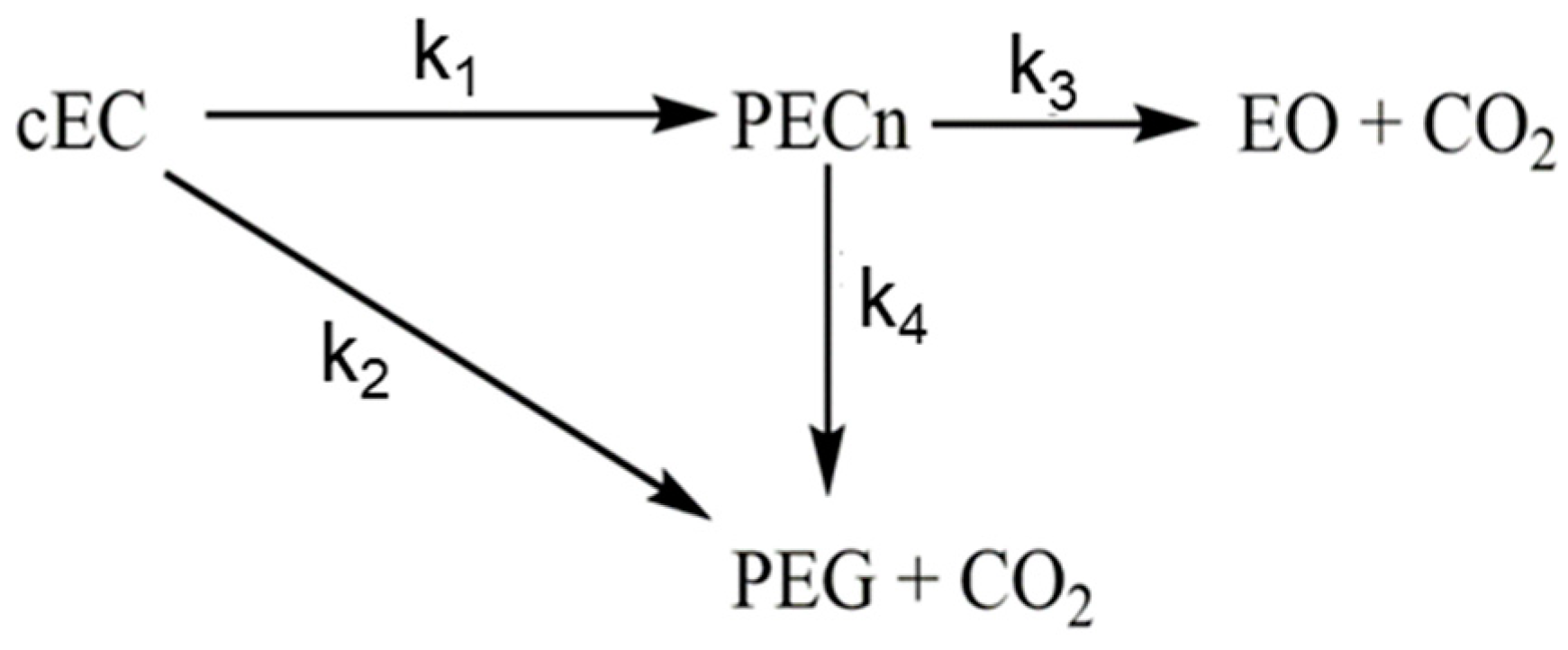
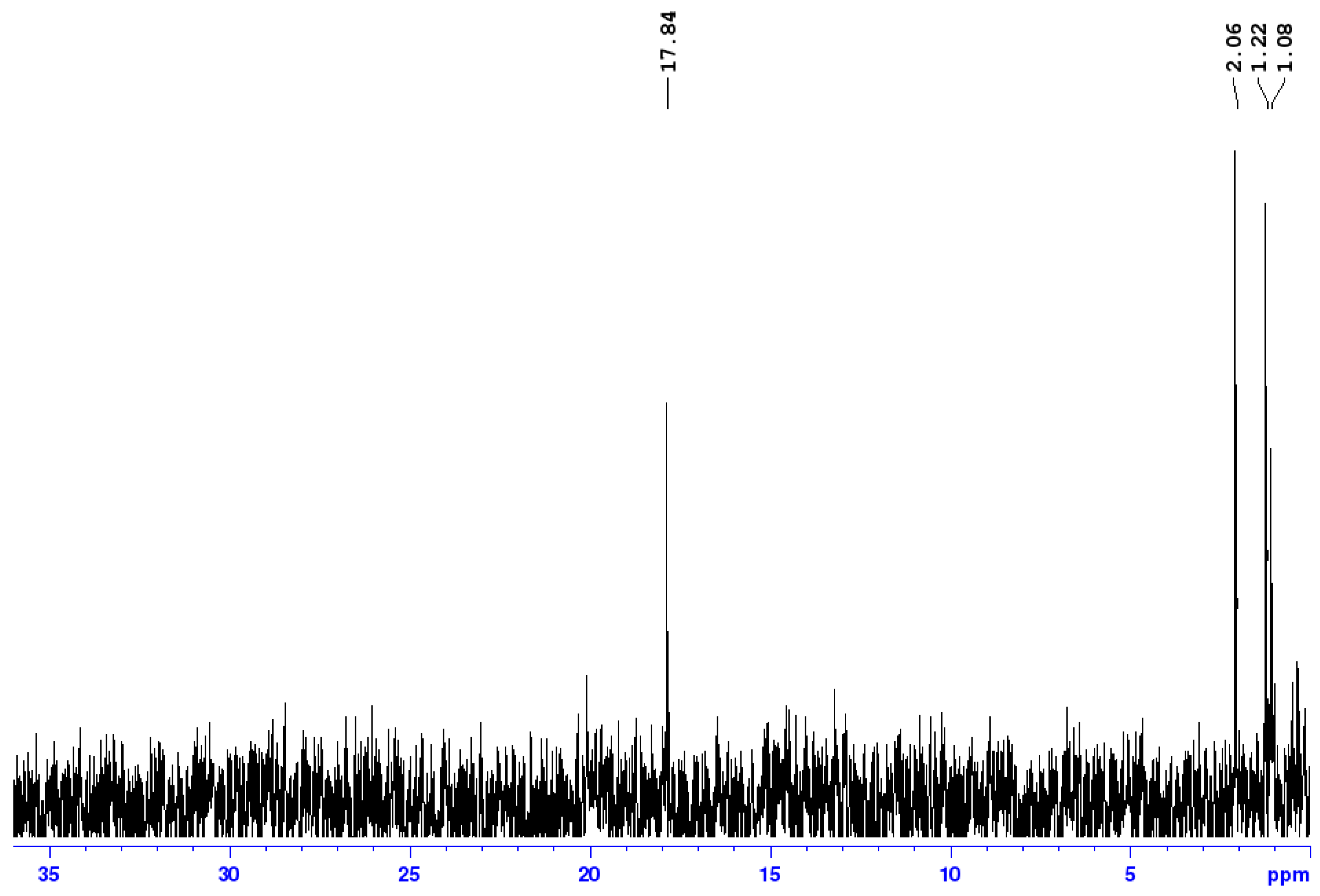
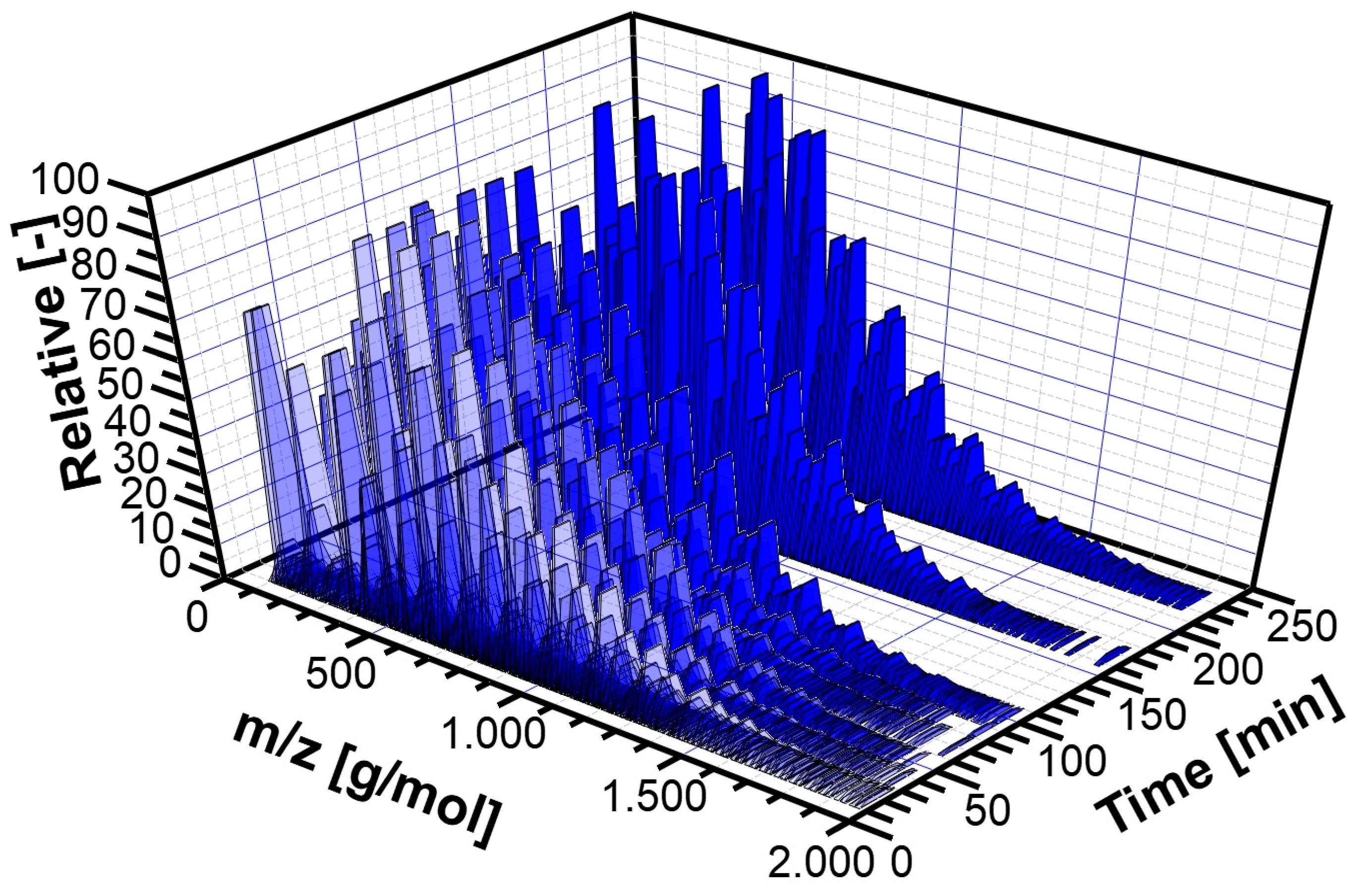
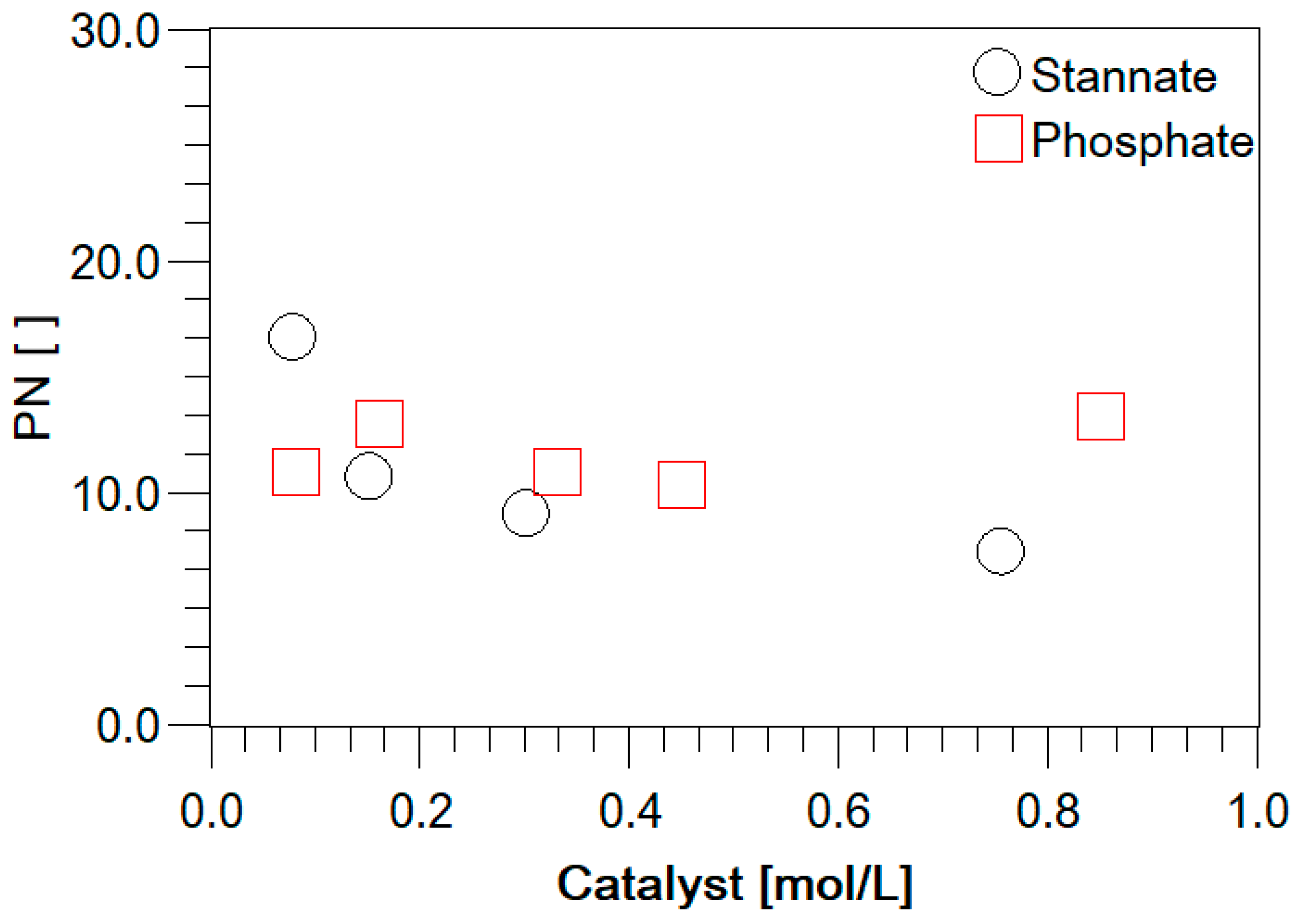
3.2. Experimental Analysis of the Kinetics of Polymerisation of cEC, including CO2 Release
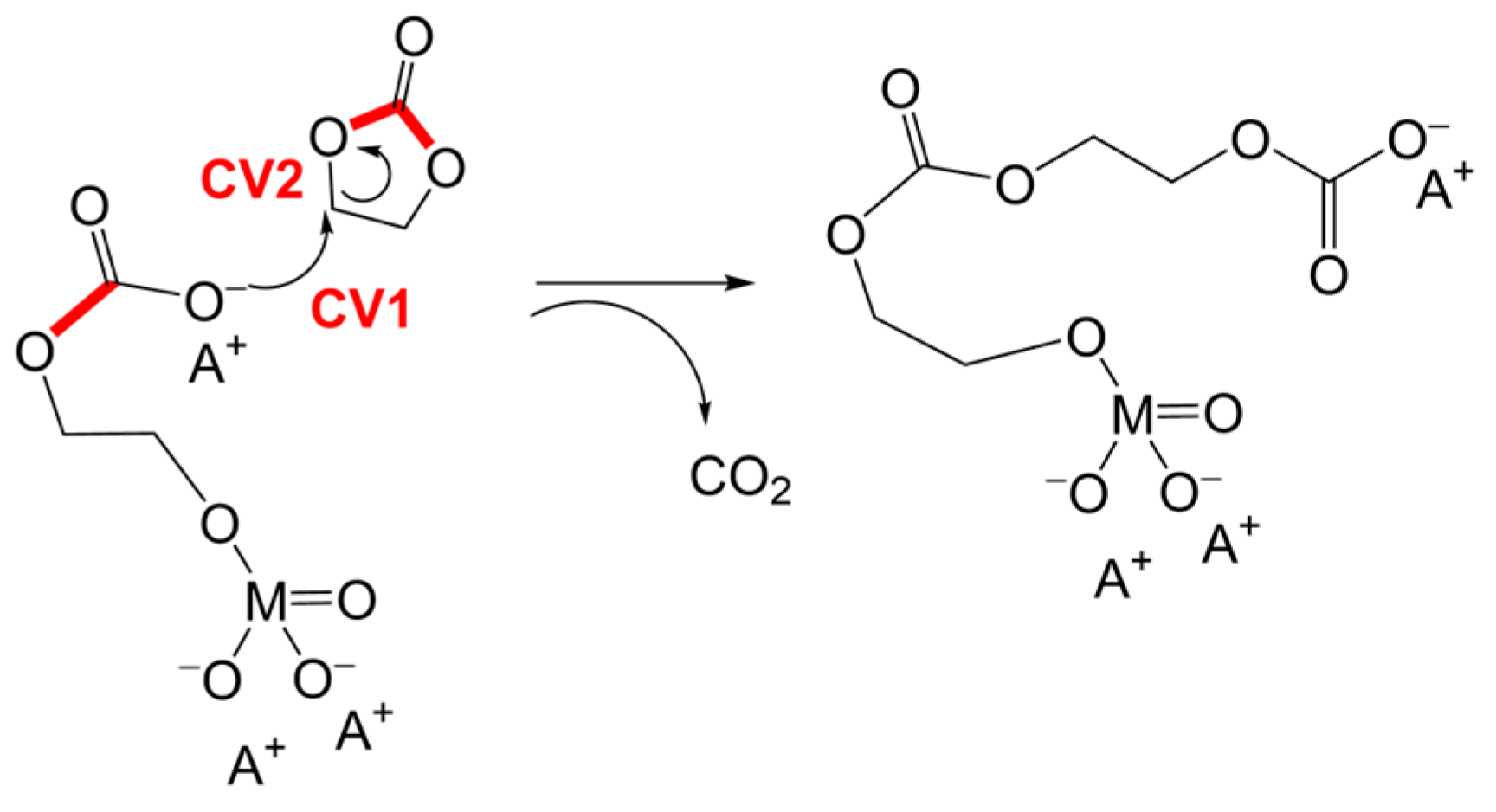
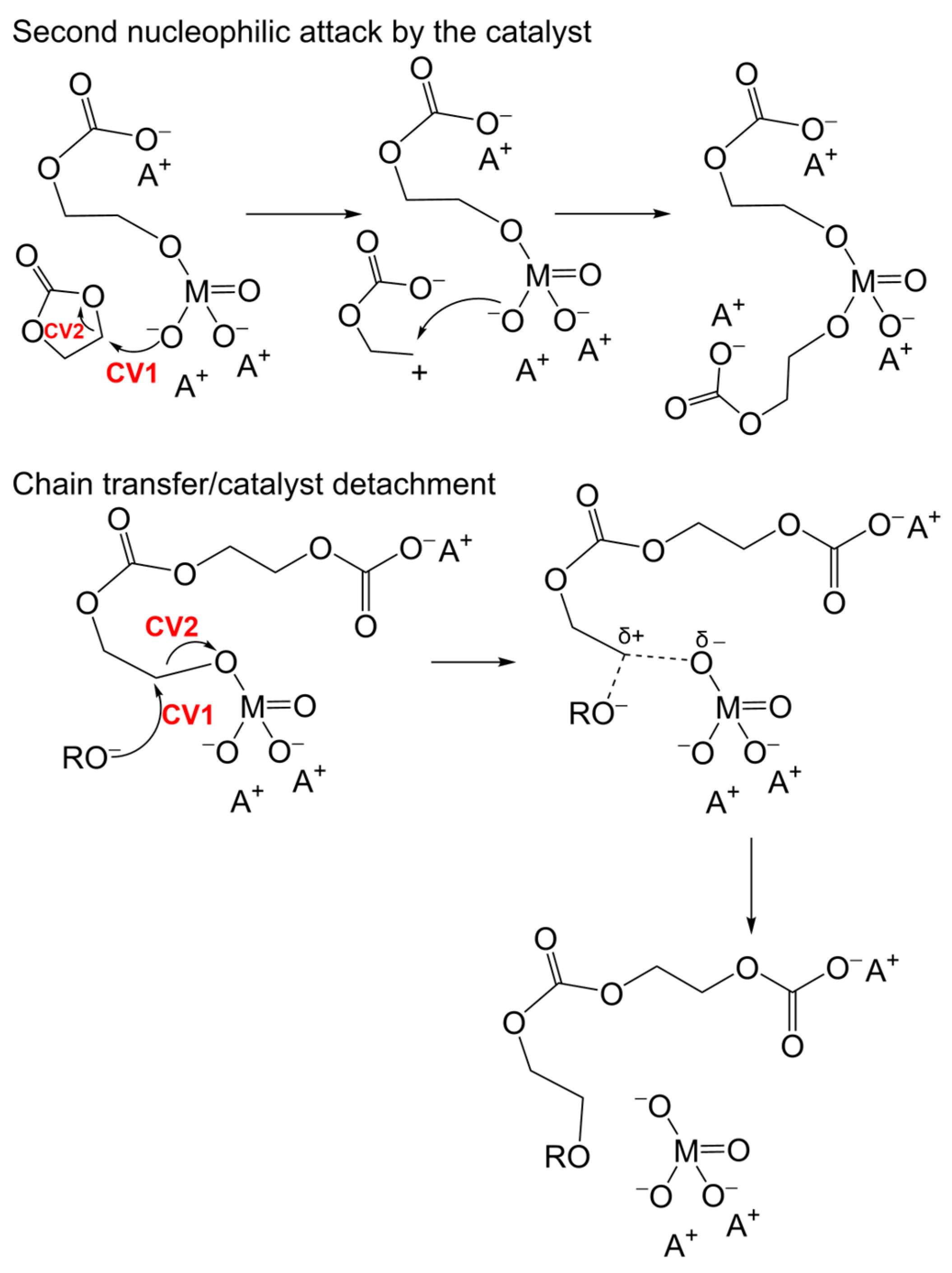
3.3. Alternative Pathways of Chain Growth Revealed by Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quadrelli, E.A.; Armstrong, K.; Styring, P. Carbon Dioxide Utilisation, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 9780444627469. [Google Scholar]
- Machat, M.R.; Marbach, J.; Schumacher, H.; Raju, S.; Lansing, M.; Over, L.C.; Adler, L.; Langanke, J.; Wolf, A.; Leitner, W.; et al. Turning CO/CO2-containing industrial process gas into valuable building blocks for the polyurethane industry. React. Chem. Eng. 2022, 7, 580–589. [Google Scholar] [CrossRef]
- Langanke, J.; Wolf, A.; Hofmann, J.; Böhm, K.; Subhani, M.A.; Müller, T.E.; Leitner, W.; Gürtler, C. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 2014, 16, 1865–1870. [Google Scholar] [CrossRef]
- Wolf, A.; Grasser, S.; Gürtler, C.; Hofmann, J. Verfahren zur Herstellung von Polyethercarbonatpolyolen Mit Doppelmetallcyanidkatalysatoren und in Gegenwart von Metallsalzen. WO Patent WO2011101403, 25 August 2011. [Google Scholar]
- Gürtler, C.; Hofmann, J.; Müller, T.E.; Wolf, A.; Grasser, S.; Köhler, B. Verfahren zur Herstellung von Polyethercarbonatpolyolen. WO Patent WO2011117332, 29 September 2011. [Google Scholar]
- Rokicki, G. Aliphatic cyclic carbonates and spiroorthocarbonates as monomers. Prog. Polym. Sci. 2000, 25, 259–342. [Google Scholar] [CrossRef]
- Rokicki, G.; Piotrowska, A. A new route to polyurethanes from ethylene carbonate, diamines and diols. Polymer 2002, 43, 2927–2935. [Google Scholar] [CrossRef]
- Takata, T.; Endo, T. Recent advances in the development of expanding monomers: Synthesis, polymerization and volume change. Prog. Polym. Sci. 1993, 18, 839–870. [Google Scholar] [CrossRef]
- Bailey, W.J.; Sun, R.L. Polymer Preprints (American Chemical Society, Division of Polymer Chemistry); American Chemical Society, Division of Polymer Chemistry: Dallas, TX, USA, 1972; pp. 281–286. ISBN 0032-3934. [Google Scholar]
- Brady, R.F. Volume Change in the Polymerization of Spirocyclic Monomers: Causes, Effects, and Applications. J. Macromol. Sci. Part C Polym. Rev. 1992, 32, 135–181. [Google Scholar] [CrossRef]
- Sadhir, R.; Luck, R. Expanding monomers. In Expanding Monomers; CRC Press: Boca Raton, FL, USA, 1992; p. 152. ISBN 9780849351563. [Google Scholar]
- Lopes, E.; Ribeiro, A.; Martins, L. New Trends in the Conversion of CO2 to Cyclic Carbonates. Catalysts 2020, 10, 479. [Google Scholar] [CrossRef]
- von Seggern, N.; Schindler, T.; Naumann, S. Dual Catalytic Ring-Opening Polymerization of Ethylene Carbonate for the Preparation of Degradable PEG. Biomacromolecules 2020, 21, 2661–2669. [Google Scholar] [CrossRef]
- Harris, R.F. Structural features of poly(alkylene ether carbonate) diol oligomers by capillary gas chromatography. J. Appl. Polym. Sci. 1989, 37, 183–200. [Google Scholar] [CrossRef]
- Harris, R.F. Effect of catalyst on the molecular weight advancement of poly(ethylene ether carbonate) polyols. J. Appl. Polym. Sci. 1990, 40, 1265–1279. [Google Scholar] [CrossRef]
- Yadav, N.; Seidi, F.; Crespy, D.; D’Elia, V. Polymers Based on Cyclic Carbonates as Trait d’Union Between Polymer Chemistry and Sustainable CO2 Utilization. ChemSusChem 2019, 12, 724–754. [Google Scholar] [CrossRef]
- Lee, J.-C.; Litt, M.H. Ring-Opening Polymerization of Ethylene Carbonate and Depolymerization of Poly(ethylene oxide-co-ethylene carbonate). Macromolecules 2000, 33, 1618–1627. [Google Scholar] [CrossRef]
- Kiesewetter, M.K.; Shin, E.J.; Hedrick, J.L.; Waymouth, R.M. Organocatalysis: Opportunities and Challenges for Polymer Synthesis. Macromolecules 2010, 43, 2093–2107. [Google Scholar] [CrossRef]
- Jang, J.H.; Ha, J.H.; Kim, I.; Baik, J.H.; Hong, S.C. Facile Room-Temperature Preparation of Flexible Polyurethane Foams from Carbon Dioxide Based Poly(ether carbonate) Polyols with a Reduced Generation of Acetaldehyde. ACS Omega 2019, 4, 7944–7952. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, P.; Ag, C.D. Bauen Mit CO2-Technologie Wird Möglich. Available online: https://www.covestro.com/press/de/bauen-mit-co-technologie-wird-moeglich/ (accessed on 4 October 2023).
- Zhang, S.-G.; Wei, Y.-D.; Yin, S.-F.; Luo, S.-L.; Au, C.-T. Superbasic sodium stannate as catalyst for dehydrogenation, Michael addition and transesterification reactions. Appl. Catal. A Gen. 2011, 406, 113–118. [Google Scholar] [CrossRef]
- Haba, O.; Tomizuka, H.; Endo, T. Anionic Ring-Opening Polymerization of Methyl 4,6-O-Benzylidene-2,3-O-carbonyl-α-d-glucopyranoside: A First Example of Anionic Ring-Opening Polymerization of Five-Membered Cyclic Carbonate without Elimination of CO2. Macromolecules 2005, 38, 3562–3563. [Google Scholar] [CrossRef]
- Tezuka, K.; Komatsu, K.; Haba, O. The anionic ring-opening polymerization of five-membered cyclic carbonates fused to the cyclohexane ring. Polym. J. 2013, 45, 1183–1187. [Google Scholar] [CrossRef]
- Piccini, G.; Lee, M.-S.; Yuk, S.F.; Zhang, D.; Collinge, G.; Kollias, L.; Nguyen, M.-T.; Glezakou, V.-A.; Rousseau, R. Ab initio molecular dynamics with enhanced sampling in heterogeneous catalysis. Catal. Sci. Technol. 2022, 12, 12–37. [Google Scholar] [CrossRef]
- Daru, J.; Bakó, I.; Stirling, A.; Pápai, I. Mechanism of Heterolytic Hydrogen Splitting by Frustrated Lewis Pairs: Comparison of Static and Dynamic Models. ACS Catal. 2019, 9, 6049–6057. [Google Scholar] [CrossRef]
- Heshmat, M. Alternative Pathway of CO2 Hydrogenation by Lewis-Pair-Functionalized UiO-66 MOF Revealed by Metadynamics Simulations. J. Phys. Chem. C 2020, 124, 10951–10960. [Google Scholar] [CrossRef]
- Liu, L.; Lukose, B.; Ensing, B. A Free Energy Landscape of CO2 Capture by Frustrated Lewis Pairs. ACS Catal. 2018, 8, 3376–3381. [Google Scholar] [CrossRef]
- Ensing, B.; Laio, A.; Parrinello, M.; Klein, M.L. A Recipe for the Computation of the Free Energy Barrier and the Lowest Free Energy Path of Concerted Reactions. J. Phys. Chem. B 2005, 109, 6676–6687. [Google Scholar] [CrossRef] [PubMed]
- Stirling, A.; Iannuzzi, M.; Parrinello, M.; Molnar, F.; Bernhart, V.; Luinstra, G.A. β-Lactone Synthesis from Epoxide and CO: Reaction Mechanism Revisited. Organometallics 2005, 24, 2533–2537. [Google Scholar] [CrossRef]
- Piccini, G.; Polino, D.; Parrinello, M. Identifying Slow Molecular Motions in Complex Chemical Reactions. J. Phys. Chem. Lett. 2017, 8, 4197–4200. [Google Scholar] [CrossRef] [PubMed]
- Ghoussoub, M.; Yadav, S.; Ghuman, K.K.; Ozin, G.A.; Singh, C.V. Metadynamics-Biased ab Initio Molecular Dynamics Study of Heterogeneous CO2 Reduction via Surface Frustrated Lewis Pairs. ACS Catal. 2016, 6, 7109–7117. [Google Scholar] [CrossRef]
- Dong, Y.; Ghuman, K.K.; Popescu, R.; Duchesne, P.N.; Zhou, W.; Loh, J.Y.Y.; Jelle, A.A.; Jia, J.; Wang, D.; Mu, X.; et al. Tailoring Surface Frustrated Lewis Pairs of In2O3−x(OH)y for Gas-Phase Heterogeneous Photocatalytic Reduction of CO2 by Isomorphous Substitution of In3+ with Bi3+. Adv. Sci. 2018, 5, 1700732. [Google Scholar] [CrossRef]
- Dong, Y.; Duchesne, P.; Mohan, A.; Ghuman, K.K.; Kant, P.; Hurtado, L.; Ulmer, U.; Loh, J.Y.Y.; Tountas, A.A.; Wang, L.; et al. Shining light on CO2: From materials discovery to photocatalyst, photoreactor and process engineering. Chem. Soc. Rev. 2020, 49, 5648–5663. [Google Scholar] [CrossRef]
- Brüggemann, D.; Shojamejer, T.; Tupinamba Lima, M.; Zukova, D.; Marschall, R.; Schomäcker, R. The Performance of Carbonate-Modified Nonionic Surfactants in Microplastic Flotation. Water 2023, 15, 1000. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, M.; Parrinello, M. Rethinking Metadynamics: From Bias Potentials to Probability Distributions. J. Phys. Chem. Lett. 2020, 11, 2731–2736. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hediger, H.J. Infrarotspektroskopie: Grundlagen, Anwendungen, Interpretation mit Zahlreichen Tabellen und 32 Spektren; Akademische Verlagsgesellschaft: Frankfurt, Germany, 1971; pp. 1–25, 29–169, 173–195. ISBN 3-400-00052-3. [Google Scholar]
- Cammann, K. Infrarotspektroskopie. In Instrumentelle Analytische Chemie; Verfahren, Anwendungen, Qualitätssicherung; Spektrum Akademischer Verlag: Heidelberg/Berlin, Germany, 2001; pp. 5–51. ISBN 3-8274-0057-0. [Google Scholar]
- Fox, T. Hesse-Meier-Zeeh Infrarot- und Ramanspektren. In Spektroskopische Methoden in der Organischen Chemie; Bienz, S., Bigler, L., Fox, T., Meier, H., Eds.; George Thieme Verlag: Stuttgart, Germany, 2016; pp. 37–74. ISBN 978-3-13-576109-1. [Google Scholar]
- Schwedt, G. Infrarot- und Raman-Spektroskopie. In Taschenatlas der Analytik; George Thieme Verlag: Stuttgart, Germany, 1992; pp. 114–121. ISBN 3-13-759301-8. [Google Scholar]
- Heshmat, M.; Leven, M.; Linker, O.; Sebastian, M.; Gürtler, C.; Machat, M.R. A DFT-metadynamics study disclosing key properties of ring-opening polymerization catalysts to produce polyethercarbonate polyols from cyclic ethylene carbonate as part of an emerging CCU technology. Phys. Chem. Chem. Phys. 2023, 25, 20485–20494. [Google Scholar] [CrossRef]
- Reich, H.; Hans Reich’s Collection. NMR Spectroscopy. Available online: https://organicchemistrydata.org/hansreich/resources/nmr/?index=nmr_index%2F31P_shift (accessed on 24 April 2023).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | ΔG≠ [kcal/mol] I/II |
|---|---|
| Na2SnO3 | 5.8/11.2 |
| K3VO4 | 5.5/7.7 |
| K3PO4 | 4.5/6.9 |
| Catalyst | ΔG≠ H+ Abstraction [kcal/mol] | ΔG≠ cEC Opening [kcal/mol] | Exp. CO2 Content [%] |
|---|---|---|---|
| H3PO4 | 22.3 | 50.0 | 3.0 |
| Li3PO4 | 12.8 | 39.1 | 5.0 |
| Na3PO4 | 7.6 | 5.2 | 11.0 |
| K3PO4 | 4.2 | 4.0 | 15.0 |
| Na2SnO3 | 6.2 | 11.2 | 26.0 |
| K2SnO3 | 6.5 | 10.8 | 22.0 |
| Na3VO4 | 4.8 | 4.5 | 26.0 |
| K3VO4 | 5.8 | 7.7 | 25.0 |
| T [K] | k1 [1/s] | k2 [1/s] | k3 [1/s] | k4 [1/s] | k2/k1 [−] | CO2 [%] |
|---|---|---|---|---|---|---|
| 413 | 0.029 | 0.026 | 5.4 × 10−7 | 1.0 × 10−5 | 0.89 | 26 |
| 423 | 0.029 | 0.028 | 6.8 × 10−6 | 7.4 × 10−6 | 0.96 | 26 |
| 433 | 0.038 | 0.034 | 1.4 × 10−6 | 2.3 × 10−4 | 0.89 | 26 |
| 443 | 0.032 | 0.034 | 1.3 × 10−5 | 4.9 × 10−4 | 1.06 | 24 |
| T [K] | k1 [1/s] | k2 [1/s] | kdes [1/s] | k2/k1 [−] | CO2 [%] |
|---|---|---|---|---|---|
| 395 | 0.021 | 0.023 | 0.020 | 1.09 | 17.4 |
| 405 | 0.0036 | 0.0028 | 0.024 | 0.78 | 27.9 |
| 415 | 0.0054 | 0.032 | 0.017 | 5.93 | 7.31 |
| 425 | 0.0071 | 0.026 | 0.029 | 3.66 | 10.64 |
| Catalyst/ΔG≠ [kcal/mol] | Na2SnO3 [kcal/mol] | K3VO4 [kcal/mol] | K3PO4 [kcal/mol] |
|---|---|---|---|
| 413 K | 34.0 | 36.0 | 28.1 |
| 423 K | 32.3 | 33.5 | 27.2 |
| 433 K | 31.8 | 30.8 | 26.0 |
| 443 K | 28.9 | 29.2 | 24.0 |
| Catalyst/ΔG≠ [kcal/mol] T = 423 K | K3PO4 [kcal/mol] | Na2SnO3 [kcal/mol] | K3VO4 [kcal/mol] |
|---|---|---|---|
| Second nucleophilic attack | 20.0 | 27.6 | 30.0 |
| Chain transfer—catalyst detachment | 42.7 | 34.0 | 36.1 |
| K2SnO3 | Na3VO4 | |||||
|---|---|---|---|---|---|---|
| T (K) | k1 | k2 | ktrans | k1 | k2 | Ktrans |
| 413 | 0.029 | 0.026 | 0.0037 | 0.0349 | 0.0346 | 0.0061 |
| 423 | 0.029 | 0.028 | 0.0034 | 0.0524 | 0.0534 | 0.0092 |
| 433 | 0.038 | 0.034 | 0.0052 | 0.1070 | 0.1157 | 0.0196 |
| 443 | 0.032 | 0.034 | 0.0073 | 0.2071 | 0.2431 | 0.0411 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brüggemann, D.; Machat, M.R.; Schomäcker, R.; Heshmat, M. Catalytic Ring-Opening Polymerisation of Cyclic Ethylene Carbonate: Importance of Elementary Steps for Determining Polymer Properties Revealed via DFT-MTD Simulations Validated Using Kinetic Measurements. Polymers 2024, 16, 136. https://doi.org/10.3390/polym16010136
Brüggemann D, Machat MR, Schomäcker R, Heshmat M. Catalytic Ring-Opening Polymerisation of Cyclic Ethylene Carbonate: Importance of Elementary Steps for Determining Polymer Properties Revealed via DFT-MTD Simulations Validated Using Kinetic Measurements. Polymers. 2024; 16(1):136. https://doi.org/10.3390/polym16010136
Chicago/Turabian StyleBrüggemann, Daniel, Martin R. Machat, Reinhard Schomäcker, and Mojgan Heshmat. 2024. "Catalytic Ring-Opening Polymerisation of Cyclic Ethylene Carbonate: Importance of Elementary Steps for Determining Polymer Properties Revealed via DFT-MTD Simulations Validated Using Kinetic Measurements" Polymers 16, no. 1: 136. https://doi.org/10.3390/polym16010136