

Biomass Valorization through Catalytic Pyrolysis Using Metal-Impregnated Natural Zeolites: From Waste to Resources

 , , ,
, , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Biomass
2.1.2. Catalyst
2.2. Methods
2.2.1. Catalyst Modification
2.2.2. Biomass Characterization
2.2.3. Physico-Chemical Characterization of Natural and Modified Zeolite Samples
2.2.4. Catalytic Fast Pyrolysis of Biomass
3. Results
3.1. Biomass Characterization
3.2. Physical-Chemical Characterization of Natural and Modified Zeolite Samples
3.2.1. Crystallinity
3.2.2. Chemical Composition and Surface Morphology
3.2.3. Textural Properties
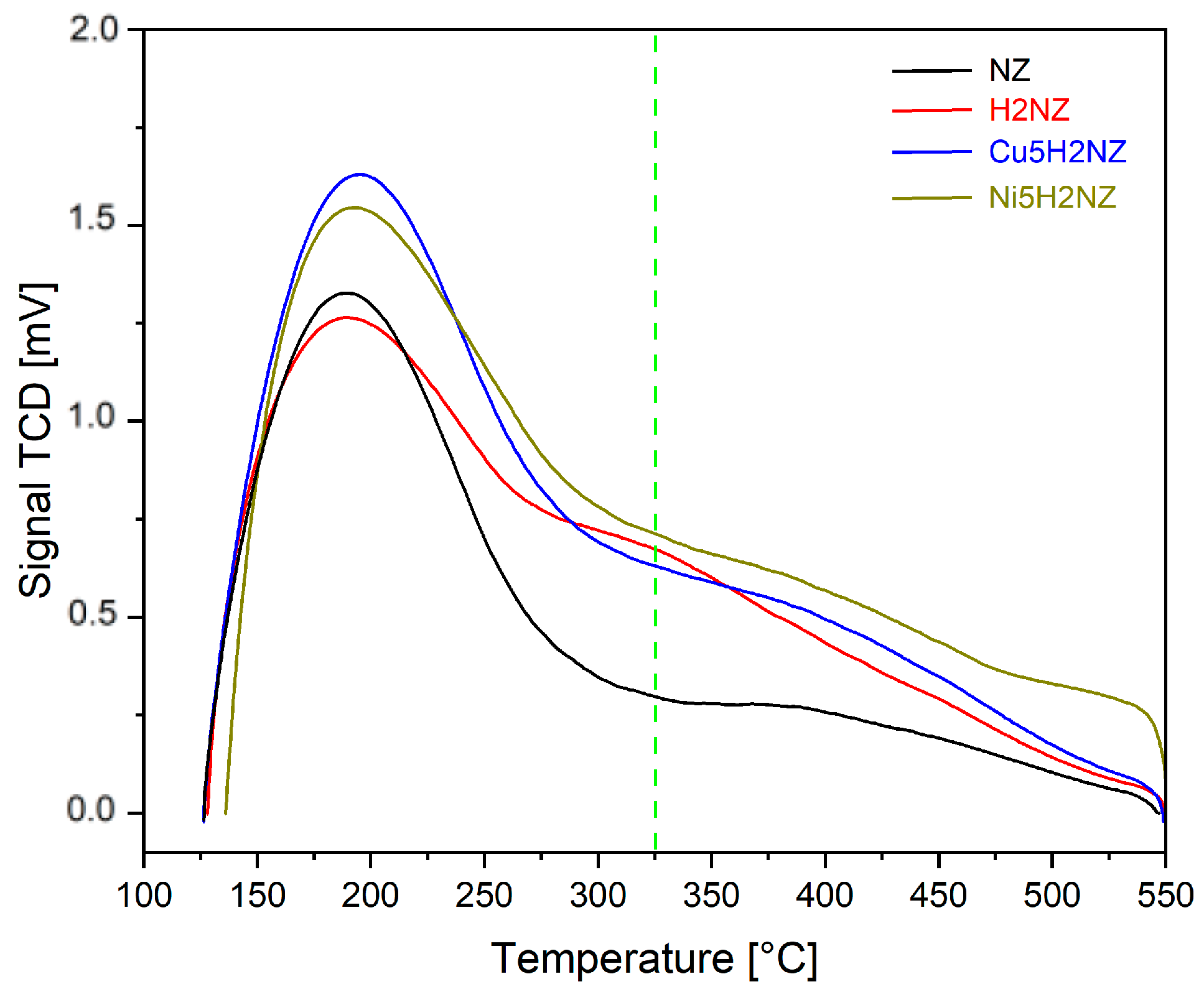
3.2.4. Thermal Analysis
3.2.5. Acid Site Strength
3.3. Catalytic Fast Pyrolysis of Biomass
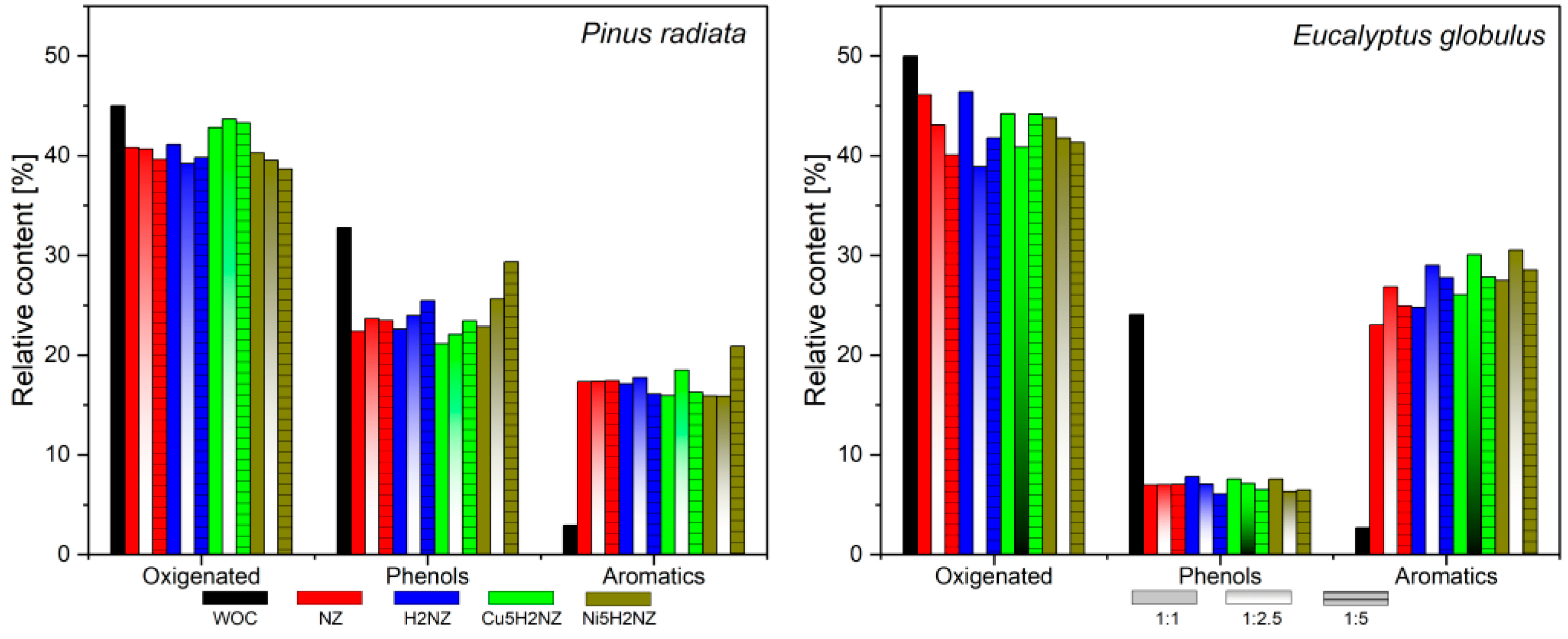
3.3.1. Family Compounds Formed during PR and EG Catalytic Pyrolysis
3.3.2. Reaction Pathways
3.3.3. Aromatics Compounds

4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- BP. Full Report—Statistical Review of World Energy 2021; BP p.l.c.: Londong, UK, 2021. [Google Scholar]
- Deng, W.; Feng, Y.; Fu, J.; Guo, H.; Guo, Y.; Han, B.; Jiang, Z.; Kong, L.; Li, C.; Liu, H.; et al. Catalytic Conversion of Lignocellulosic Biomass into Chemicals and Fuels. Green Energy Environ. 2023, 8, 10–114. [Google Scholar] [CrossRef]
- Hu, X.; Gholizadeh, M. Biomass Pyrolysis: A Review of the Process Development and Challenges from Initial Researches up to the Commercialisation Stage. J. Energy Chem. 2019, 39, 109–143. [Google Scholar] [CrossRef]
- Álvarez González, V.; Poblete Hernández, P.; Soto Aguirre, D.; Caselli, J.G.; Kahler González, C.; Pardo Velásquez, E.; Carlos, J.; Munita, B.; Rocha, D.B. Anuario Forestal Chilean Statistical Yearbook of Forestry 2022. Anu. For. INFOR 2022, 187, 1–280. [Google Scholar] [CrossRef]
- Arteaga-Pérez, L.; Segura, C.; Diéguez, K. Procesos de Torrefacción Para Valorización de Residuos Lignocelulósicos. Análisis de Posibles Tecnologías de Aplicación En Sudamérica. Afinidad 2016, 573, 60–68. [Google Scholar]
- Xiu, S.; Shahbazi, A. Bio-Oil Production and Upgrading Research: A Review. Renew. Sustain. Energy Rev. 2012, 16, 4406–4414. [Google Scholar] [CrossRef]
- Chaihad, N.; Karnjanakom, S.; Abudula, A.; Guan, G. Zeolite-Based Cracking Catalysts for Bio-Oil Upgrading: A Critical Review. Resour. Chem. Mater. 2022, 1, 167–183. [Google Scholar] [CrossRef]
- Kumar, R.; Strezov, V.; Lovell, E.; Kan, T.; Weldekidan, H.; He, J.; Jahan, S.; Dastjerdi, B.; Scott, J. Enhanced Bio-Oil Deoxygenation Activity by Cu/Zeolite and Ni/Zeolite Catalysts in Combined in-Situ and Ex-Situ Biomass Pyrolysis. J. Anal. Appl. Pyrolysis 2019, 140, 148–160. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.J. Lignocellulosic Biomass Pyrolysis: A Review of Product Properties and Effects of Pyrolysis Parameters. Renew. Sustain. Energy Rev. 2016, 57, 1126–1140. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.; Steele, P.H. Pyrolysis of Wood/Biomass for Bio-Oil: A Critical Review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Huo, X.; Xiao, J.; Song, M.; Zhu, L. Comparison between In-Situ and Ex-Situ Catalytic Pyrolysis of Sawdust for Gas Production. J. Anal. Appl. Pyrolysis 2018, 135, 189–198. [Google Scholar] [CrossRef]
- Galadima, A.; Muraza, O. In Situ Fast Pyrolysis of Biomass with Zeolite Catalysts for Bioaromatics/Gasoline Production: A Review. Energy Convers. Manag. 2015, 105, 338–354. [Google Scholar] [CrossRef]
- Wang, K.; Johnston, P.A.; Brown, R.C. Comparison of In-Situ and Ex-Situ Catalytic Pyrolysis in a Micro-Reactor System. Bioresour. Technol. 2015, 173, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Sarker, M.; Rahman, M.M.; Li, C.; Chai, M.; Nishu; Cotillon, R.; Scott, N.R. Multi-Scale Complexities of Solid Acid Catalysts in the Catalytic Fast Pyrolysis of Biomass for Bio-Oil Production—A Review. Prog. Energy Combust. Sci. 2020, 80, 100852. [Google Scholar] [CrossRef]
- Barbosa, A.S.; Siqueira, L.A.M.; Medeiros, R.L.B.A.; Melo, D.M.A.; Melo, M.A.F.; Freitas, J.C.O.; Braga, R.M. Renewable Aromatics through Catalytic Flash Pyrolysis of Pineapple Crown Leaves Using HZSM-5 Synthesised with RHA and Diatomite. Waste Manag. 2019, 88, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Alejandro-Martín, S.; Acaricia, A.M.; Cerda-Barrera, C.; Pérez, H.D. Influence of Chemical Surface Characteristics of Ammonium-Modified Chilean Zeolite on Oak Catalytic Pyrolysis. Catalysts 2019, 9, 465. [Google Scholar] [CrossRef]
- Dehmani, Y.; Ba Mohammed, B.; Oukhrib, R.; Dehbi, A.; Lamhasni, T.; Brahmi, Y.; El-Kordy, A.; Franco, D.S.P.; Georgin, J.; Lima, E.C.; et al. Adsorption of Various Inorganic and Organic Pollutants by Natural and Synthetic Zeolites: A Critical Review. Arab. J. Chem. 2024, 17, 105474. [Google Scholar] [CrossRef]
- Shekarchi, M.; Ahmadi, B.; Azarhomayun, F.; Shafei, B.; Kioumarsi, M. Natural Zeolite as a Supplementary Cementitious Material—A Holistic Review of Main Properties and Applications. Constr. Build. Mater. 2023, 409, 133766. [Google Scholar] [CrossRef]
- Serrano, D.P.; Melero, J.A.; Morales, G.; Iglesias, J.; Pizarro, P. Progress in the Design of Zeolite Catalysts for Biomass Conversion into Biofuels and Bio-Based Chemicals. Catal. Rev. Sci. Eng. 2018, 60, 1–70. [Google Scholar] [CrossRef]
- Gurevich Messina, L.I.; Bonelli, P.R.; Cukierman, A.L. In-Situ Catalytic Pyrolysis of Peanut Shells Using Modified Natural Zeolite. Fuel Process. Technol. 2017, 159, 160–167. [Google Scholar] [CrossRef]
- Bhoi, P.R.; Ouedraogo, A.S.; Soloiu, V.; Quirino, R. Recent Advances on Catalysts for Improving Hydrocarbon Compounds in Bio-Oil of Biomass Catalytic Pyrolysis. Renew. Sustain. Energy Rev. 2020, 121, 109676. [Google Scholar] [CrossRef]
- Veses, A.; Puértolas, B.; Callén, M.S.; García, T. Catalytic Upgrading of Biomass Derived Pyrolysis Vapors over Metal-Loaded ZSM-5 Zeolites: Effect of Different Metal Cations on the Bio-Oil Final Properties. Microporous Mesoporous Mater. 2015, 209, 189–196. [Google Scholar] [CrossRef]
- Rajić, N.; Logar, N.Z.; Rečnik, A.; El-Roz, M.; Thibault-Starzyk, F.; Sprenger, P.; Hannevold, L.; Andersen, A.; Stöcker, M. Hardwood Lignin Pyrolysis in the Presence of Nano-Oxide Particles Embedded onto Natural Clinoptilolite. Microporous Mesoporous Mater. 2013, 176, 162–167. [Google Scholar] [CrossRef]
- Cruz, N.; Bustos, C.; Aguayo, M.G.; Cloutier, A.; Castillo, R. THM Densification of Wood. Bioresources 2018, 13, 2268–2282. [Google Scholar]
- Alejandro, S.; Valdés, H.; Manero, M.H.; Zaror, C.A. BTX Abatement Using Chilean Natural Zeolite: The Role of Brønsted Acid Sites. Water Sci. Technol. 2012, 66, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Alejandro-Martín, S.; Valdés, H.; Zaror, C.A. Natural Zeolite Reactivity towards Ozone: The Role of Acid Surface Sites. J. Adv. Oxid. Technol. 2011, 14, 182–189. [Google Scholar] [CrossRef]
- Soled, S.L.; Malek, A.; Miseo, S.; Baumgartner, J.; Kliewer, C.; Afeworki, M.; Stevens, P.A. Supported Metal Catalysts: Some Interesting New Leads in an Old Field. In Studies in Surface Science and Catalysis; Elsevier Inc.: Amsterdam, The Netherlands, 2006; Volume 162, pp. 103–110. ISBN 044452827X. [Google Scholar]
- Chaihad, N.; Anniwaer, A.; Karnjanakom, S.; Kasai, Y.; Kongparakul, S.; Samart, C.; Reubroycharoen, P.; Abudula, A.; Guan, G. In-Situ Catalytic Upgrading of Bio-Oil Derived from Fast Pyrolysis of Sunflower Stalk to Aromatic Hydrocarbons over Bifunctional Cu-Loaded HZSM-5. J. Anal. Appl. Pyrolysis 2021, 155, 105079. [Google Scholar] [CrossRef]
- Persson, H.; Duman, I.; Wang, S.; Pettersson, L.J.; Yang, W. Catalytic Pyrolysis over Transition Metal-Modified Zeolites: A Comparative Study between Catalyst Activity and Deactivation. J. Anal. Appl. Pyrolysis 2019, 138, 54–61. [Google Scholar] [CrossRef]
- ASTM D5373-08; Standard Test Methods for Instrumental Determination of Carbon, Hydrogen, and Nitrogen in Laboratory Samples of Coal. ASTM International: West Conshohocken, PA, USA, 2017.
- ASTM D3172; Standard Practice for Proximate Analysis of Coal and Coke. ASTM International: West Conshohocken, PA, USA, 2013.
- TAPPI T 280 Wd-06; Acetone Extractives of Wood and Pulp. TAPPI Press: Peachtree Corners, GA, USA, 2015.
- Aguayo, M.G.; Quintupill, L.; Castillo, R.; Baeza, J.; Freer, J.; Mendonça, R.T. Determination of Differences in Anatomical and Chemical Characteristics of Tension and Opposite Wood of 8-Year Old Eucalyptus Globulus. Maderas Cienc. Y Tecnol. 2010, 12, 241–251. [Google Scholar] [CrossRef]
- Venegas-Vásconez, D.; Arteaga-Pérez, L.E.; Aguayo, M.G.; Romero-Carrillo, R.; Guerrero, V.H.; Tipanluisa-Sarchi, L.; Alejandro-Martín, S. Analytical Pyrolysis of Pinus Radiata and Eucalyptus Globulus: Effects of Microwave Pretreatment on Pyrolytic Vapours Composition. Polymers 2023, 15, 3790. [Google Scholar] [CrossRef]
- Sihombing, J.L.; Gea, S.; Wirjosentono, B.; Agusnar, H.; Pulungan, A.N.; Herlinawati, H.; Yusuf, M. Characteristic and Catalytic Performance of Co and Co-Mo Metal Impregnated in Sarulla Natural Zeolite Catalyst for Hydrocracking of MEFA Rubber Seed Oil into Biogasoline Fraction. Catalysts 2020, 10, 121. [Google Scholar] [CrossRef]
- ASTM D5357-19; Standard Test Method for Determination of Relative Crystallinity of Zeolite Sodium A by X-ray Diffraction. American Society for Testing and Materials (ASTM): West Conshohocken, PA, USA, 2019.
- Galarza, E.D.; Fermanelli, C.S.; Pierella, L.B.; Saux, C.; Renzini, M.S. Influence of the Sn Incorporation Method in ZSM-11 Zeolites in the Distribution of Bio-Oil Products Obtained from Biomass Pyrolysis. J. Anal. Appl. Pyrolysis 2021, 156, 105116. [Google Scholar] [CrossRef]
- Zhang, C.; Kwak, G.; Park, H.G.; Jun, K.W.; Lee, Y.J.; Kang, S.C.; Kim, S. Light Hydrocarbons to BTEX Aromatics over Hierarchical HZSM-5: Effects of Alkali Treatment on Catalytic Performance. Microporous Mesoporous Mater. 2019, 276, 292–301. [Google Scholar] [CrossRef]
- Alejandro-Martín, S. Estudio de La Reacción Ozono-Compuestos Orgánicos Volátiles a Temperatura Ambiente En Presencia de Zeolita Natural Modificada. Ph.D. Thesis, Universidad de Concepción, Concepción, Chile, 2013. [Google Scholar]
- Valdés, H.; Solar, V.A.; Cabrera, E.H.; Veloso, A.F.; Zaror, C.A. Control of Released Volatile Organic Compounds from Industrial Facilities Using Natural and Acid-Treated Mordenites: The Role of Acidic Surface Sites on the Adsorption Mechanism. Chem. Eng. J. 2014, 244, 117–127. [Google Scholar] [CrossRef]
- Yogalakshmi, K.N.; Poornima Devi, T.; Sivashanmugam, P.; Kavitha, S.; Yukesh Kannah, R.; Varjani, S.; AdishKumar, S.; Kumar, G.; Rajesh Banu, J. Lignocellulosic Biomass-Based Pyrolysis: A Comprehensive Review. Chemosphere 2022, 286, 131824. [Google Scholar] [CrossRef]
- Hubble, A.H.; Childs, B.A.; Pecchi, M.; Sudibyo, H.; Tester, J.W.; Goldfarb, J.L. Role of in Situ (in Contact with Biomass) and Ex Situ (in Contact with Pyrolysis Vapors) Transition Metal Catalysts on Pyrolysis of Cherry Pits. Fuel 2023, 352, 129062. [Google Scholar] [CrossRef]
- Huang, M.; Xu, J.; Ma, Z.; Yang, Y.; Zhou, B.; Wu, C.; Ye, J.; Zhao, C.; Liu, X.; Chen, D.; et al. Bio-BTX Production from the Shape Selective Catalytic Fast Pyrolysis of Lignin Using Different Zeolite Catalysts: Relevance between the Chemical Structure and the Yield of Bio-BTX. Fuel Process. Technol. 2021, 216, 106792. [Google Scholar] [CrossRef]
- Tian, H.; Chen, L.; Huang, Z.; Cheng, S.; Yang, Y. Increasing the Bio-Aromatics Yield in the Biomass Pyrolysis Oils by the Integration of Torrefaction Deoxygenation Pretreatment and Catalytic Fast Pyrolysis with a Dual Catalyst System. Renew. Energy 2022, 187, 561–571. [Google Scholar] [CrossRef]
- Niu, Q.; Ghysels, S.; Wu, N.; Rousseau, D.P.L.; Pieters, J.; Prins, W.; Ronsse, F. Effects of Demineralization on the Composition of Microalgae Pyrolysis Volatiles in Py-GC–MS. Energy Convers. Manag. 2022, 251, 114979. [Google Scholar] [CrossRef]
- Arteaga-Pérez, L.E.; Segura, C.; Espinoza, D.; Radovic, L.R.; Jiménez, R. Torrefaction of Pinus Radiata and Eucalyptus Globulus: A Combined Experimental and Modeling Approach to Process Synthesis. Energy Sustain. Dev. 2015, 29, 13–23. [Google Scholar] [CrossRef]
- Wang, Y.; Akbarzadeh, A.; Chong, L.; Du, J.; Tahir, N.; Awasthi, M.K. Catalytic Pyrolysis of Lignocellulosic Biomass for Bio-Oil Production: A Review. Chemosphere 2022, 297, 134181. [Google Scholar] [CrossRef]
- Trisunaryanti, W. Characteristics of Metal Supported-Zeolite Catalysts for Hydrocracking of Polyethylene Terephthalat. IOSR J. Appl. Chem. 2013, 3, 29–34. [Google Scholar] [CrossRef]
- Kadarwati, S.; Wahyuni, S.; Trisunaryanti, W.; Triyono, T. Preparation, Characterizacion and Catalytic Activity Test of Ni-Mo/Natural Zeolite on Pyridine Hydrodenitrogenation. Indones. J. Chem. 2010, 10, 327–333. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Y.; Sekyere, D.T.; Wang, W.; Tian, Y. Catalytic Fast Pyrolysis of Waste Pine Sawdust over Solid Base, Acid and Base-Acid Tandem Catalysts. Bioresour. Technol. 2024, 394, 130294. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, F.; Yang, X.; Huang, Y.; Liu, C.; Zheng, Z.; Gu, J. Study on Aromatics Production via the Catalytic Pyrolysis Vapor Upgrading of Biomass Using Metal-Loaded Modified H-ZSM-5. J. Anal. Appl. Pyrolysis 2017, 126, 169–179. [Google Scholar] [CrossRef]
- Seyed Mousavi, S.A.H.; Sadrameli, S.M.; Saeedi Dehaghani, A.H. Catalytic Pyrolysis of Municipal Plastic Waste over Nano MIL-53 (Cu) Derived @ Zeolite Y for Gasoline, Jet Fuel, and Diesel Range Fuel Production. Process Saf. Environ. Prot. 2022, 164, 449–467. [Google Scholar] [CrossRef]
- Alshameri, A.; Ibrahim, A.; Assabri, A.M.; Lei, X.; Wang, H.; Yan, C. The Investigation into the Ammonium Removal Performance of Yemeni Natural Zeolite: Modification, Ion Exchange Mechanism, and Thermodynamics. Powder Technol. 2014, 258, 20–31. [Google Scholar] [CrossRef]
- Pabalan, R.T.; Bertetti, F.P. Cation-Exchange Properties of Natural Zeolites. Rev. Mineral. Geochem. 2001, 45, 453–517. [Google Scholar] [CrossRef]
- Ates, A.; Hardacre, C. The Effect of Various Treatment Conditions on Natural Zeolites: Ion Exchange, Acidic, Thermal and Steam Treatments. J. Colloid Interface Sci. 2012, 372, 130–140. [Google Scholar] [CrossRef]
- Ünaldi, T.; Mizrak, I.; Kadir, S. Physicochemical Characterisation of Natural K-Clinoptilolite and Heavy-Metal Forms from Gördes (Manisa, Western Turkey). J. Mol. Struct. 2013, 1054–1055, 349–358. [Google Scholar] [CrossRef]
- Alejandro, S.; Valdés, H.; Manéro, M.H.; Zaror, C.A. Oxidative Regeneration of Toluene-Saturated Natural Zeolite by Gaseous Ozone: The Influence of Zeolite Chemical Surface Characteristics. J. Hazard Mater. 2014, 274, 212–220. [Google Scholar] [CrossRef]
- Trisunaryanti, W.; Triyono; Falah, I.I.; Widyawati, D.; Yusniyanti, F. The Effect of Oxalic Acid and NaOH Treatments on the Character of Wonosari Natural Zeolite as Ni, Cu, and Zn Metal Support Catalyst for Hydrocracking of Castor Oil. Biomass Convers. Biorefinery 2024, 14, 5637–5649. [Google Scholar] [CrossRef]
- Alejandro-Martín, S.; Valdés, H.; Manero, M.H.; Zaror, C.A. Catalytic Ozonation of Toluene Using Chilean Natural Zeolite: The Key Role of Brønsted and Lewis Acid Sites. Catalysts 2018, 8, 211. [Google Scholar] [CrossRef]
- Veses, A.; Puértolas, B.; López, J.M.; Callén, M.S.; Solsona, B.; García, T. Promoting Deoxygenation of Bio-Oil by Metal-Loaded Hierarchical ZSM-5 Zeolites. ACS Sustain. Chem. Eng. 2016, 4, 1653–1660. [Google Scholar] [CrossRef]
- Perraki, T.; Orfanoudaki, A. Mineralogical Study of Zeolites from Pentalofos Area, Thrace, Greece. Appl. Clay Sci. 2004, 25, 9–16. [Google Scholar] [CrossRef]
- Sprynskyy, M.; Golembiewski, R.; Trykowski, G.; Buszewski, B. Heterogeneity and Hierarchy of Clinoptilolite Porosity. J. Phys. Chem. Solids 2010, 71, 1269–1277. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Xie, S.; Xu, L. Promoted Metal Utilization Capacity of Alkali-Treated Zeolite: Preparation of Zn/ZSM-5 and Its Application in 1-Hexene Aromatization. Appl. Catal. A Gen. 2009, 360, 8–16. [Google Scholar] [CrossRef]
- Wibowo, S.; Efiyanti, L.; Pari, G. Catalytic and Thermal Cracking of Bio-Oil from Oil-Palm Empty Fruit Bunches, in Batch Reactor. Indones. J. Chem. 2020, 20, 1000–1009. [Google Scholar] [CrossRef]
- Valizadeh, S.; Pyo, S.; Kim, Y.; Hakimian, H.; Park, Y. Production of Aromatics Fuel Additives from Catalytic Pyrolysis of Cow Manure over HZSM-5, HBeta, and HY Zeolites. Chem. Eng. J. 2022, 450, 137971. [Google Scholar] [CrossRef]
- Wang, H.; Male, J.; Wang, Y. Recent Advances in Hydrotreating of Pyrolysis Bio-Oil and Its Oxygen-Containing Model Compounds. ACS Catal. 2013, 3, 1047–1070. [Google Scholar] [CrossRef]
- Iliopoulou, E.F.; Stefanidis, S.D.; Kalogiannis, K.G.; Delimitis, A.; Lappas, A.A.; Triantafyllidis, K.S. Catalytic Upgrading of Biomass Pyrolysis Vapors Using Transition Metal-Modified ZSM-5 Zeolite. Appl. Catal. B 2012, 127, 281–290. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of Hemicellulose, Cellulose and Lignin Pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Cheng, Y.T.; Huber, G.W. Chemistry of Furan Conversion into Aromatics and Olefins over HZSM-5: A Model Biomass Conversion Reaction. ACS Catal. 2011, 1, 611–628. [Google Scholar] [CrossRef]
- Zhang, J.; Fidalgo, B.; Kolios, A.; Shen, D.; Gu, S. Mechanism of Deoxygenation in Anisole Decomposition over Single-Metal Loaded HZSM-5: Experimental Study. Chem. Eng. J. 2018, 336, 211–222. [Google Scholar] [CrossRef]
- Iliopoulou, E.F.; Triantafyllidis, K.S.; Lappas, A.A. Overview of Catalytic Upgrading of Biomass Pyrolysis Vapors toward the Production of Fuels and High-Value Chemicals. Wiley Interdiscip. Rev. Energy Environ. 2019, 8, e322. [Google Scholar] [CrossRef]
- Chen, W.H.; Cheng, C.L.; Lee, K.T.; Lam, S.S.; Ong, H.C.; Ok, Y.S.; Saeidi, S.; Sharma, A.K.; Hsieh, T.H. Catalytic Level Identification of ZSM-5 on Biomass Pyrolysis and Aromatic Hydrocarbon Formation. Chemosphere 2021, 271, 129510. [Google Scholar] [CrossRef]
- Williams, P.T.; Horne, P.A. The Influence of Catalyst Type on the Composition of Upgraded Biomass Pyrolysis Oils. J. Anal. Appl. Pyrolysis 1995, 31, 61. [Google Scholar] [CrossRef]
- Stefanidis, S.D.; Karakoulia, S.A.; Kalogiannis, K.G.; Iliopoulou, E.F.; Delimitis, A.; Yiannoulakis, H.; Zampetakis, T.; Lappas, A.A.; Triantafyllidis, K.S. Natural Magnesium Oxide (MgO) Catalysts: A Cost-Effective Sustainable Alternative to Acid Zeolites for the in Situ Upgrading of Biomass Fast Pyrolysis Oil. Appl. Catal. B 2016, 196, 155–173. [Google Scholar] [CrossRef]
- Mante, O.D.; Rodriguez, J.A.; Senanayake, S.D.; Babu, S.P. Catalytic Conversion of Biomass Pyrolysis Vapors into Hydrocarbon Fuel Precursors. Green Chem. 2015, 17, 2362–2368. [Google Scholar] [CrossRef]
- Lazaridis, P.A.; Fotopoulos, A.P.; Karakoulia, S.A.; Triantafyllidis, K.S. Catalytic Fast Pyrolysis of Kraft Lignin with Conventional, Mesoporous and Nanosized ZSM-5 Zeolite for the Production of Alkyl-Phenols and Aromatics. Front. Chem. 2018, 6, 295. [Google Scholar] [CrossRef] [PubMed]
- Tawalbeh, M.; Al-Othman, A.; Salamah, T.; Alkasrawi, M.; Martis, R.; El-Rub, Z.A. A Critical Review on Metal-Based Catalysts Used in the Pyrolysis of Lignocellulosic Biomass Materials. J. Environ. Manag. 2021, 299, 113597. [Google Scholar] [CrossRef]
- Chen, X.; Che, Q.; Li, S.; Liu, Z.; Yang, H.; Chen, Y.; Wang, X.; Shao, J.; Chen, H. Recent Developments in Lignocellulosic Biomass Catalytic Fast Pyrolysis: Strategies for the Optimisation of Bio-Oil Quality and Yield. Fuel Process. Technol. 2019, 196, 106180. [Google Scholar] [CrossRef]
- Grams, J.; Jankowska, A.; Goscianska, J. Advances in Design of Heterogeneous Catalysts for Pyrolysis of Lignocellulosic Biomass and Bio-Oil Upgrading. Microporous Mesoporous Mater. 2023, 362, 112761. [Google Scholar] [CrossRef]
- Alcazar-Ruiz, A.; Sanchez-Silva, L.; Dorado, F. Enhancement of BTX Production via Catalytic Fast Pyrolysis of Almond Shell, Olive Pomace with Polyvinyl Chloride Mixtures. Process Saf. Environ. Prot. 2022, 163, 218–226. [Google Scholar] [CrossRef]
- Huang, M.; Ma, Z.; Zhou, B.; Yang, Y.; Chen, D. Enhancement of the Production of Bio-Aromatics from Renewable Lignin by Combined Approach of Torrefaction Deoxygenation Pretreatment and Shape Selective Catalytic Fast Pyrolysis Using Metal Modified Zeolites. Bioresour. Technol. 2020, 301, 122754. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Plots | Ref | ||
|---|---|---|---|---|
| Catalysts | NZ | H2NZ | Cu5H2NZ | Ni5H2NZ |
| Relation B/C | 1/1 | 1/2.5 | 1/5 | |
| Biomass | PR | EG |
| Proximate Analysis | Ultimate Analysis | Chemical Composition | ||||||
|---|---|---|---|---|---|---|---|---|
| PR | EG | PR | EG | PR | EG | |||
| Moisture (%) | 7.75 | 5.51 | Carbon (%) | 48.02 | 47.76 | Hemicellulose (%) | 28.6 ± 0.8 | 26.3 ± 0.3 |
| Volatiles (%) | 76.73 | 77.13 | Hydrogen (%) | 5.90 | 6.32 | Cellulose (%) | 43.1 ± 0.1 | 53.0 ± 0.2 |
| Fixed carbon (%) | 14.68 | 16.85 | Nitrogen (%) | 0.29 | 0.09 | Lignin (%) | 26.6 ± 1.6 | 23.9 ± 2.1 |
| Ash (%) | 0.83 | 0.51 | Sulphur (%) | 0.10 | 0.05 | Extractives (%) | 1.8 ± 0.3 | 1.9 ± 0.0 |
| Oxygen (%) | 45.69 | 45.77 | ||||||
| Sample | D (nm) | Crystallinity |
|---|---|---|
| NZ | 98.38 | |
| H2NZ | 100.00 | |
| Cu5H2NZ | 7.25 | 95.77 |
| Ni5H2NZ | 9.49 | 94.38 |
| Sample | Al | Si | Na | K | Mg | Ca | Ti | Mn | Fe | Cu | Ni | Si/Al |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NZ | 6.43 | 31.09 | 0.89 | 0.87 | 0.57 | 1.82 | 0.26 | 0.04 | 2.31 | nd | nd | 4.83 |
| H2NZ | 6.45 | 31.90 | 0.49 | 0.82 | 0.54 | 1.12 | 0.26 | 0.04 | 2.33 | nd | nd | 4.95 |
| Cu5H2NZ | 6.25 | 32.58 | nd | 0.80 | 0.36 | 1.00 | 0.22 | 0.03 | 2.34 | 4.66 | nd | 5.21 |
| Ni5H2NZ | 5.13 | 27.76 | 0.38 | 0.73 | 0.61 | 0.97 | 0.21 | 0.05 | 2.37 | nd | 5.93 | 5.42 |
| Sample | SBET (m2·g−1) | Pore Size (nm) | Vmicropore (cm3·g−1) | Vmesopore (cm3·g−1) |
|---|---|---|---|---|
| NZ | 160.59 | 1.77 | 0.063 | 0.13 |
| H2NHZ | 111.73 | 1.71 | 0.042 | 0.11 |
| Cu5H2NZ | 99.34 | 1.55 | 0.047 | 0.10 |
| Ni5H2NZ | 132.30 | 1.55 | 0.052 | 0.10 |
| Area [μmol·g−1] | Peak Temperature [°C] | ||||
|---|---|---|---|---|---|
| Sample | Weak Sites | Strong Sites | Total Sites | Weak Sites | Strong Sites |
| NZ | 148.42 | 50.40 | 198.82 | 190.1 | 382.6 |
| H2NZ | 165.48 | 98.59 | 264.07 | 193.2 | 338.8 |
| Cu5H2NZ | 197.82 | 92.54 | 290.30 | 194.0 | 390.3 |
| Ni5H2NZ | 190.51 | 124.80 | 315.31 | 195.3 | 397.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venegas-Vásconez, D.; Orejuela-Escobar, L.; Valarezo-Garcés, A.; Guerrero, V.H.; Tipanluisa-Sarchi, L.; Alejandro-Martín, S. Biomass Valorization through Catalytic Pyrolysis Using Metal-Impregnated Natural Zeolites: From Waste to Resources. Polymers 2024, 16, 1912. https://doi.org/10.3390/polym16131912
Venegas-Vásconez D, Orejuela-Escobar L, Valarezo-Garcés A, Guerrero VH, Tipanluisa-Sarchi L, Alejandro-Martín S. Biomass Valorization through Catalytic Pyrolysis Using Metal-Impregnated Natural Zeolites: From Waste to Resources. Polymers. 2024; 16(13):1912. https://doi.org/10.3390/polym16131912
Chicago/Turabian StyleVenegas-Vásconez, Diego, Lourdes Orejuela-Escobar, Alfredo Valarezo-Garcés, Víctor H. Guerrero, Luis Tipanluisa-Sarchi, and Serguei Alejandro-Martín. 2024. "Biomass Valorization through Catalytic Pyrolysis Using Metal-Impregnated Natural Zeolites: From Waste to Resources" Polymers 16, no. 13: 1912. https://doi.org/10.3390/polym16131912
APA StyleVenegas-Vásconez, D., Orejuela-Escobar, L., Valarezo-Garcés, A., Guerrero, V. H., Tipanluisa-Sarchi, L., & Alejandro-Martín, S. (2024). Biomass Valorization through Catalytic Pyrolysis Using Metal-Impregnated Natural Zeolites: From Waste to Resources. Polymers, 16(13), 1912. https://doi.org/10.3390/polym16131912